
Abstract
By following one-pot synthesis, an acridinediones derivative compound 3,3,6,6-tetramethyl-9-(4-methoxyphenyl)-10-(4-chlorophenyl)-1,8-dioxodecahydroacridine dimethyl sulfoxide, C30H32O3ClNS·C2H6OS has been synthesized using a green protocol and its crystal structure is determined by X-ray structure analysis. The crystals are Triclinic with space group P\(\bar {1}\), Z = 2. The crystal structure was solved by direct method and refined by full matrix least squares procedure to a final R value of 0.0587 for 4137 observed reflections. The pyridine ring adopts flattened boat conformation whereas both cyclohexene adopts envelope conformations. The crystal structure is stabilized by C–H⋅⋅⋅O and C–H⋅⋅⋅π hydrogen bonds involving the DMSO solvent molecule which are further understood through Hirshfeld surface analysis.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
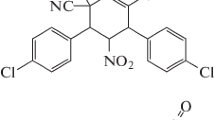
Acridinediones containing 1,4-dihydropyridine nucleus along with remarkable physical and chemical properties exhibits large variety of biological activity and finds utility in pharmaceutical and dye industry. This 1,4-dihydropyridine unit shows biological activities such as neuroprotectant, antihypertension, antidiabetics, anticancer, geroprotective [1]. In addition they are calcium channels blockers and also act as therapeutic agents [2–6]. It is also used as laser dyes having very high efficiencies of photo initiators [7, 8]. In view of the significant biological activity associated with the acridine derivatives, various methods have been reported for their synthesis. Most common among these involve the one-pot three component condensation of cyclic 1,3-dicarbonyls, arylaldehydes and ammonium acetate or anilines. For example, Shen et al. have reported one-pot three-component synthesis of 1,8-dioxo-9,10-diaryl-decahydroacridines in water in the presence of the Brønsted acidic imidazolium salts containing perfluoroalkyl tails [9], Aday et al. have reported the synthesis of acridinedione derivatives via a one-pot multi-component condensation of dimedone, various aromatic aldehydes, and various aromatic amines using highly monodisperse platinum nanoparticles supported onto reduced graphene oxide (Pt NPs@rGO) as a recyclable heterogeneous catalyst [10] and Vaid et al. have prepared MPTMS-functionalized silica immobilized with biphenyl-2,2'-dioic acid as an efficient catalyst for the one-pot synthesis of acridine-1,8-dione derivatives [11]. Here, we wish to report the synthesis, characterization and crystal structure of a fully functionalized acridine derivative, namely 3,3,6,6-tetramethyl-9-(4-methoxyphenyl)-10-(4-chlorophenyl)-1,8-dioxo-decahydroacridine dimethyl sulfoxide (Fig. 1) using a green procedure which involve the one-pot reaction of 4-methoxybenzaldehyde, dimedone and 4-chloroaniline in presence of CStar@SiO2-TfOH [12]. This method have several advantages such as low catalyst loading, use of non-toxic, highly active, stable and green solid acid catalyst, cleaner reaction profile, simple work-up procedure, good to excellent yield, recyclability and use of environmentally benign solvent.

Chemical structure of 3,3,6,6-tetramethyl-9-(4-methoxyphenyl)-10-(4-chlorophenyl)-1,8-dioxodecahydroacridine dimethyl sulfoxide (I).
EXPERIMENTAL
Synthesis
In a typical experimental procedure, a mixture of 4-methoxybenzaldehyde (1 mmol), dimedone (2 mmol), 4-chloroaniline (1 mmol) and CStar@SiO2-TfOH (0.1 g) were dispersed in water (5 mL) and stirred at 80°C for an appropriate time. After completion of the reaction as monitored by TLC, the catalyst was separated by filtration and washed with ethyl acetate (3 × 10 mL) and deionized water (3 × 10 mL). It was dried in an oven at 80°C for 24 h. The reaction mixture was cooled to room temperature, diluted with ethyl acetate (20 mL) and washed with brine solution followed by drying over anhydrous Na2SO4. Finally, the product was obtained after removal of the solvent under reduced pressure followed by crystallization in ethyl acetate (yield 92%; m.pt. 254–255°C).
Single crystals suitable for X-ray diffraction were obtained by slow evaporation of DMSO solution.
Characterization
Yellow coloured crystals; m.pt. 254–255°C; (lit. m.pt. 255–257°C); [10] 1H NMR (400 MHz, DMSO-d6): δ 0.72 (s, 6H, 2CH3), 0.89 (s, 6H, 2CH3), 1.74–1.78 (d, 2H, J = 16 Hz, CH2), 1.98–2.02 (d, 2H, J = 16 Hz, CH2), 2.16–2.21 (m, 4H, 2CH2), 3.69 (s, 3H, OCH3), 4.96 (s, 1H, CH), 6.79–6.81 (d, 3H, J = 8 Hz, ArH), 7.19–7.21 (d, 3H, J = 8 Hz, ArH), 7.67–7.69 (d, 2H, J = 8 Hz, ArH); 13C NMR (100 MHz, DMSO-d6): δ 26.4, 29.8, 32.1, 32.4, 41.4, 50.1, 113.1, 117.4, 128.5, 130.1, 130.8, 139.1, 145.8, 151.1, 156.8, 195.9; MS (ESI): 488.24 [M – 1], 491.24 [M + 2]+.
Crystal Structure Determination and Refinement
The crystallographic data are summarized in Table 1. The molecular structure solution was obtained by direct method procedure as using SHELXT [13]. The cell dimensions were determined by least-squares fit of angular settings of 2544 reflections in the θ range 2.1170° to 27.3560°. The value of Rint = 0.0215 and Rσ = 0.0473 shows satisfactory quality of the data. Five cycles of full-matrix least-squares refinement was carried out and it brought the final R-factor to 0.0587. All non-hydrogen atoms of the molecule were located in the best E-map and refined in anisotropic approximation using SHELXS [13]. The position of all the Hydrogen atoms bonded to C atoms were geometrically fixed and allowed to ride on the corresponding non-H atoms (C–H = 0.93–0.96 Å, and Uiso(H) = 1.5Ueq of the attached C atoms for methyl groups and 1.2Ueq(C) for other H atoms. The residual electron density in the final difference Fourier map between –0.491 < ∆ρ < 0.773. The geometry of the title molecule was calculated using WinGX [14], PARST [15], and PLATON [16] software. Hirshfeld surfaces are mapped using dnorm, the shape index and curvature presented in this paper were generated using Crystal Explorer 17 [17].
Crystallographic information has been deposited with Cambridge Crystallographic Data Centre, CCDC no. 2009450. The data can be obtained from through www.ccdc.cam.ac.uk/data_request/cif by e-mailing data request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallography Data Centre, 12 Union Road, Cambridge, CB2 IEZ, UK. Fax: +44(0) 1223-336033.
RESULTS AND DISCUSSION
The molecular structure containing atomic labeling of the asymmetric unit, C30H32O3ClNS·C2H6OS is shown in Fig. 2 (ORTEP) [18] and the packing diagram as generated using PLATON [16] is shown in Fig. 3. X-ray structure analysis has shown that the asymmetric unit is one molecule of the compound and one molecule of solvent DMSO. Most of the geometrical parameter are normal [19] and are in close agreement with the reported solvent-free polymorph structure (C30H32ClNO3) [20]. An overlay diagram for (C30H32ClNO3) (II) and for molecule (I) is shown in Fig. 4. Significant bond lengths and bond angles are listed in Table 2.

The structure of the crystal (I), displacement ellipsoids are drawn at 50% probability level.

Packing view of molecules down to b-axis (noninteracting hydrogen atoms are omitted for clarity).

Overlay diagram for (C30H32ClNO3) (II) and molecule of (I).
The molecule (I) consists of two cyclohexene rings A and B bridged with pyridine unit through double bond lengths of C12–C13 = 1.354(3) Å and C11–C14 = 1.356(3) Å. In these cyclohexene rings A and B, the bond length of C1–O19 = 1.233(3) Å and C8–O20 = 1.227(3) Å are of typical C=O bond lengths. The geometrical parameters of nitrogen atom in the pyridine unit are N10–C13 = 1.400(3) Å, N10–C14 = 1.402(3) Å, N10–C22 = 1.449(3) Å; C13–N10–C14 = 120.0(2)° which on comparison with the reported polymorph shows quiet closeness (1.405(4) Å, 1.398(4) Å, 120.1(3)°, respectively). Moreover, the exocyclic bond angles at the ring junctions N10–C13–C4 = 116.7(2)°, N10–C14–C5 = 117.1(2)°, C9–C11–C8 = 118.4(2)°, and C9–C12–C1 = 118.6(2)° are significantly lesser than ideal value of 120°. All the endocyclic bond angles of the anisole and chlorobenzene rings lies between 113.7(2)° to 123.3(2)° and 108.0(2)° to 123.1(2)°, respectively. Whereas in case of pyridine ring all the bond angles lies in the range of 119.7(2)° to 121.6(2)°. The oxymethane group attached C30 atom of anisole ring have O34–C30 = 1.370(3) Å and O34–C35 = 1.432(4) Å which are quite similar to bond lengths of 1.387(4), 1.420(4) Å in reported structure (C30H32ClNO3) (II) [19]. In the chlorobenzene ring C25–Cl33 bond length is 1.741(3) Å. In the title molecule, the orientation of anisole and chlorobenzene rings with respect to the attached pyrazole and pyran rings is characterized by torsion angles C13–N10–C22–C27 = 92.3(3)° and C11–C9–C21–C32 = –65.6(3)° both in (+sc) configuration.
Here the central pyridine ring and both the cyclohexene rings A and B are non-planar having total puckering amplitudes, QT, of 0.2932(2), 0.4660(1), 0.4607(2) Å, respectively. The central pyridine ring shows flattened boat conformation, where atom C9 shows maximum deviation of –0.5509(3) from plane of mean deviation. Both the cyclohexene rings A and C shows envelope conformation with maximum deviation for C7 and C2 of 0.5429(3) and 0.5468(3) from planarity. The puckering parameters are ϕ = 179.31(2)° and θ = 73.57(2)° for pyridine ring, ϕ = 469.60(9)° and θ = 56.52(3)° for cyclohexene ring A, and ϕ = 250.37(6)° and θ = 123.48(2)° for cyclohexene ring B [21]. Both anisole ring and chlorobenzene ring are in perpendicular configuration with respect to the acridine ring system, as can be seen from the dihedral angle of 89.70(6)° and 89.02(6)°, respectively.
The DMSO solvent molecule plays an important role in the stability of the crystal structure. The crystal structure is stabilized by hydrogen bonds involving the solvent molecule, where carbon atom C52 of DMSO solvent acts as donor via H52A and H52B to oxygen atom O20 of the anisole ring, in C52–H52A⋅⋅⋅O32 and C52–H52B⋅⋅⋅O31 interaction. In addition the molecular packing is also stabilized with the help of weak C–H⋅⋅⋅π and Vander Waal’s forces. Fig. 3 shows the molecular packing in the unit cell viewed down the b-axis (PLATON program) [16]. The geometry of these interactions is presented in Table 3.
HIRSHFELD SURFACE ANALYSIS
In order to carry out the Hirshfeld surface analysis CrystalExplorer 17 program was used, for which Crystallographic information file (CIF) was used as input. The molecular Hirshfeld surfaces are created by splitting space in the crystal into a number of regions based on electronic distribution of atoms along the crystal. Figure 5 shows a comparison of Hirshfeld 3D surfaces of dnorm, shape index, curvedness in addition to 2D fingerprint plots for crystal (I) and (II), which are achieved by mapping dnorm over the Hirshfeld surface in the range –0.2377 to 1.5625. It indicates intermolecular interactions with the solvent DMSO molecule for (I). Transparent surfaces are shown to visualize functional groups present within the surface. Color areas symbolize closer contacts with a negative dnorm value, exactly the comparable distance when separating a van der Waals contact and zero or more distant contacts with a positive dnorm value, respectively. Large circular spots on dnorm surfaces indicate hydrogen bonds and other spots indicate bonding between other atoms. These regions correspond to positive and negative electrostatic potentials, respectively, which reveal the contribution of donor and acceptor interactions. The shape index on the Hirshfeld surface can be used to recognize complementary bumps and hollows, where the bump-shape corresponds to donor, and the hollow represents the acceptor of intermolecular interactions, and these stains here represents the regional molecular surfaces having contact and C–H⋅⋅⋅π interactions present in the crystal structure [22, 23].

A comparision of Hirshfeld surfaces mapped with dnorm for (I) (a), dnorm for (II) (b), shape index for (I) (c), shape index for (II) (d), curvedness for (I) (e), curvedness for (II) (f) and 2D finger print plots for (I) (g), 2D finger print plots for (II) (h).
2D fingerprint graphs are plotted by accumulating (di, de) pairs. The coloring for each collection has been taken as a function of the fraction over surface points, varying from several points to average and numerous points. A sketch of the full fingerprint is shown in light color [24]. The comparison of percentage contribution to the total Hirshfeld surface area, are shown in Table 4, where the H⋅⋅⋅H interaction is followed by O⋅⋅⋅H/H⋅⋅⋅O interaction as leading intermolecular interactions, in both crystals (I) and (II).
CONCLUSIONS
In summary, 3,3,6,6-tetramethyl-9-(4-methoxyphenyl)-10-(4-chlorophenyl)-1,8-dioxodecahydroacridine was synthesized via one-pot three component reaction in water at 80°C using CStar@SiO2-TfOH as a highly active, stable and green solid acid catalyst. The synthesized compound was characterized by using various spectroscopic techniques such as 1H-NMR, 13C-NMR and mass spectral analysis. Suitable single crystals were obtained by slow evaporation in DMSO solution, whose strucutre was elucidated using X-ray diffraction. The compound (I) crystallizes in the triclinic system with space group P\(\bar {1}\). The pyridine and cyclohexene rings adopt boat and envelope conformation respectively. Solvent molecules played an incomparable role in the formation of supramolecular structure through hydrogen bonded network which was understood in detail through Hirshfeld surface analysis.
REFERENCES
A. Rajacka, K. Yuvaraju, C. Praveen, et al., J. Mol. Catal. A 370, 197, (2013).
B. Wysocka-Skrzela and A. Ledochowski, Rocz. Chem. 50, 127 (1976).
A. Nasim and T. Brychey, Mutat. Res. 65, 261 (1979).
U. Thull and B. Testa, Biochem. Pharmacol. 47, 2307 (1994).
E. Reil, M. Scoll, K. Masson, et al., Biochem. Soc. Trans. 22, 62s (1994).
Y. Mandi, K. Regely, I. Ocsovszky, et al., Anticancer Res. 14, 2633 (1994).
S. Kumar and K. N. Singh, J. Heterocycl. Chem. 48, 69 (2011).
X. Fan, Y. Li, X. Zhang, G. Qu, et al., Heteroatom Chem. 18, 786 (2007).
W. Shen, L. M. Wang, H. Tian, et al., J. Fluor. Chem. 130, 522 (2009).
B. Aday, Y. Yildiz, R. Ulus, et al., New J. Chem. 40, 748 (2016).
R. Vaid, M. Gupta, and V. K. Gupta, J. Iran. Chem. Soc. 14, 2199 (2017).
S. Sharma, H. Sharma, S. Sharma, S. Paul, et al., ChemistrySelect 5, 2201 (2020).
G. M. Sheldrick, Acta Crystallogr. A 71, 3 (2015).
L. J. Farrugia, J. Appl. Crystallogr. 45, 849 (2012).
M. Nardelli, J. Appl. Crystallogr. 28, 659 (1995).
A. L. Spek, Acta Crystallogr. D 65, 148 (2009).
M. J. Turner, J. J. McKinnon, S. K. Wolff, et al., CrystalExplorer17 (Univ. of Western Australia, 2017).
L. J. Farrugia, J. Appl. Crystallogr. 30, 565 (1997).
F. H. Allen, O. Kennard, D. G. Watson, et al., J. Chem. Soc., Perkin Trans. 2, S1 (1987).
Y. Chen, W.-J. Hao, Z.-Q. Tang, B. Jiang, and C.-M. Li, Acta Crystallogr. E 63, o3934 (2007).
D. Cremer and A. J. Pople, J. Am. Chem. Soc. 97, 1354 (1975).
M. A. Spackman and D. Jayatilaka, CrystEngCommun 11, 19 (2009).
R. Hoffmann Inc. (New York, 1988).
J. J. McKinnon, A. S. Mitchell, and M. A. Spackman, Chemistry A 4, 2136 (1998).
ACKNOWLEDGMENTS
One of the authors (VKG) is thankful to University of Jammu, Jammu, India for financial support under RUSA 2.0.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Sharma, V., Sharma, S., Paul, S. et al. Synthesis, Characterization, and Crystal Structure of 3,3,6,6-Tetramethyl-9-(4-Methoxyphenyl)-10-(4-Chlorophenyl)-1,8-Dioxodecahydroacridine Dimethyl Sulfoxide. Crystallogr. Rep. 66, 1227–1232 (2021). https://doi.org/10.1134/S1063774521070154
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1063774521070154