
Abstract
Zinc and iron-based phosphate Zn3Fe4(PO4)6 was synthesized by solid-state reaction and characterized by single-crystal X-ray diffraction and energy dispersive X-ray spectroscopy. The structure of the title compound belongs to the Howardevansite family and crystallizes in the triclinic system, space group \(P\bar {1}\) with the following unit cell parameters a = 6.3447(2) Å, b = 7.8838(2) Å, c = 9.3019(2) Å, α = 104.726(1)°, β = 108.408(1)°, γ = 101.245(1)°, and Z = 1. The proposed structural model was supported by both charge distribution and bond valence sum validation methods. The crystal structure is built up from kinked chains of edge-sharing polyhedra following the sequence [Fe(1)2O10]–[Zn(1)O5]–[Fe(2)2O10]. These chains are held together through PO4 tetrahedra forming a stacked layers perpendicular to [010]. The resulting 3D framework defines tunnels running along [100], in which some Zn2+ cations are located.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
The research of new mixed metal phosphate with open-framework architectures is currently a subject of perpetual development in solid-state chemistry. Especially iron phosphate which are of particular interest because of their highly attractive potentials as a promising functional material for many practical applications including phosphate glass fibers (PGFs) engineering [1], catalysis [2], corrosion inhibition [3], biosensing [4], composites materials [5], electrode for Li-ion batteries [6], and immobilization of nuclear waste [7]. Moreover, these kinds of compounds exhibit fascinating structural chemistry, with a large variety of chemical compositions due to the tendency of iron (Fe2+or Fe3+) to adopt different coordinate geometries FeOn (n = 4, 5, or 6), as well as a versatile structural arrangements [8]. In fact, their impressive crystal structures lead generally to noteworthy, physical and/or chemical properties [9]. Typical examples are phases with an original magnetic behavior [10] or characterized by a spin-order-induced ferroelectricity and magnetoelectric effect [11], important electronic and optical attributes [12], Intervalence Charge Transfer (IVCT) phenomenon [13], and offering a facile redox reactions [14].
Based on our previous studies concerning iron phosphate materials, a number of new compounds with distinctive topological networks have been elaborated and structurally characterized. For instance we cite Ag1.655Co1.64Fe1.36(PO4)3 [15], Na2Co2Fe(PO4)3 [16], Na1.67Zn1.67Fe1.33(PO4)3 [17], and (M,Mn)3Fe(PO4)3 (M = Ca , Cd) [18] with an alluaudite-like structure or M'Ni2Fe(PO4)3 (M' = Ca [19], Sr [20]) adopting the α-CrPO4 structure type. Additional phases such M''Mn2Fe(PO4)3 (M '' = Ba, Sr) [21] and CaZn2Fe(PO4)3 [22] were also reported. During the investigation of BaO–ZnO–Fe2O3–P2O5 pseudo-quaternary system, and in an attempt to obtain the new compound BaZn2Fe(PO4)3 belonging to one of the latter structure types, Zn3Fe4(PO4)6 was successfully isolated. Its structure is similar to that of the natural Howardevansite NaCuFe2(VO4)3 [23]. Since the first description of the crystal structure of mixed-valence Fe-phosphate \({\text{Fe}}_{3}^{{2 + }}\)\({\text{Fe}}_{4}^{{3 + }}\)(PO4)6 [24], the number of the papers devoted to the Howardevansite-type \(A_{{3 + n}}^{{2 + }}B_{{4-n}}^{{3 + }}{{({\text{P}}{{{\text{O}}}_{{\text{4}}}})}_{{6-n}}}{{({\text{HP}}{{{\text{O}}}_{{\text{4}}}})}_{n}}\) (n ≤ 4) [25], has been increased significantly. As a result, multiple compounds covering a wide range of substitution exist e.g., AxBy(XO4)6 with (A = Fe3+, Mg2+, Cu2+, Zn2+, Co2+, Na+, and Ag+; B = Fe3+, Ti4+, Mg2+, Cu2+, Zn2+, Co2+; X = PV, VV, MoVI) and 6 ≤ x + y ≤ 8 [26], and \(M_{7}^{{{\text{II}}}}\)(PO4)2(HPO4)4 (MII = Mg [27], Fe [28], Mn [29], Co [30]).
Herein we report the synthesis of Zn3Fe4(PO4)6 via the classical solid-state route and its characterization, through single-crystal X-ray diffraction (XRD) supported by the energy-dispersive X-ray spectroscopy (EDS). The resulting structural model has been supported by means of bond-valence-sum (BVS) [31] and charge-distribution (CHARDI) [32] validation tools.
EXPERIMENTAL
Synthesis and Chemical Composition
Single crystals of Zn3Fe4(PO4)6 were grown by conventional solid-state reaction in air. In the first step, appropriate amounts of the starting precursors: Ba(NO3)2, Zn(NO3)2⋅6H2O, Fe(NO3)3⋅9H2O, and NH4H2PO4 were weighted in molar ratios Ba : Zn : Fe : P = 1 : 2 : 1 : 3 and subsequently dissolved in concentrated nitric acid. The resulting mixture was stirred at room temperature for 24 h and then evaporated under moderate stirring until dryness. The dry was carefully milled in an agate mortar, placed in a platinum crucible and preheated at 723 K for 24 h, to remove gaseous products. After regrinding, the residue was melted for 1 h at 1150 K and cooled down slowly to 950 K at rate of 5 K/h and then rapidly to room temperature by turning off the furnace. Finally, green crystal of Zn3Fe4(PO4)6 with parallelepiped shape and optimal size have been picked up under polarizing microscope for the X-ray diffraction (XRD) and the energy dispersive X-ray spectroscopy (EDS) experiments.
The qualitative analysis and morphology of the synthetized single crystals were performed using JEOL JSM-IT100 InTouchScope™ scanning electron microscope (SEM) equipped with energy dispersive X-ray spectrometer (EDS), operating under 20 kV accelerating voltage. The EDS analysis unambiguously confirms the exclusive presence of Zn, Fe, P, and O atoms. Furthermore, the Zn/P and Fe/P ratios are in agreement with its nominal composition resulting from single crystal X-ray diffraction study. The SEM micrograph of Zn3Fe4(PO4)6 crystal and EDS spectrum are given in Fig. 1. Elemental mapping SEM-EDS analysis was also carried out to appraise the elemental spatial repartition. As shown in Fig. 2 Zn, Fe, P, and O are evenly distributed in the sample.

SEM micrographs and EDS spectrum of Zn3Fe4(PO4)6.

EDS elemental mapping images showing the distribution of chemical elements in Zn3Fe4(PO4)6 crystal.
X-ray Diffraction Study
A suitable single crystal of dimensions 0.36 × 0.15 × 0.08 mm3, was selected for X-ray data collection at 296 K. The specimen was glued to a thin glass fiber and mounted on a Bruker X8 APEXII diffractometer equipped with an Apex II CCD area detector and a sealed-tube X-ray source (MoKα radiation, λ = 0.71073 Å). The APEX2 software was used for data collection over a full sphere of reciprocal space with φ and ω scan. Cell refinement and data reduction were carried out using SAINT program. A total number of 25997 reflections were measured in the range of 2.5° ≤ θ ≤ 35.6° of which 3740 were independent and 3638 reflections with the statistical criterion I > 2σ(I). Complementary absorption corrections were introduced semi-empirically from equivalent reflections on the basis of multi-scans using SADABS program [33]. The structure was solved by Direct Methods in the space group \(P\bar {1}\) (no. 2) and refined anisotropically by full-matrix least-squares on F 2 using SHELXT 2014/7 [34] and SHELXL2018/3 [35] programs implemented in the software package WinGX [36]. Following the proposed structural model, the final refinement cycle including all atomic positions and their anisotropic displacement parameters converged to the reduced reliability factors R1 = 0.017 and wR2 = 0.043. At this stage the remaining electron densities in the last Fourier-difference map are Δρmin = −1.195 and Δρmax = 0.615 eÅ−3 located at 0.30 Å from the Zn(2) site and 0.73 Å from the P(2) one, respectively.
Crystallographic data, X-ray data collection and structure refinement details are compiled in Table 1. The coordinates of the basic atoms and their displacement parameters are listed in Table 2. Selected interatomic distances are summarized in Table 3.
RESULTS AND DISCUSSION
Validation of Structural Model
The powerful analysis tools (BVS) and (CHARDI), were chosen to confirm the validity as well as to prove the adequacy of the proposed structural model of Zn3Fe4(PO4)6. Firstly, the (BVS) concept was introduced successfully to predict the bond lengths, estimates the quality of the crystal structure determination as well as the bond strain. In this model all, the neighboring cation-anion distances are taken into account and considered as bonds, with unequal strengths. The bond valence or bond strength Sij for a bond set up between two ions i and j can be calculated by the following relation:
where tij is the bond length between two ions i and j, while r0 and B (usually B = 0.37 Å), are empirical constants tabulated and determined for each pair of cation-anion. In practice, the valence or oxidation state V(i) of ions i coordinated by the k nearest counter-ions j obey to the relation [37]:
The CHARDI analysis is fundamentally differs from BVS calculations by exploiting the true bond distances in a self-consistent computation, instead of employing empirical curves (R – s) requiring empirical parameters. This method adopt a Madelung-type description of crystal structures in which each atom is described as a point charge, where the formal charge corresponds to the oxidation number [38]. The CHARDI analysis method gives the distribution of calculated ECoN (effective coordination number) of a central atom among all the neighbouring ligands [39]. The calculation of this number is directly correlated to the distribution of atomic charges in crystalline structures. The correctness of the structure (cation ratio) and of the degree of over or under bonding (OUB) (anion ratio) is performed via the evaluation of the internal criterion q(i)/Q(i) (where q(i) is the formal oxidation number, and Q(i) represents the computed charge) [40]. For a correctly refined structure, the calculated valences V(i) giving by the (BVS) approach and the computed charge Q(i) according to the CHARDI concept must be in close agreement with the oxidation number of each atoms. The CHARDI computations were carried out with the CHARDI2015 Fortran program [41], while the BVS calculation was performed using EXPO2014 program [42] (Table 4). The crystal structure of the title compound shows no statistical disorder of the seven metallic sites. Furthermore, the repartition of the electric charges at the nineteen independent crystallographic sites of the asymmetric unit shows that the Zn2+, Fe3+, and P5+ cations occupy only 7 sites. Otherwise, balancing electrical charges requirement imply the repartition of additional 12 oxygen atoms in the remaining 2i sites. A close examination of the obtained results (Table 4), reveals that the obtained values of Q(i) charges and V(i) bond-valence-sums of all cations are very close to the corresponding weighted oxidation number q(i).sof(i). Some minor fluctuations were also reported, revealing the distortion of the accommodate sites. Despite these irregularities, all the values of the internal criterion q(i)/Q(i) are very close to unity, supporting the validity of the proposed structural model.
For a structure described as built on classical cation-centered, the Mean Absolute Percentage Deviation (MAPD) was successfully introduced to evaluate the convergence of the CHARDI model. The MAPD measures the agreement between q(i) and Q(i) for the whole sets of PC atoms (polyhedron-centering atoms) and of V atoms (the vertex atoms) [43].
where N is the number of polyhedron-centering or vertex atoms in the asymmetric unit. For the PC atoms a MAPD > 10% results in an incorrect analysis or bad accuracy of refinement, while for the V atoms a MAPD < 5% indicates a low OUB effect, which can increase proportionally to MAPD. In the structure, such effect is noticeable for some oxygen atoms labelled O(1), O(3), O(5), O(6), O(8), O(9), and O(11), which deviate slightly from the ideal value of –2 (Table 5). Following the current distribution scheme, the obtained value of MPAD for the positive charges is only 0.8 and 2.9% for the negative ones, which proves once again the applicability and adequacy of the experimental model. In order to verify the chemical plausibility of the crystal structure we have also looked into the Global Instability Index (GII) [44]:
The GII index evaluates the rationality of the structure and measures the deviation of the bond-valence sums from the formal valence V(i) averaged over all N atoms of the asymmetric unit.
Indeed, for an unstrained structure GII < 0.1 v.u. and may approach 0.2 v.u. in a structure with lattice-induced strains. Values larger than 0.2 v.u. indicates intrinsic strains strong enough to involve instability of the crystal structure. In our case, we found a very good (GII) index converging to 0.03 v.u, which proves the high stability and rigidity of the structure.
Crystal Structure Description
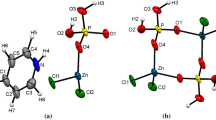
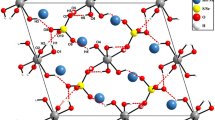
The basic building units of Zn3Fe4(PO4)6 belonging to the Howardevansite are more or less distorted polyhedra (FeO6, ZnO6, ZnO5) and nearly regular PO4 tetrahedra, as depicted in Fig. 3. In the current structural model, all atoms are located in Wyckoff position 2i, except Zn(2) which is on Wyckoff position 1h of the \(P\bar {1}\) space group. This structure consists of infinite kinked chains of edge-sharing [FeO6] octahedra, linked into two kinds of dimers ([Fe(1)2O10] and [Fe(2)2O10]) which are in turn interconnected via a common edge to [Zn(1)O5] group, as shown in Fig. 4. These chains are held together by the apices of the PO4 tetrahedra giving rise to a stacked layers perpendicular to [010] (Fig. 5). Inside of the layers the junction of Fe(2)O6 and Zn(1)O5 polyhedra by common vertices of P(2)O4 and P(3)O4 tetrahedra built an open three-dimensional framework that delimits tunnels parallel to [100], hosting the Zn(2)2+ cations (Fig. 6).

The principal building units in the structure of the title compound. Displacement ellipsoids are drawn at the 60% probability level. Symmetry codes: (i) x + 1, y, z; (ii) x, y – 1, z; (iii) –x + 2, –y + 1, –z + 2; (iv) –x + 1, –y + 1, –z + 1; (v) –x + 1, –y + 1, –z + 2; (vi) x – 1, y, z; (vii) –x + 1, –y + 2, –z + 2; (viii) x + 1, y + 1, z; (ix) –x + 1, –y + 2, –z + 1; (x) x – 1, y – 1, z; and (xi) x, y + 1, z.

Edge-sharing [Zn(1)O5] polyhedral and [Fe2O10] dimers forming an infinite zigzag chain running parallel to [101].

A view along [010], showing a layer resulting from the connection of chains via vertices of [PO4] units.

Three-dimensional view of Zn3Fe4(PO4)6 crystal structure showing tunnels parallel to [100].
As we have pointed out, the (CHARDI) method extends to the analysis of the geometric aspect of each hetero polyhedron building a crystal structure using the number (ECoN). A careful inspection of the ratios ECoN (Fe(1))/CN(Fe(1)) = 5.49/6 and ECoN (Fe(2))/CN(Fe(2)) =5.67/6 reveals an increased distortion in the FeO6 units ranging from the Fe(2)O6 to Fe(1)O6 polyhedra. Indeed both Fe(1)O6 and Fe(2)O6 octahedral groups are slightly distorted and weakly compressed. This tendency to deviates from the regularity is due to the junction based on edge sharing within the [Fe2O10] dimers, which induces metal-metal repulsion forces. Despite this electrostatic stress, the extremely large void space (VFe(1) ≃ 10.50 Å and VFe(2) ≃ 10.61 Å3) in the unit cell fully supports such deformations of both octahedral units. The interatomic distances Fe–O are ranging from 1.8896(9) to 2.1819(9) Å for Fe(1) Å and varying between 1.862 (1) and 2.0892 (9) Å for Fe(2). The corresponding average distances 〈Fe(1)–O〉 = 2.0141 Å and 〈Fe(2)–O〉 = 2.0202 Å are close to 2.03 Å, reported by Shannon for Fe3+ cations within octahedral coordination [45]. The first zinc atom is surrounded by five neighboring O atoms, resulting from four long Zn(1)–O ≃ 2 Å and a short one Zn(1)–O(5) (Zn(1)–O(5) = 1.958(1) Å), forming a highly deformed trigonal bipyramid. The related distortion was evidenced trough the ECoN(Zn(1))/CN(Zn(1)) = 4.86/5 ratio as well as by measuring the degree of trigonality using the τ5 index which reach 0.504 v.u. [46]. The octahedral coordination sphere of the Zn2+(2) cation is quite distorted as seen from the Zn(2)–O bond lengths in the range 2.0071(9)–2.2575(9) Å with a mean value of 2.1567 Å. The remaining P–O interatomic distances within the PO4 tetrahedra units vary between 1.494(1) and 1.5605(9) Å. Their mean distances 〈P(1)–O〉 = 1.5400 Å, 〈P(2)–O〉 = 1.5330 Å, and 〈P(3)–O〉 = 1.5433 Å, are in a good accordance with the 〈P–O〉 length often encountered in monophosphate groups [47].
REFERENCES
I. Ahmed, C. A. Collins, M. P. Lewis, et al., Biomaterials 25, 3223 (2004).
M. Ai, Catal. Today. 52, 65 (1999).
E. Dudrová, M. Kabátová, R. Oriňaková, and P. Hvizdoš, Int. J. Electrochem. Sci. 11, 1751 (2016).
Y. Yin, H. Zhang, P. Wu, B. Zhou, et al., Nanotechnology 21, 425504 (2010).
A. H. Taghvaei, H. Shokrollahi, K. Janghorban, and H. Abiri, Mater. Des. 30, 3989 (2009).
F. Croce, A. D’Epifanio, J. Hassoun, et al., Electrochem. Solid-State Lett. 5, 47 (2002).
G. Hazra, P. Mitra, and T. Das, Trans. Indian Ceram. Soc. 78, 69 (2019).
M. Hidouri, N. Sendi, A. Wattiaux, and M. B. Amara, J. Phys. Chem. Solids 69, 2555 (2008).
R. Essehli, B. El Bali, S. Benmokhtar, et al., J. Alloys Compd. 509, 1163 (2011).
D. Rénald, A. Pautrat, D. Filimonov, et al., J. Am. Chem. Soc. 135, 13023 (2013).
A. V. Koshelev, K. V. Zakharov, A. P. Pyatakov, et al., Phys. Rev. Appl. 10, 034008 (2018).
K. Zaghib, A. Mauger, J. B. Goodenough, et al., Chem. Mater. 19, 3740 (2007).
G. Amthauer and G. R. Rossman, Phys. Chem. Miner. 11, 37 (1984).
B. I. Lazoryak, V. A. Morozov, A. A. Belik, et al., J. Solid State Chem. 122, 15 (1996).
A. Bouraima, T. Makani, A. Assani, et al., Acta Crystallogr. E 73, 890 (2017).
A. Bouraima, A. Assani, M. Saadi, et al., Acta Crystallogr. E 71, 558 (2015).
J. Khmiyas, A. Assani, M. Saadi, and L. El Ammari, Acta Crystallogr. E 71, 690 (2015).
G. Alhakmi, A. Assani, M. Saadi, and L. El Ammari, MATEC Web Conf. 149, 01084 (2018).
S. Ouaatta, A. Assani, M. Saadi, and L. El Ammari, Acta Crystallogr. E 73, 893 (2017).
S. Ouaatta, A. Assani, M. Saadi, and L. El Ammari, Acta Crystallogr. E 71, 1255 (2015).
G. Alhakmi, A. Assani, M. Saadi, and L. El Ammari, Acta Crystallogr. E 73, 767 (2017).
J. Khmiyas, A. Assani, M. Saadi, and L. El Ammari, Acta Crystallogr. E 72, 1260 (2016).
J. M. Hughes, J. W. Drexler, C. F. Campana, and M. L. Malinconico, Am. Mineral. 73, 181 (1988).
Y. A. Gorbunov, B. A. Maksimov, Y. K. Kabalov, et al., Dokl. Akad. Nauk SSSR 254, 873 (2020).
M. Gruß and R. Glaum, Z. Krist. 212, 510 (1997).
M. A.Lafontaine, J. M. Grenéche, Y. Laligant, and G. Férey, J. Solid State Chem. 108, 1 (1994).
A. Assani, M. Saadi, M. Zriouil, and L. El Ammari, Acta Crystallogr. E 67, i52 (2011).
B.-C. Zhou, Y.-W. Yao, and R.-J. Wang, Acta Crystallogr. C 58, i109 (2002).
A. Riou, Y.Cudennec, and Y.Gerault, Acta Crystallogr. C 43, 821 (1987).
P. Lightfoot and A. K. Cheetham, Acta Crystallogr. C 44, 1331 (1988).
D. Altermatt and I. D. Brown, Acta Crystallogr. B 41, 240 (1985).
M. Nespolo, Acta Crystallogr. B 72, 51 (2016).
L. Krause, R. Herbst-Irmer, G. M. Sheldrick, and D. Stalke, J. Appl. Crystallogr. 48, 3 (2015).
G. M. Sheldrick, Acta Crystallogr. A 71, 3 (2015).
G. M. Sheldrick, Acta Crystallogr. C 71, 3 (2015).
L. J. Farrugia, J. Appl. Crystallogr. 45, 849 (2012).
N. E. Brese and M. O’Keeffe, Acta Crystallogr. B 47, 192 (1991).
J.-G. Eon and M. Nespolo, Acta Crystallogr. B 71, 34 (2015).
R. Hoppe, Z. Krist. 150, 23 (1979).
M. Nespolo, G. Ferrarisb, and O. Haruo, Acta Crystallogr. B 55, 902 (1999).
M. Nespolo and B. Guillot, J. Appl. Crystallogr. 49, 317 (2016).
A. Altomare, C. Cuocci, C. Giacovazzo, et al., J. Appl. Crystallogr. 46, 1231 (2013).
M. Nespolo, Acta Crystallogr. B 72, 51 (2016).
A. Salinas-Sanchez, J. L. Garcia-Muñoz, J. Rodriguez-Carvajal, et al., J. Solid State Chem. 100, 201 (1992).
R. D. Shannon, Acta Crystallogr. A 32, 751 (1976).
A. W. Addison and T. N. Rao, J. Chem. Soc. Dalt. Trans., p. 1349 (1984).
W. H. Baur, Acta Crystallogr. B 30, 1195 (1974).
ACKNOWLEDGMENTS
The authors thank the Unit of Support for Technical and Scientific Research (UATRS, CNRST) for the X-ray measurements.
Funding
Funding for this research was provided by Mohammed V University, Rabat, Morocco.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Khmiyas, J., Assani, A., Saadi, M. et al. Crystal Growth, Structural Elucidation, and Accurate (CHARDI/BVS) Investigations of Zn3Fe4(PO4)6 with Howardevansite-like Structure. Crystallogr. Rep. 66, 1216–1222 (2021). https://doi.org/10.1134/S1063774521070087
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1063774521070087