
Abstract
In order to extract structural and electronic properties of BxIn1 – xAs ternary alloys and enrich the database of materials based on boron and indium, we have used full-potential augmented plane wave (FP-LAPW) method through the density function theory (DFT) and within generalized gradient approximation (GGA), local density approximation (LDA), and Tran–Blaha modified Becke–Johnson approximation (TB–mBJ). We have optimized the cohesive energy of our binary compound and ternary alloys versus volume of the unit cell firstly, and we have found that the optimum volume, lattice parameter, and the bulk modulus vary for different boron concentrations. Using DFT–mBJ calculations, we found that InAs possess direct band-gap energy and an indirect gap semiconductor for BAs and B0.75In0.25As. However, B0.25In0.75As and B0.5In0.5As ternary alloys have a metallic and semi metallic characters, respectively. We also studied the optical properties of our BAs and InAs binary and B0.75In0.25As ternary semiconductors and their behaviors are also investigated under the application of hydrostatic pressure in a range of 0 to 25 GPa. In summary, we conclude that the incorporation of boron atom in InAs increase its hardness and affects the band-gap energy considerably, and therefore provides a novel research perspective. We note that InAs binary compound loses its semiconductor character and becomes semi-metal at 5 GPa.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 INTRODUCTION
Researchers were found compelled to meet the increasing requirements of companies and enterprises in different field, in the recent few years. Study, characterize, analyze, predict, and develop new materials for new applications are the most important preoccupation of laboratories by means of theoretical and experimental works.
In the parallel with the progress made in technology and growing techniques of semiconductors, researchers are invited to follow this evolution and must devote many works to predict new materials with structural and electronic properties that can enhance the quality of various devices.
Semiconductor materials containing the atom of indium (In) or that of arsenic (As) find an important place in current research [1–5], for their attractive properties in many applications [6–9].
Among this group, InAs and BAs have different structural and electronic properties that we can combine in ternary alloy BxIn1 – xAs, with the objective to obtain new materials with controllable properties. These will serve as another alternative for optoelectronic devices and extend application domain.
In this work, we focus on determining the structural, electronic, and optical properties of BAs, InAs compounds, and their ternary BxIn1 – xAs alloys, using a full-potential linearized augmented plan-wave method (FP-LAPW). The behavior of these materials under hydrostatic pressure has also been studied in a pressure range from 0 to 25 GPa.
2 METHOD OF CALCULATION
In this contribution, we have used the density functional theory (DFT) via a full-potential linear augmented plane wave implemented in WIEN2k code [10], to calculate the structural and electronic properties of BxIn1 – xAs. About the exchange-correlation energy, we have employed the generalized approximation (GGA) of Perdew–Burke–Enzerhof (PBE98) and the local density approximation (LDA) for structural properties. In addition to these two approximations, the modified Becke–Johnson approximation has been applied for electronic properties. The space is divided into two regions, an interstitial region (IR) and a muffin tin region centered at the atomic sites. In the first region, the basis sets consists of plane waves. In the second one, the basis sets are described by radial solutions of the one particle Schrödinger equation. For the matrix size, we used a parameter RMTKmax = 7, where RMT denotes the minimum radius of the sphere muffin tin in unit cell, and Kmax gives the magnitude of the largest K vector in the plane wave expansion. The muffin tin radii of B, In, and As are chosen to be 1.45, 2.1, and 2 Bohr radius, respectively.
3 RESULTS AND DISCUSSIONS
3.1 Structural Properties
Firstly, we start by optimizing the cohesive energy of our binary compounds (BAs and InAs) and their ternary alloys BxIn1 – xAs in zinc-blende (ZB) structure with GGA and LDA approximations. Our compound have been modeled for the concentrations x = 0, 0.25, 0.5, 0.75, 1 with periodically repeated super-cells of eight atoms per unit cell.
We explored the structural optimization by minimizing the total energy with respect to the parameters and the atomic positions, in order to obtain the equilibrium lattice parameter and the bulk modulus for the binary compounds and our ternary alloys.
The total energy ET, given by our calculation, as a function of unit cell volume was fitted by Murnaghan’s equation of state [11]:
The results compared with other available experimental and theoretical works are cited in Table 1. Our GGA results of binary are in best agreement with values given in [12–20].
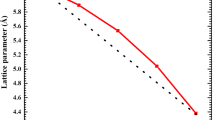
Figure 1 shows the variation of the calculated equilibrium lattice parameter as a function of concentration x for BxIn1 – xAs alloys, which indicates a small deviation from Vegard’s law with an upward bowing parameter equal to 1.344 Å for the GGA approximation and 1.312 Å for the LDA one.

Composition dependence of the calculated lattice constants of ZB BxIn1 – xAs alloys.
The bowing parameter is determined by fitting the calculated values with a polynomial function. The important difference between the lattice parameters of our binary compounds (BAs and InAs) is at the origin of such deviation.
In Fig. 2 that indicates the composition dependence of the bulk modulus of BxIn1 – xAs alloys, the values of the bulk modulus increase with the boron concentration, meaning that the addition of boron to InAs improves their hardness in good agreement with others works.

Composition dependence of the calculated bulk modulus of ZB BxIn1 – xAs alloys.
For this parameter, we can remark a considerable deviation from LCD (linear composition dependence) with downward bowing equal to –16.2777 and ‒22.605 GPa with GGA and LDA, respectively. The important bowing values are also mainly due to the significant mismatch of the bulk modulus of BAs and InAs.
3.2 Electronic Properties
3.2.1. Band structure. We have performed a first-principle calculation for electronic properties, in order to study the electronic band structure of our binary compounds (InAs and BAs) and their ternary alloys (BxIn1 – xAs) in ZB structure. We have used the generalized gradient and the modified Becke–Johnson approximations using in full-potential linearized augmented plane wave (FP-LAPW) implemented in WIEN2k code, with the aim to know more opportunity to improve the electronic characteristics of optoelectronic devices using this category of materials and extend their applications fields. It should be noted that we have found difficulties to compare our results with few works in the literature devoted to study the el-ectronic properties of this class of materials [1, 10, 12, 21].
After full optimization and the calculation of the structural parameters, the electronic band structures computed along some high-symmetry directions in the Brillouin zone have been shown in Figs. 3–7.

Non-relativistic mBJ band structure of ZB InAs compound.

Non-relativistic mBJ band structure of ZB B0.25In0.75As.

Non-relativistic mBJ band structure of ZB B0.5In0.5As.

Non-relativistic mBJ band structure of ZB B0.75In0.25As.

Non-relativistic mBJ band structure of ZB BAs compound.
The zero energy is selected to coincide with the top of valence band, for our studied compounds. We can note that the calculations indicate that the valence band maximum (VBM) is located at Γ point and the conduction band minimum (CBM) at Γ point for x = 0, at R point for x = 0.75, and at X point for x = 1, which means a direct gap semiconductor for InAs, and an indirect gap semiconductor for BAs in good agreement with [22] in the direction Γ–X, and an indirect gap semiconductor for B0.75In0.25As in the direction Γ–R.
We found that our compound has a metallic and semi-metallic behavior for B0.25In0.75As and B0.50In0.50As, respectively. In order to obtain the bowing parameter, we have fitted the non-linear variation of the calculated direct and indirect band gaps in terms of concentration with polynomial function, which obey to the following equations:
Figures 8 and 9 show the variation of the fitted direct and indirect band gaps.

Composition dependence of the mBJ band gaps for BxIn1 – xAs alloys.

Composition dependence of the fitted mBJ band gaps for BxIn1 – xAs alloys.
It should be noted that the values of Eg represent a deviation from the linear behavior about 7.95 and 1.34 eV for Γ–Γ and Γ–X band gap, respectively. We can also note that the addition of a weak percentage of boron can significantly increase the width of the gap.
Our computed results compared with the few available data are summarized in Table 2, which indicates a good agreement.
3.2.2. Density of states. By using FP-LAPW method with GGA approximation, we have also calculated the total and partial density of states for BAs, InAs, and B0.75In0.25As. The density of states function gives the number of states that are available in a system and is essential for determining the carrier concentrations and energy distributions of carriers, and from this function we can clearly understand the contribution of each atom compared to the total density.
Because the calculations of the density of states request a high degree of precision with use of K-points mesh in the first Brillouin zone. In fact, we used a k‑mesh of 250 special K-points for B0.75In0.25As ternary alloy and 1000 special K-points for binary BAs and InAs compounds. We have distinguished the B (1s2), In (1s22s22p63s23p63d104s24p6), and As (1s2 2s22p63s23p6) inner-shell electrons from valence electrons of B (2s22p1), In (4d105s25p1), and As (3d104s24p3) shells.
The spectrum of density of states of the binary BAs, shown in Fig. 10, present three regions, two in valence region below the Fermi level EF and one in conduction band (CB) above EF.

Total and partial DoS of BAs.
The lower valence band (LVB) is dominated by As‑4s, the As-4p participates in upper valence band (UVB), with a few contribution of B-2s and B-2p. CB is mainly formed by B-2s and As-4p with a small contribution of As-3d.
For InAs, the DOS spectrum, displayed in Fig. 11, indicates also three main regions, LVB principally dominated by As-4s with a few participation of In-4d. In-5s and In-5p contribute in the other valence region.

Total and partial DoS of B0.75In0.25As.
The third region is formed by a mixture of In-5p, As-4p, In-4d, and As-3d.
Our B0.75In0.25As ternary alloy possess a DoS with a similar topography, composed of three region as shown on Fig. 12, with two valence regions (LVB and UVB) below the Fermi level and one in CB above EF.

Total and partial DoS of InAs.
From left to right, in the first region (LVB) all the different atoms contribute with a comparable order. The next region (UVB), the orbitals B-2s, In-5s, and As-4s are at the origin of its formation, with a contribution of the orbital p of B, In, and As atoms toward the end of this region. In CB, the last region, we have a contribution of all orbitals with a comparable order. We can see that the addition of the boron atom can changed the topography of the different contribution, and at 75% boron concentration, the gap of our alloy increase considerably compared to that of InAs, which means that our alloy can be very useful for various applications in future.
3.3 Optical Properties
To understand the behavior of electrons and ions of our binary compounds and our B0.75In0.25As ternary alloy in interaction with an electromagnetic wave, we will be interested on optical properties. The dielectric function is expressed as ε(ω) = ε1(ω) + iε2(ω), where ε2(ω), the imaginary part, can be calculated from the momentum matrix elements combining the occupied and unoccupied states, which is given by
The integral is over the first Brillouin zone. The momentum dipole elements are given as
where δ is the potential vector defining the electric field. The Mcv(k) are matrix elements for direct transitions between the valence band, uvk(r) and conduction band, uck(r) states.
Using the Kramers–Kronig relationships, we can deduct the real part ε1(ω) of the dielectric function, which is given by the following formula:
where P implies the principal value of the integral.
Using a FP-LAPW calculation, we can calculate the real and imaginary parts of the frequency-dependent dielectric function, and we can easily deduce the refractive index of our binary compounds BAs and InAs and our B0.75In0.25As ternary alloy by using of the following expression:
Figures 13 and 14 show the real and the imaginary parts of the dielectric function as well as refractive index spectrum for a radiation up to 14 eV for binary InAs, BAs compounds and their ternary B0.75In0.25As alloy.

The real and imaginary parts of the dielectric function of the ZB InAs, BAs, and B0.75In0.25As compounds.

The refractive index of the ZB InAs, BAs, and B0.75In0.25As compounds.
From these figures, we can clearly see that the optical spectrum doesn’t vary remarkably.
We can see from Fig. 13, for the real part, that the energy of the main peak in the ultraviolet region shifts toward lowers energy values in the visible regions, from 3.75 eV for BAs to 2.32 eV for B0.75In0.25As to finally reach the value of 1.75 eV for InAs. The origin of this decrease is related to the electronic band structures of the material.
The frequencies, for which ε1(ω) = 0, correspond to a maximum of absorption and absence of dispersion, and are located in the ultraviolet region. We noticed a decrease from 5.76 to 4.56 to 4.09 eV for BAs, B0.75In0.25As, and InAs, respectively. For higher frequencies corresponding to ε1(ω) < 0, the electro-magnetic waves don’t propagate, and our compounds have an important reflectivity inducing a metallic behavior.
From our calculations results, we can also notice that ε1(0) increases from BAs to B0.75In0.25As to InAs, which is due to the fact that the energy gap decreases from BAs to B0.75In0.25As to InAs.
The imaginary part values of the dielectric function based on the photon energy are also shown in Fig. 13. The critical energy point occurs at about 3.11 and 1.51 eV for BAs and B0.75In0.25As, respectively. These values correspond to the threshold for direct optical interband transitions at the r point from the top of the valence band to the bottom of the conduction band. In addition, the main peaks are observed at energies of 5.65, 4.18, and 4.15 eV for the BAs, B0.75In0.25As, and InAs compounds, respectively.
The refractive index displayed in Fig. 14, which is defined as a dimensionless number, is obtained from ε1 and ε2 values. At the frequency ω = 0, the static refractive index can be obtained using the formula [30]
In order to compare our results with others approaches, the refractive index has been also obtained using other empirical models related to the fundamental energy band gap:
(1) Herve and Vandamme’s empirical relation [31]:
with A = 13.6 eV and B = 3.4 eV.
(2) The expression proposed by Ravindra et al. [32]:
where α = 4.084 and β = 0.62 eV–1.
In Table 3, the values of the refractive index and the zero-frequency real part, ε1(0) of the dielectric function, are given, which can be considered as a measure of the interaction force between the electronic states of the valence and conduction band under an external electric field, of BAs, InAs, and B0.75In0.25As compounds, along with the available experimental and theoretical results.
Our obtained results are in reasonable agreement with other theoretical works. However, for B0.75In0.25As there is no experimental data in the literature for comparison.
For InAs compound, the first peak of the n(ω) spectrum is located at 1.80 eV with a magnitude of 4.17 and a second major peak is located at around 3.82 eV that corresponds to a magnitude of 3.82, a small peak is also observed at energy of 5.40 eV with a magnitude of 1.92. However, the spectrum of the refractive index increases from its static value at zero frequency, for BAs, until 5.45 eV that corresponds to a magnitude of 4.48. Beyond this energy, the magnitude of refractive index decrease dramatically. For the B0.75In0.25As compound, the refractive index display two minor peaks located at energies of 3.08 and 4.26 eV are observed where the corresponding magnitudes are 4.30 and 3.98, respectively.
3.4 Pressure Effect on Electronic and Optical Properties
The arrangement of atoms in a material can be changed under the application of an external perturbation, for example, when we apply hydrostatic pressure to a solid with a given crystal structure, the arrangement of these atoms is modified and probably we pass from a crystalline phase to another. Therefore, the variation of the inter-atomic distance under pressure generally affects its behavior in relation to these electronic properties, structural phase transitions and optical characteristics, etc.
We have studied the effects of pressure on the properties of our binary compound and their ternary alloys using the FP-LAPW method within the WC-GGA and mBJ-GGA approaches and varying the pressure from 0 to 25 GPa with a step size of 5 GPa.
The lattice parameter can be expressed from the relationship given by [39]:
where B is the bulk modulus, B' is the pressure derivative of the bulk modulus, and a(P) is the lattice parameter at pressure P.
Using the same method, we have also calculated the band gap energies and the results of direct and indirect gaps for each compound under pressure which are shown in Figs. 15a, 15b, and 15c.

The direct and indirect band gaps energies versus pressure of the BxIn1 – xAs alloy compound at concentrations (a) x = 0, (b) x = 1.00, and (c) x = 0.75.
From these figures, we can see that the band gap energies EΓ–X decrease by increasing the applied pressure. However, concerning the EΓ–Γ direct gap, our three alloys have a different behavior under the effect of pressure; the gap direct of B0.75In0.25As ternary rapidly decreases under the applied pressure. On the other hand, the value of direct gap of BAs compound decreases in the pressure range from 0 to 5 GPa and then it slowly increases when the applied pressure increases. Furthermore, the direct gap of our InAs binary equals zero at 5 GPa, and therefore it will have a behavior of a semi-metal material (Eg = 0 eV); beyond this pressure, it increases to reach 0.70 eV at 25 GPa.
The first important result is that the applying of a pressure in the range of 0–5 GPa significantly reduced the direct gap of InAs. Consequently, it changes its behavior and it has a semi-metallic behavior. Secondly, our semiconductor alloy B0.75In0.25As remains a direct band gap semiconductor under the range considered for pressure, where the value of its gap decreases with increasing applied pressure.
In Table 4, we summarize the calculated values of the direct and indirect gaps of our BAs and InAs binary compounds and B0.75In0.25As ternary alloys under hydrostatic pressure in the interval from 0 to 25 GPa.
The pressure dependence of the principal band gap energies is given by the empirical formula [39]
where Eg(0) is the band gap energy at zero pressure, α and β are the first- and second-order pressure coefficients, respectively.
Using a non-linear quadratic function, we fitted the results of the direct and indirect band gap of our BAs and InAs binary compounds and B0.75In0.25As ternary alloys.
The quadratic functions for fitting the band gap energies, for each compound, are given as follows:
In order to study the effect of pressure on the optical properties, in particular on the dielectric function and the refractive index, we also used the same method FP-LAPW. For the different concentrations x, we varied the lattice parameter which is also a function of the applied pressure. Figure 16 shows the calculated real and imaginary parts of the dielectric function under different values of pressure for the BAs, InAs compounds and B0.75In0.25As alloy.

The calculated real and imaginary parts of the dielectric function under different values of pressure for the BAs, InAs compounds and B0.75In0.25As alloy.
For the real part ε1(ω), as shown in Figs. 16a, 16b, and 16c, we can clearly notice that the spectra at higher pressures keep similar forms to those at ambient pressure with a small displacement towards the higher energies when the pressure increases. For B0.75In0.25As, we can see the appearance of a small peak for high pressure observed at energy of 4.56 eV.
We can also notice that ε1(0) decrease significantly when the pressure applied increases for InAs, which is due to the fact that the energy gap decreases in the same conditions.
For the imaginary parts of the dielectric function, as shown in Figs. 16a', 16b', and 16c', similar forms with a small displacement towards the highest energies when the applied pressure increased was noted, where the amplitude of the spectrum was relatively higher than that with ambient pressure. The energy levels relative to the maximum of the valence band and the minimum of the conduction band under pressure have been displaced, consequently the optical transitions between these energy levels will also be changed, which can be considered at the origin of such changes.
The variation in the refractive index at ambient pressure and under high pressure is also calculated and shown in Fig. 17 spectra that are also of similar forms with the exception of InAs for which there is a remarkable decrease in the static refractive index under pressure; that can be justified by the fact that the pressure considerably affects the band gap energies and affect the optical transitions.

The calculated refractive index under different values of pressure for the BAs, InAs compounds and B0.75In0.25As alloy.
4 CONCLUSIONS
We have presented a FP-LAPW simulation in order to explore structural, electronic, and optical properties of BAs and InAs binary compounds and their BxIn1 ‒ xAs ternary alloys under hydrostatic pressure. Our main results agree very well with other experimental and theoretical data. We can note that the most important results are:
• The incorporation of boron in InAs increases its hardness and affects the band gap energy considerably.
• The addition of low concentration of boron (lower than 25%) reduces the gap, and for the concentrations of more than 50%, the value of the direct gap increases significantly.
• BxIn1 – xAs have an indirect band gap for concentration of boron superior to 77%.
• There has been a significant reduction in the gap of InAs in the range from 0 to 5 GPa and it becomes semi-metal at 5 GPa with a decrease in its refractive index in the same range.
• BxIn1 – xAs alloy may be an alternative choice for optoelectronic applications at certain concentrations, for example from 0 to 25% of boron at ambient pressure and for 75% under hydrostatic pressure up to 25 GPa.
REFERENCES
N. Chimot, J. Even, H. Folliot, and S. Loualiche, Phys. B (Amsterdam, Neth.) 364, 263 (2005).
F. El Haj Hassan and H. Akbarzadeh, Mater. Sci. Eng. B 121, 171 (2005).
M. Guemou, B. Bouhafs, A. Abdiche, R. Khenata, Y. Al Douri, and S. Bin Omran, Phys. B (Amsterdam, Neth.) 407, 1292 (2012).
R. Ahmed, S. J. Hashemifar, H. Akbarzadeh, M. Ahmed, and F. Aleem, Comput. Mater. Sci. 39, 580 (2007).
M. Ferhat, A. Zaoui, M. Certier, and H. Aourag, Phys. B (Amsterdam, Neth.) 252, 229 (1998).
R. Bhat, P. S. Dutta, and S. Guha, J. Cryst. Growth 310, 1910 (2008).
V. N. Brudnyi, N. G. Kolin, and A. I. Potapov, Semiconductors 37, 390 (2003).
J. A. Perri, S. Laplaca, and B. Post, Acta Crystallogr. 11, 310 (1958).
T. L. Chu and A. E. Hyslop, J. Appl. Phys. 43, 276 (1972).
P. Blaha, K. Schwarz, G. K. H. Madsen, D. Kvasnicka, and J. Luitz, WIEN2k, An Augmented Plane Wave Plus Local Orbital Program for Calculating Crystal Properties (Vienna Univ. Technol., Vienna, Austria, 2001).
F. D. Murnaghan, Proc. Natl. Acad. Sci. U. S. A. 30, 244 (1947).
Y. Yan, Q. Wang, W. Shu, Z. Jia, X. Ren, X. Zhang, and Y. Huang, Phys. B (Amsterdam, Neth.) 407, 4570 (2012).
R. W. G. Wyckoff, Crystal Structures, 2nd ed. (Krieger, Malabar, 1986).
K. H. Hellwege and O. Madelung, Landolt–Börnstein, New Series, Group III (Springer, Berlin, 1982), Vol. 17, Pt. A.
M. Vubcevich, Phys. Status Solidi B 54, 219 (1972).
H. Meradji, S. Labidi, S. Ghemid, S. Drablia, and B. Bouhafs, Phys. Proc. 2, 933 (2009).
H. Bross and R. Bader, Phys. Status Solidi B 191, 369 (1995).
O. Madelung, Landolt-Börnstein, New Series, Group III (Springer, Berlin, 1982), Vol. 17a.
R. Wentzcovitch, M. L. Cohen, and P. K. Lam, Phys. Rev. B 36, 6058 (1987).
M. Briki, M. Abdelouhab, A. Zaoui, and M. Ferhat, Superlatt. Microstruct. 45, 80 (2009).
N. Tayebi, K. Benkabou, and F. Z. Aoumeur-Benkabou, Phys. B (Amsterdam, Neth.) 407, 2739 (2012).
P. P. Paskov, J. Appl. Phys. 81, 1890 (1997).
T. Hofmann, M. Schubert, G. Leibiger, and V. Gottschalch, Appl. Phys. Lett. 90, 182110 (2007).
S. Adachi, J. Appl. Phys. 61, 4869 (1987).
H. Baaziz, Z. Charifi, and N. Bouarissa, Mater. Chem. Phys. 68, 197 (2001).
M. Merabet, D. Rached, R. Khenata, S. Benalia, B. Abidri, N. Bettahar, and S. BinOmran, Phys. B (Amsterdam, Neth.) 406, 3247 (2011).
A. Zaoui and F. El Haj Hassan, J. Phys.: Condens. Matter 13, 253 (2001).
V. M. Daniel’tsev, N. V. Vostokov, Yu. N. Drozdov, M. N. Drozdov, A. V. Murel, D. A. Pryakhin, O. I. Khrykin, and V. I. Shashkin, J. Surf. Invest. 2, 514 (2008).
M. Rabah, B. Abbar, Y. Al-Douri, B. Bouhafs, and B. Sahraoui, Mater. Sci. Eng. B 100, 163 (2003).
F. Tran and P. Blaha, Phys. Rev. Lett. 102, 226401 (2009).
P. J. L. Hervé and L. K. J. Vandamme, J. Appl. Phys. 77, 5476 (1995).
N. M. Ravindra, S. Auluck, and V. K. Srivastava, Phys. Status Solidi B 93, 155 (1979).
D. E. Aspnes and A. A. Studna, Phys. Rev. B 27, 985 (1983).
M. Othman, E. Kasap, and N. Korozlu, J. Alloys Compd. 496, 230 (2010).
N. Amrane and M. Benkraouda. J. Adv. Phys. 13, 5041 (2017).
M. I. Ziane, Z. Bensaad, B. Labdelli, and H. Bennacer, Sens. Transducers 27 (Spec. Iss.), 374 (2014).
R. Wentzcovitch, M. L. Cohen, and P. K. Lam, Phys. Rev. B 36, 6058 (1987).
H. Meradji, S. Labidi, S. Ghemid, S. Drablia, and B. Bouhafs, Phys. Proc. 2, 933 (2009).
S. Adachi, Properties of Group IV, III–V, and II–VI Semiconductors (Wiley, New York, 2005), Chap. 2.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of interest.
Rights and permissions
About this article

Cite this article
Guemou, M., Khelil, M. & Abdiche, A. Pressure Effect Study on the Electronic and Optical Properties of BxIn1 – xAs Alloys Using DFT Calculation. Phys. Solid State 62, 1815–1829 (2020). https://doi.org/10.1134/S106378342010011X
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S106378342010011X