
Abstract
The second and final part of the review. Provides general information about sub- and supercritical extraction (pressurized liquid extraction, subcritical water extraction, supercritical fluid extraction), matrix solid-phase dispersion and the QuEChERS method. Based on an analysis of review works, information on the features of sample preparation using these methods is systematized, experimental parameters affecting extraction efficiency are considered, and examples of using these methods for isolating organic compounds in the analysis of solid environmental samples, food products, and plants are given.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Extraction of organic compounds from solid samples is the most important, most complex, and time-consuming stage of chemical analysis before their determination. Until now, classical methods of liquid–liquid extraction from solid matrices—extraction by mechanical shaking, extraction in a Soxhlet apparatus, or ultrasound-assisted extraction—are widely used for this purpose [1–4]. The disadvantages of these methods include long extraction time; the use of large amounts of organic solvents, often very toxic; low recoveries, difficulties in automation, and the risk of the destruction of heat-sensitive compounds. In addition, in some cases further purification and preconcentration are required after sample preparation using the above methods.
The current trend in the development of sample preparation methods in general, as well as sample preparation of solid samples in particular, is associated with the use of “more environmentally friendly” approaches with reducing the number of toxic solvents used or their complete elimination and replacement by so-called “green” solvents, automation and miniaturization of the equipment, reducing the number analytical operations, and minimizing negative impacts on the environment and human health [5–9]. In addition, one of notable trends is a combination of various sample preparation methods in one analytical cycle [10, 11], as well as the online combination of sample preparation methods with subsequent determination methods [12].
Most modern methods of liquid–liquid extraction from solid matrices use elevated temperatures and pressures, as well as environmentally friendly solvents, to shorten extraction times and reduce solvent consumption. As was noted in the reviews cited above, the principles of “green analytical chemistry” [13] correspond to pressurized liquid extraction, subcritical water extraction, and supercritical fluid extraction. In addition, “green” sample preparation methods include matrix solid-phase dispersion and the QuEC-hERS method. The development of the last two methods has been greatly facilitated by the ability of performing extraction and purification in a single step, which significantly reduces the time of analysis and the number of solvents used. In addition, these methods do not require special equipment, are simple to implement, and are characterized by low cost.
The second part of this review summarizes review articles on pressurized liquid extraction, subcritical water extraction, supercritical fluid extraction, matrix solid-phase dispersion, and QuEChERS; a general description of the methods is given, methods for their implementation are considered, experimental parameters affecting the efficiency of the extraction of organic compounds are listed, and examples of the practical application of methods to the preparation of various samples are given. The first part of the review summarized review articles describing traditional methods for extraction of organic compounds from solid samples: shaking solid–liquid extraction, Soxhlet extraction, ultrasound-assisted extraction, and microwave extraction [14].
SUB- AND SUPERCRITICAL EXTRACTION
Modern sub- and supercritical extraction methods include pressurized liquid extraction, also known as accelerated solvent extraction, subcritical water extraction, and supercritical fluid extraction [15–17]. The common feature of these methods is that they are carried out under high pressure and at elevated temperatures, which significantly changes the physicochemical characteristics of the extraction solvents. The advantages of the methods include a sharp reduction in the amount of organic solvents and extraction time, as well as high productivity and a possibility of the automation of the sample preparation process. In addition, the use of environmentally friendly subcritical water and supercritical CO2 as solvents allows one to classify these methods as “green” sample preparation methods. In the last five years, there has been a boom in the use of these methods in technological processes for the isolation of biologically active substances (phenolic compounds, lignans, carotenoids, oils and lipids, essential oils and other nutraceuticals) from various plant materials and food products [18–25]. Despite the high cost of laboratory equipment required for extraction, these methods have not lost their relevance in chemical analysis, as evidenced by the number of reviews devoted to the use of pressurized liquid extraction [26–44], subcritical water extraction [45–57], and supercritical fluid extraction [58–78] for the extraction of organic compounds from natural and biological solids, as well as from food products (Table 1). An important argument in favor of these expensive methods is also the reduction in manual labor through process automation.
Pressurized liquid extraction (PLE) is an automated sample preparation method that uses elevated temperature and pressure to increase the efficiency of solvent extraction, applicable to solid and semi-solid matrices. Table 1 lists reviews on the use of pressurized liquid extraction for the isolation of organic compounds from solid samples in the chronological order [26–44].
A few words about the confusion that still exists regarding the name of the method [26, 29, 40]. The method and the corresponding equipment were developed and patented by Dionex (United States) under the commercial name “accelerated solvent extraction, ASE”, the first publication appeared in 1996. As in the first few years after the appearance of the method, the only commercially available extractor for its implementation was the ASE1 200 extraction unit, many authors in their studies used the name of the method proposed by the company. Over time, alternative names for the method appeared, which began to supplant its commercial name, which did not reflect the essence of the method. In the United States, this method is known as “pressurized fluid extraction”. The American Chemical Society has introduced the acronym PFE in its journals. This term and abbreviation are also used by the U.S. Environmental Protection Agency in EPA Method 3545, “Extraction of Volatile and Moderately Volatile Compounds from Soils, Clays, Sediments, Silts, and Solid Wastes by the Use of Solvents at High Pressures and Temperatures.” In addition, this method was also named pressurized hot solvent extraction (PHSE), pressurized liquid extraction (PLE), high-pressure solvent extraction (HPSE), and sub-critical solvent extraction (SSE). In the scientific literature, in particular in the journals published by Elsevier Science, the term “pressurized liquid extraction” and the abbreviation PLE are most often used. Despite the fact that, in Russian literature, this method is better known under the commercial name “Accelerated solvent extraction,” in this review we use the term “Pressurized liquid extraction,” which is used by the authors of most publications cited in this review.
The principle of the method was discussed in detail in the reviews [30, 35, 39, 40, 44]. Pressurized liquid extraction is carried out in the interval between the boiling point of the solvent and its critical temperature at a pressure slightly higher than the equilibrium vapor pressure of the solvent. The combined use of high pressure and temperature reduces viscosity and surface tension, allowing the solvent to more effectively penetrate into the matrix structure, enhancing the extraction of the target compounds. The use of high temperature increases the solubility of the analytes and the rate of mass transfer. Elevated pressure keeps solvents in a liquid state for safe and rapid extraction, while automated instrumentation allows for the development of a less labor-intensive methods and improves reproducibility. In addition, the ability of combining extraction and purification steps by incorporating an adsorbent layer retaining interfering compounds directly into the extraction cells makes this method extremely versatile and selective. The advantages and disadvantages of pressurized liquid extraction compared to other methods of the sample preparation of solid samples in terms of the consumption of organic solvents, process time, and equipment costs were given in more detail in the review [44].
The pressurized liquid extraction procedure has been described in detail in several reviews [26, 28, 30, 33, 35, 36, 44]. It involves the dispersion of a sample with an inert material; placement of the mixed sample in a steel extraction cell; filling the cell with an organic solvent; heating the vessel (usually up to 75–200°C), and increasing pressure to a necessary value (usually up to 100 atm); the extraction of the target analytes over a period of time; the transfer of the extract to a collection bottle and cleaning the sample with a fresh solvent; the removal of the residual solvent from the sample by purging the cell with nitrogen gas (Fig. 1). The configuration of the basic equipment for performing PLE depends on whether the process is static, using a fixed volume of an extractant, or dynamic, in which the extractant is fed continuously through an extraction cell throughout the entire extraction time. Most equipment operating in an automatic mode allows you to load up to 24 cells, the volume of which can vary from 1 to 100 mL, with cells of a volume of 33 mL being most often used. Pressurized liquid extraction systems are currently available from three suppliers: Thermo Scientific (United States) (accelerated solvent extraction system, ASE, formerly Dionex); Fluid Management Systems, Inc. (United States) (FMS) (PLE system); and “Büchi” (Switzerland) (Speed Extractor system) [40].

Scheme of pressurized liquid extraction [41].
The block diagram of PLE installations for carrying out extraction under static or dynamic conditions is given in the reviews [26, 28, 41, 44]. Technical solutions combining static and dynamic pressurized liquid extraction with other stages of the analytical process (preconcentration, derivatization, filtration, chromatographic separation, and detection) are given in the reviews [29, 42]. Thus, the review [42] analyzed publications devoted to the analysis of herbal medicines using online pressurized liquid extraction and subsequent HPLC–MS/MS determination. The issues of the sequential combination of pressurized liquid extraction with derivatization, as well as simultaneous extraction and derivatization in situ, were discussed in the review [37], which noted that the conditions created in the PLE method allow derivatization with a smaller amount of a derivatizing agent.
Experimental parameters affecting the completeness of the separation of organic compounds from solid matrices by the PLE method have been systematized in a number of reviews [26, 27, 30, 33–36, 39–41, 44]. Conventionally, these parameters can be divided into three groups: varied at the stage of sample preparation; varied at the extraction stage; and varied after extraction [30, 33, 35, 44]. Sample pretreatment usually involves sample homogenization and sieving, because the diffusion of analytes from the sample to the solvent can be significantly increased by reducing particle size. To prevent the aggregation of small sample particles, in most methods a crushed sample is mixed with a dispersant, which is quartz sand or diatomaceous earth. Samples with high water content are dried in vacuum ovens or sublimated from the frozen state during sample preparation. In some cases, pre-purified drying agents, such as anhydrous sodium sulfate, are added to the analyzed sample. Another parameter is the mass of a sample, which varies depending on the type of the sample and cell volume [33]. Typically, the range of masses used is 0.2–5 g.
Parameters varied at the extraction stage include the nature of the solvent and of the modifying additives, temperature, pressure, extraction time, and the number of cycles [26, 27, 30, 33–36, 44]. When choosing a solvent, one should take into account the solubility of the target analytes in it, as well as some physicochemical properties of the solvent, i.e., boiling point, polarity, specific gravity (affects penetration into the sample matrix), as well as toxicity (creates hazard in the workplace). In addition, it is important for the extract obtained using the selected solvent to be compatible with the subsequent purification and determination steps. Ideally, it is advisable to select a solvent such that the target analytes are dissolved in it as much as possible and other compounds are dissolved to a minimal extent. This method often uses methanol, dichloromethane, hexane, acetone, toluene, ethyl acetate, acetonitrile, as well as their mixtures, often in proportions 1 : 1 (by volume): hexane–dichloromethane, toluene–acetone, hexane–acetone, acetone–dichloromethane, dichloromethane–acetonitrile, acetonitrile–water, methanol–water [41]. In addition, in some cases, various modifiers are added to the solvents, such as surfactants, which can change the physicochemical properties of the solvent at elevated temperatures and affect solubility [35, 36, 44]. More detailed examples of solvents that have found application to the extraction of a wide variety of organic compounds from soils or sewage sludge can be found in the reviews [26–28, 33, 39, 41]; from food products, in the reviews [28, 30, 36, 39, 41, 44]; and from plants, in the reviews [35, 36].
Temperature and pressure are the most important parameters influencing the duration, efficiency, and selectivity of sample preparation using the PLE method. With increasing temperature, the properties of the solvent—viscosity, surface tension, diffusion coefficient— change, which ensures a better penetration of the solvent into the pores and between the particles of the matrix and increases the solubility of the analytes. As an example, we can cite data from the review [27]: with increasing temperature from 50 to 150°C, the solubility of anthracene increases by 13 times; diffusion rate, by 2−10 times; and the viscosity of 2-propanol decreases by 9 times. However, an increase in temperature can cause the degradation of thermolabile analytes, especially when combined with long extraction times, and can also reduce extraction selectivity due to the co-extraction of some interfering substances. The temperature range used in this method ranges from 50 to 150°C, and the most commonly used temperature is 100°C [41]. This temperature is above the boiling point of most known organic solvents and is low enough to avoid the degradation of analytes and/or co-extractables. The primary function of pressurization is to maintain the solvent in a liquid state at elevated temperatures well above its boiling point. An increase in pressure also promotes the penetration of the solvent into the matrix pores, which it usually not reached under normal conditions [33, 36]. Equipment for pressurized liquid extraction allows you to change pressure in the range from 35 to 200 atm [41, 44]. A comparison of the data presented in the tables in the reviews [33, 36, 41, 44] indicated that, for each sample and group of analytes, pressure is selected individually, but in most cases it is set at 100 atm.
In performing PLE in a static mode, interrelated parameters that also greatly influence extraction efficiency are extraction time and the number of cycles [33, 41, 44]. Extraction time is defined as the time for which the solvent is in contact with the matrix at a selected pressure, temperature, and flow rate; it is optimized depending on the matrix, the extracted analytes, and extraction mode [44]. Typically, this parameter is set at 3–15 min. The number of cycles is the number of times for which fresh solvent enters the cell and contacts with the samples [33]. The equipment is usually capable of performing up to five cycles, but most often no more than three cycles are used, because, after several cycles, the selectivity of extraction may decrease. Dynamic mode provides a continuous flow of an extraction solvent at an appropriate rate through a cell, resulting in short contact times between the sample and the solvent, thereby improving mass transfer. However, this type of extraction is rarely used, mainly because of the higher solvent consumption compared to the static process [44].
As follows from the published data, three more parameters are recorded before the start of an extraction cycle and after its completion, despite the fact that they do not directly affect extraction efficiency. These are preheating time, washing volume, and purging time [33, 36, 44]. Preheating time is the amount of time for which the cell is held in an oven at a selected temperature before adding the solvent; to ensure a fixed cell temperature, 5 min is usually sufficient. The wash volume is the percentage of fresh volume introduced into the cell after the extraction time required to move the analytes into the collection vessel. In most cases, this volume is 60% of the solvent volume used at the extraction step. Finally, the third parameter is the time of purging the cell with nitrogen gas; this time varies between 30–300 s.
As mentioned above, the selectivity of the extraction of organic compounds in pressurized liquid extraction is low; therefore, a purification step is required to obtain purer extracts suitable for subsequent analysis by chromatographic methods. The simplest method in terms of time and automation is the method in which extraction and purification occur at one stage. This version of PLE was named selective pressurized liquid extraction (SPLE) [32, 38]. In this version of the method, purification is achieved by adding a layer of a sorbent/sorbents or reagents capable of retaining interfering compounds (usually fats, proteins or pigments) directly into the extraction cell. During the extraction process, the target analytes are extracted from the sample and the non-target substances remain in the cell on these solid phases. The most widely used cleaning materials are Florisil, silica gel (silica) in various forms (acidic, basic, neutral, activated or not, modified with octadecyl groups, treated with silver nitrate or copper), alumina, and graphitized carbon. Examples of using selective pressurized liquid extraction were given in the reviews [32, 38, 40, 41, 43]. Other purification methods, described in detail in the reviews [41, 42], include off-line purification by column chromatography, as well as solid-phase extraction (SPE), solid-phase microextraction, and a number of other methods that have been used for the purification of liquid samples.
An analysis of the review papers carried out in this review indicates that pressurized liquid extraction, an automated method for the rapid sample preparation of solid or semi-solid samples, has found widespread use in serial chemical analysis. This method is used to isolate non-volatile and moderately volatile organic compounds from environmental samples [26–28, 31, 33, 34, 39–41, 43], food products [27, 30, 34–36, 39, 41, 44] and plants [35, 42]. Noteworthy is the fact that, in the initial period of development, this method was used mainly for the extraction of organochlorine pesticides, polycyclic aromatic hydrocarbons (PAHs), polychlorinated biphenyls (PCBs), dibenzofurans, dibenzo-p-dioxins, and other persistent organic pollutants (POPs) [26–28, 30, 31]. Over time, along with the extraction of POPs, this method has increasingly been used for the extraction of various classes of pesticides and medicinal substances, personal hygiene products, mycotoxins, flame retardants, and many other organic compounds [34–36, 39, 41, 44]. The tables given in the reviews cited above indicate not only the analyzed samples and the analytes, but also provide information on the pre-extraction sample treatment and the preparation of extraction cells, extraction solvent and its volume, methods for purifying the extracts, as well as on such specified parameters as temperature, pressure, time, number of cycles, volume of flushing solvent, and nitrogen purge time.
Subcritical water extraction (SWE), also known as pressurized hot water extraction (PHWE) or superheated water extraction, is a type of pressurized liquid extraction in which water heated to 100–300°C under a pressure of 30–50 atm is used as a solvent, i.e. in subcritical conditions. Reviews [45–57] were devoted to this method of sample preparation (Table 1).
At elevated temperatures and sufficient pressure to maintain water in a liquid state, dramatic changes in its physicochemical properties are observed [45, 46, 50–55]. These changes manifested themselves in a decrease in the dielectric constant, viscosity, and surface tension. For example, the dielectric constant (ε) of water upon changing temperature from 25 to 250°C at a pressure of 50 atm decreased from 80 to 27 and became comparable with the dielectric constants of methanol (ε = 33) and ethanol (ε = 24) at 25°C [50]. Under these conditions, water, like organic solvents, could dissolve various organic compounds. In addition, by reducing surface tension and viscosity, subcritical water better wetted and deeper penetrated into the solid samples, improving the kinetics of the diffusion and mass transfer of the analyte [53]. The review [47] emphasized that subcritical water is an ideal environmentally friendly solvent and is used in many laboratories both for extraction and as an eluent for reversed-phase HPLC.
Subcritical water was first used to extract PAHs, PCBs, phenols, pesticides, and other substances from sediments, soils, and suspended matter in the mid-1990s; references to these first works are given in the reviews [28, 45]. Already in the first works it was shown that the dielectric constant of subcritical water, and, therefore, its dissolving ability, can be changed in a wide range by varying temperature and pressure, which makes it possible to use this method for the extraction of both polar and non-polar analytes. Over time, works have appeared on the mechanism of subcritical extraction; they were systematized in the reviews [48, 50, 52, 54, 55]. It is assumed that the extraction process with subcritical water includes several successive stages: the desorption of analytes from various active centers of the sample matrix under the conditions of elevated pressure and temperature; their diffusion into the extract; and the elution of solutes from the extraction cell. The extraction rate is limited by the slowest step. It has been shown that the extraction of components from many solid samples is determined primarily by the desorption of microcomponents from the surface of a solid matrix. In the static mode, during an experiment, an equilibrium is established in the “sample–solvent” system, which can be described within the framework of a simple thermodynamic model, according to which recovery primarily depends on the distribution coefficient of the microcomponent. The extraction mechanism under dynamic conditions is described within the framework of the theory of frontal chromatography, which includes two stages of mass transfer: the desorption of a microcomponent from the matrix surface and its elution.
Subcritical water extraction is carried out mainly in laboratory installations created on the basis of serial chromatographic equipment. Block diagrams of laboratory installations for subcritical extraction under static or dynamic conditions were given in the reviews [48–50, 54, 55, 57]. They consist of heated elements, an extraction cell and an inlet capillary for heating water to the working temperature (its length is usually 1.5 m or more), which are placed in an oven. In most cases, an oven included in a gas chromatograph is used. At the outlet of the cell, outside the furnace, a pressure limiter is installed, which maintains water in a liquid state at temperatures above 100°C. To more effectively cool the extract, the outlet capillary is placed in a container with cold water or in another cooling device. In addition, to carry out extraction in a static mode, you can use commercial equipment for pressurized liquid extraction, e.g., a Dionex ASE 200 unit [48, 54]. Technical solutions describing possible combinations of the dynamic version of subcritical extraction with the subsequent chromatographic determination were given in the reviews [49, 54, 57].
The main parameters varied during the development of procedures were discussed in detail in the reviews [48–52, 54–56]. These include temperature, pressure, time, nature and concentration of modifier additives, dynamic flow rate, and sample particle size. As discussed above, temperature affects the dielectric constant of water, the solubility of a microcomponent, and its distribution coefficient. As temperature is increased, the viscosity of water also decreases, which leads to an increase in the rate of the diffusion of microcomponents and, accordingly, the rate of mass transfer. Reviews provide examples indicating that increasing temperature to 300°C increases the solubility and the recovery of PAHs, phenolic compounds, and pesticides [45, 49, 54]. However, some compounds, e.g., many biologically active substances, can be thermally unstable, oxidized, and decomposed in an aggressive medium of subcritical water; in this case, in choosing temperature, a compromise is made: despite the reduction in the recovery, extraction is carried out at sufficiently low temperatures [48, 51]. To maintain water in a liquid state, pressure is varied from 10 to 80 atm. For example, at 200 and 300°C, water remains in a liquid state if pressure is 15 and 85 atm, respectively. Unlike temperature, pressure does not significantly affect the recovery of the analytes. The effect of higher pressure used in subcritical extraction, compared with extraction under atmospheric pressure, is an increase in the extraction rate, because subcritical water under pressure can penetrate into hard-to-reach areas of the sample matrix. Extraction times vary depending on the temperature of the extractant, the nature of the sample matrix, and the microcomponents being extracted. In most cases, an increase in temperature reduces the time required for quantitative extraction. In dynamic extraction, an increase in the throughput rate of the extractant often increases the recovery of the microcomponents due to the maintenance of a high concentration gradient. The subcritical water transmission rate is selected based on the specified sample processing time and the desired concentration of analytes in the extract. It has been shown that increasing the rate is advisable when extraction is limited by the solubility of the extracted substances, the diffusion of the extractant into the sample matrix, and the rate of analyte transfer from the matrix surface [54]. To increase the recovery of the analytes, additives of organic solvents (methanol, ethanol, ethyl acetate) or surfactants, such as sodium dodecyl sulfate and Triton X-100, are added to water [48–50, 52, 54]. These substances are typically used to increase the solubility of analytes in water or to enhance the interaction of water with the analytes. They can also change the physicochemical properties of water at elevated temperatures and critical temperatures and pressures.
Another factor influencing recovery is sample particle size [51, 52, 55, 56]. As with pressurized liquid extraction described above, smaller particles increase the recovery of compounds, while larger particles reduce efficiency and increase extraction time. In some cases, dispersants (such as glass beads) are introduced into the sample in an extraction vessel to more evenly distribute the sample and the extractant and increase extraction efficiency. In contrast, the geometry of the extraction cell or vessel and the direction of water flow have only a minor effect on the recovery of the target analytes.
Because of its non-toxicity, non-flammability, and availability, subcritical water has recently found increasing use for the extraction of various biologically active substances from natural products [48, 51–53, 55–57]. The authors of one of the latest reviews [57] analyzed more than 200 publications for the period from 2004 to 2021 on this topic, of which about 160 were published in the last decade. The review provides conditions for the extraction of flavonoids, polyphenols, organic acids, glycosides, carbohydrates, essential oils, alkaloids, quinones, terpenes, lignans, and steroids from various samples of plant origin (medicinal herbs, vegetables, fruits, algae, tea leaves, grains and seeds). Subcritical extraction of the above-mentioned biologically active compounds is carried out at temperatures from 120 to 200°C in both static and dynamic modes, followed by the determination of the isolated analytes by various chromatographic methods. The conditions for the extraction of PAHs, PCBs, chlorophenols, pesticides from soils, sediments and other environmental samples can be found in the reviews [28, 45, 49, 50, 54].
Supercritical fluid extraction (SFE) is based on the use of supercritical fluids as extractants. The principle of the method and the features of its application to chemical analysis were covered in the reviews [56, 58–78] (Table 1). To obtain supercritical fluids, this method uses carbon dioxide, nitrogen oxide (I), ethane, propane, n-pentane, ammonia, fluoroform, sulfur hexafluoride, or various freons. Their properties were described in the reviews [58, 60, 61, 69–71]. Of all the substances studied, carbon dioxide still remains the most commonly used extractant in SFE, which is associated with a number of its unique properties. In addition to a fairly low critical temperature (31°C) and pressure (73 atm) [61], CO2 is non-flammable, non-explosive, non-toxic, available at low cost, and has high purity. It is also important that CO2 is a gas under normal conditions. After extraction, the purification of the product from the solvent is achieved by simply releasing pressure. Supercritical CO2 then passes into the gas phase and evaporates, thereby relieving an analyst of the need in carrying out the long-term evaporation of the extract after isolation, which often leads to a partial thermal destruction of the target substances. In addition, CO2 has low surface tension and viscosity, but its diffusion coefficient is two to three times higher than those of other liquids. In terms of polarity, CO2 in the supercritical region is similar to pentane and is, therefore, suitable for the extraction of lipophilic substances. The main disadvantage of CO2 is the low recovery of polar substances, but this disadvantage is easily eliminated by adding various modifiers [56, 58, 60, 65, 69, 76].
Supercritical fluid extraction has many similarities to pressurized liquid extraction and subcritical water extraction discussed above, except for that this method uses a supercritical fluid as the extractant rather than organic solvents or water. The main advantage of SFE is, as noted above, that after the extraction, the extraction solvent turns into a gas, and the analytes are conveniently preconcentrated in a solid-phase trap or a liquid, which was described in detail in the review [66]. In addition, this method is distinguished by higher selectivity, which, depending on the purpose of sample preparation, can be attributed to both the advantages and disadvantages of the method. Other challenges associated with SFE include the high cost of automated instrumentation, relatively small sample sizes, and a more complex procedure development process [58, 61].
Supercritical fluid extraction, as a rule, is carried out in a flow or in a periodic flow-stationary mode. A typical block diagram of equipment for SFE is given in the reviews [59, 68, 69, 72]. It includes an extraction cell, which is equipped with temperature controls and pressure valves on both ends to maintain the desired extraction conditions, a pump for supplying CO2 to the extraction system, sometimes a pump for supplying a modifier, and one or more separators, named fractionation cells, in which the extract is collected and pressure is released to remove the solvent.
The list of experimental parameters affecting the efficiency of SFE is quite long; it includes characteristics of the supercritical fluid (with and without modifiers) and of the solid matrix; thermodynamic and kinetic conditions for extraction (temperature, pressure, density and flow rate); water content; sample particle size; and physical and chemical properties of the target analytes [59–62, 68–72, 74]. Theoretical models describing the SFE of substances from various solid matrices were considered in the reviews [68, 70, 73, 75].
In analytical practice, SFE has found application to the isolation of PAHs, dioxins, pesticides, drugs, biologically active compounds, and many other organic compounds from soils and sediments [59, 60, 63, 66, 68, 70], food products [62, 63, 66, 70, 72, 77], plants [62, 63, 72, 74, 77], samples of forensic medical examination [64, 72, 76], as well as for extracting substances from sorption tubes, cartridges or membrane disks after sorption preconcentration [59]. The features of the online combination of sample preparation using SFE with supercritical fluid chromatography and other chromatographic methods were discussed in the reviews [59, 64, 77, 78].
MATRIX SOLID-PHASE DISPERSION
Matrix solid phase dispersion (MSPD) was proposed in 1989 for the extraction of drugs from animal tissues [79]. The essence of the method was to disperse an analyzed sample in the presence of a suitable sorbent, transfer the resulting homogeneous mass to a column or a cartridge, and then the elution of the target analytes with a selected solvent. The rapid development of the method has been facilitated by the fact that it does not require special equipment and is a simple and an inexpensive sample preparation procedure under mild conditions (room temperature and atmospheric pressure), which can be easily implemented in any laboratory. Since the inception of the method, a number of reviews have been published [40, 80–95] (Table 2). Currently, matrix solid-phase dispersion is used for the extraction of organic compounds from solid and liquid samples of food products and raw materials with both high and low fat content, from fruits and plants, as well as from solid environmental samples.
The principle of the method was described in detail in the first few reviews [80, 81, 83], and its theoretical aspects were covered in the reviews [82, 84, 85]. Historical information about the development of the method in different periods was presented in [85, 94, 95]. Thus, a 2023 review [95] noted that, over the period from 1989 to June 2022, more than 840 articles were published that used the term “MSPD” in the title, abstract, or keywords, with a particularly noticeable increase in interest in the use of this method of sample preparation prior to chromatographic determination has been observed in the last 15 years.
The features of sample preparation using the original version of the method, which includes three main stages, can be found in the reviews [40, 82–84, 88, 89, 93]. The first stage consists of manually mixing a sample with a pre-selected dispersing sorbent or a mixture of sorbents. This is a mechanical step, usually performed with a glass pestle in a glass or an agate mortar, because porous materials, such as porcelain, can lead to analyte and/or sample loss [82]. In some cases, at this stage matrix modifiers and drying agents are added to a mixture, and internal standards are also introduced. During the mixing process, the structure of the sample is disrupted and its uniform distribution on the sorbent particles occurs. The procedure is carried out until a dry, a smooth, a homogeneous, and a free-flowing powder is obtained. The mixing stage usually takes 0.5–15 min. At the second stage, the resulting homogeneous powder mixture is quantitatively transferred into an empty syringe or an SPE cartridge, in the lower part of which a sorbent is preliminarily placed to purify the extract and compacted. At the final third step, the target analytes are eluted with a suitable solvent. In some cases, before using an eluent, the column or the cartridge is washed with deionized water, weak acid solutions, or buffer solutions to remove unwanted compounds, or hexane to remove fats [40]. In the original version of the method, chemically modified silicas are used as sorbents for sample dispersion: hydrophobized silicas C8 and C18, BondeSil C18, BondeSil NH2, etc., which are often used together with mixing modifiers (acids, bases, salts, or EDTA). SiO2, Al2O3, or Florisil (synthetic magnesium silicate) are used as sorbents for purification, and Na2SO4 is used as a desiccant.
Over time, the sample preparation procedure using this method has been modified through the use of additional energy sources, such as ultrasound, microwave radiation, vortex action (stirring), or a magnetic field. Modified versions of the method were named matrix ultrasound-assisted solid-phase dispersion (UA-MSPD), microwave-assisted solid-phase dispersion (MWA-MSPD), vortex-assisted solid-phase dispersion (VA-MSPD), and magnetically assisted matrix solid-phase dispersion (MA-MSPD) [91–95]. Sample preparation using modified versions of the method in some cases allows not only to reduce extraction time, but also to reduce the amount of a sample required for an analysis, as well as the consumption of the organic solvents used. Methods for sample preparation using the matrix solid-phase dispersion method according to the original and modified versions are schematically presented in Fig. 2 [92]. Single examples of combinations of matrix solid-phase dispersion with pressurized liquid extraction were given in the reviews [40, 84, 85]. Another direction of the development of the method in recent years is its miniaturization, which became possible through the use of very sensitive detection methods. Reviews [84, 92, 95] provide examples of the developed micro/mini versions of the method, in which the mass of the analyzed samples is reduced from the generally accepted 0.5 g to 25–100 mg, and in some cases to 0.3–3 mg.

Options for conducting matrix solid-phase dispersion (MSPD) [92].
Experimental parameters influencing the matrix solid-phase dispersion method have been systematized in a number of reviews [82, 84, 85, 87, 91, 93]. It was noted that the main parameters influencing the selectivity and efficiency of the sample preparation process include the nature of the dispersing sorbent and the size of its particles, the mass ratio of the sample and to the sorbent, and the nature and volume of the solvent at the elution stage. In addition, in developing procedures, other aspects should be taken into account, such as the duration of mixing, a possibility of introducing a small volume of a selected solvent at the grinding stage to facilitate the destruction of the matrix, the use of an additional sorbent for purification (the so-called cosorbent), and a possibility of using an additional stage for the further purification of the eluate.
As mentioned above, in the original version of the method, reversed-phase sorbents are used as materials for sample dispersion, such as chemically modified silicas C18 and C8 and sorbents with phenyl, cyano, and amino groups, which not only destroy the structure of the sample, but also act in as a “bound” solvent, facilitating the complete destruction and dispersion of the sample [85, 89]. Using electron microscopy, it has been proven that a special mixed phase of a thickness of about 100 μm formed on the surface of chemically modified silicas, in which matrix components were retained due to hydrophobic and hydrophilic interactions [82]. Until now, chemically modified silicas are most often used in applied research related to the sample preparation of not only biological samples and food products, but also of solid environmental samples, such as dust [93, 95].
Other traditional dispersing materials are normal-phase sorbents, such as alumina, silica and Florisil (synthetic magnesium silicate) [85, 87, 89]. It is obvious that normal-phase sorbents are unable to dissolve the sample matrix, as is the case of reversed-phase sorbents. Target, polar analytes are retained on the surface of these sorbents due to various types of sorbate–sorbent interactions, mainly the formation of hydrogen bonds [87].
In some cases, at the stage of sample homogenization, cheaper solid inert materials are used, i.e., sea sand, celite or diatomite, which cause the mechanical destruction of a sample without an ability of solubilizing or adsorbing components, as happens with reversed- or normal-phase sorbents. The selectivity of the process in using inert materials is determined only by the solubility of various components of the sample in the eluting solvent [87, 89, 93].
Recent trends in improving the matrix solid-phase dispersion method are based on the use of new, more selective and effective sorbents. A comparative description of new sorbents that have found application to matrix dispersion, including advantages, limitations, possible interactions, and areas of application, was given in the review [95]. Reviews [91–93, 95] provide numerous examples indicating that the selectivity of the method can be increased through the use of molecularly imprinted polymers. Information on the use of carbon nanotubes, graphene, and other carbon-based nanomaterials and composites can be found in the reviews [91, 93, 95]. Finally, reviews [91, 93] provide examples of the use of magnetic nanosorbents in MSPD, and review [65] provides the first examples of the use of metal-organic frameworks, boron nitride nanosheets, calixarenes, and cucurbituril.
The particle size of the sorbent influences the elution stage. Very small sorbent particles (3–20 µm) lead to increased elution times and the need in applying positive pressure or vacuum. Usually in this method it is recommended to use particles of a sorbent or another dispersing material of a size of 40–100 µm. As for the mass ratio of the sample and the sorbent, in most methods it varies from 1 : 1 to 1 : 4, and the most commonly used mass ratio is 1 : 4. In the original version of the method, the recommended mass of the sample was 0.5 g, of the sorbent-dispersant, 2 g, and of the sorbent for purification, 0.5 g [83, 85].
The choice of washing and eluting solvents is very important to achieve complete extraction and obtain pure extracts [40, 87, 93, 95]. In some cases, to remove unwanted compounds, before using the eluent, the column or cartridge is washed with 1–2 mL of a specially selected additional solvent for every 100 mg of the sorbent‒sample mixture. The choice of this solvent depends on the nature of the interfering components, if they are known to the analyst in advance. It is important to ensure that these solvents do not remove the target analytes. Highly soluble compounds are removed using deionized water or buffer solutions, basic substances by solutions of weak acids, such as acetic acid, and fats by hexane.
In choosing an elution solvent, the solubility of the target analytes in it is primarily taken into account. In addition, the elution solvent must be compatible with the subsequent determination method. For example, for the subsequent HPLC determination, it is desirable to use a water-miscible solvent, and for determination by GC, a volatile solvent. According to the data presented in the reviews [87, 93, 95], acetonitrile, methanol, and their mixtures are usually used to elute analytes from reversed-phase sorbents, and hexane, methylene chloride, acetone, ethyl acetate, and their mixtures, from normal-phase sorbents. In recent years, ionic liquids, surfactants, and deep eutectic solvents have begun to be used as alternative solvents. Little work has been done in this direction; they were cited in the reviews [40, 91, 93, 95]. Another parameter to consider during the elution step is the volume of the solvent. In the original version of the method, for eluting analytes from a column filled with 0.5 g of a sample and 2 g of a sorbent-dispersant, it was recommended to use 8 mL of an eluent [85]. Obviously, depending on the nature of the matrix and the sorbents used, this volume can be both smaller and larger.
A 2023 review [95] based on a scientometric analysis of 840 publications from the Scopus database noted that the matrix solid-phase dispersion method has found application not only to the chemical analysis of various samples (35% of publications), but also in the field of biochemistry, genetics, and molecular biology (about 20% of publications), in the field of agricultural and biological sciences (almost 10% of publications), as well as in the fields of environmental sciences, pharmacology, toxicology, engineering, materials science, and medicine (together almost 35% of publications). Separate reviews and large sections in reviews were devoted to the use of matrix solid-phase dispersion for the isolation and preconcentration of organic compounds from environmental samples [40, 83, 86, 90, 93], food products [80–84, 90, 93], and plants [83, 84, 94, 95]. In these reviews, informative tables provide information on the release conditions for pesticides [40, 81, 84, 86, 95], medicinal compounds [80–82, 84, 86, 90, 93, 94], mycotoxins [86, 90], persistent organic pollutants [84, 86, 93], endocrine disrupting compounds [95], flavonoids [91, 95], and many other organic compounds.
QuEChERS METHOD
The QuEChERS method was proposed in 2003 as a new version of the sample preparation of fruits and vegetables with high moisture contents for the determination of pesticides in them [96]. The essence of the method was in the simultaneous extraction of pesticides (up to 200 compounds) from vegetables and fruits with acetonitrile in the presence of a large amount of salts (mainly magnesium sulfate) and the subsequent purification of the extract using dispersive solid-phase extraction using the PSA amino sorbent. The abbreviation QuEChERS contains the most important advantages of the method (Quick, Easy, Cheap, Effective, Rugged, and Safe). The QuEChERS method made it possible to radically simplify the analysis of vegetables and fruits in determining pesticide residues in them, and subsequently was widely used for the extraction of a wide variety of organic compounds from food, natural, biological, and other matrices [97]. Currently, the QuEChERS method is a basis of standard official methods recommended for use in the United States, Europe, and other countries. More than 30 suppliers worldwide offer QuEChERS extraction kits. Undoubtedly, QuEChERS has become one of the most commonly used and popular analysis methods. According to Web of Science, by 2020, almost 4500 publications and a large number of reviews have been published [97–116], which are listed in chronological order in Table 2. The review [101] notes that QuEChERS should be viewed as a concept (methodology) for sample preparation rather than as a specific method.
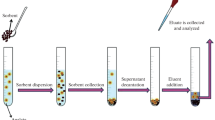
An original sample preparation procedure using the QuEChERS method, which was originally used to isolate pesticides from vegetables and fruits, includes the sequential implementation of several steps, which are illustrated in Fig. 3 [106]. In order to improve the overall efficiency of the procedure, the original QuEChERS method was subjected to several modifications, which were described in the reviews [99–101, 106, 107, 110] and are schematically presented in Fig. 4 [110]. Initially, these modifications were aimed at increasing the efficiency of the extraction of polar and non-polar pesticides belonging to different classes, and consisted of introducing acetate or citrate buffer solutions at the extraction stage. It should be noted that the acetate and citrate modifications of QuEChERS form a basis of two official methods: the AOAC 2007.01 method and the official method of the European Committee for Standardization (Standard Method UNE-EN 15662, CEN2008) (Fig. 4) [108, 110]. Adaptation of the QuEChERS procedure to foods with medium or high fat contents, highly pigmented foods, foods with a high chlorophyll content, and items with a water content of less than 75–80% has led to the need for further modifications [101]. These changes included the use of different solvents and salting out agents at the extraction stage and sorbents at the purification stage, as well as the introduction of freeze-out strategies (Fig. 4), which allowed the implementation of the QuEChERS methodology for the multi-component recovery of pesticides, mycotoxins, polyphenols, PAH, antibiotics, and other organic compounds from various matrices, including a variety of foods, environmental samples, and biological fluids [107, 110].

Main stages of the original QuEChERS method [106].

Options for improving the QuEChERS protocol. Abbreviations: d-SPE is dispersive solid phase extraction, GC is gas chromatography, HPLC is high performance liquid chromatography, DMD is diode array detector, FL is fluorescence detector, MS is mass spectrometric detector, PSA is primary-secondary amine, SCDS is sesquihydrate of dibasic sodium citrate, SCTD—tribasic sodium citrate dihydrate, Z-Sep—sorbents based on zirconium dioxide, cmin—detection limit [110].
In the process of modification, researchers varied various experimental parameters, which were described in detail in the reviews [100, 101, 103, 106, 111, 112]. Conventionally, they can be divided into parameters varied at the stages of sample preparation, extraction, and purification. Sample preparation of solid samples before the QuEChERS procedure involves thoroughly grinding them to increase the surface area. Depending on the nature of the matrix, adjustments are made to this simple procedure. For example, samples with a high fat content are frozen overnight before grinding, or dry ice is added to a sample during grinding to obtain finer fractions. As in the original QuEChERS method, most studies use samples weighing 10 g, but, according to the recent data, by increasing the sensitivity of analytical instruments, sample weight can be reduced to 5, 2, or even 1 g [103, 112]. Reviews [101, 103, 106, 112] provide information on the need to add water to samples if its content does not exceed 75–80%. The amount of the added water depends, as follows from the tables given in the reviews [101, 106], on the type of the food product and can be equal to the weight of the sample or exceed it by one and a half to two times. Water is also added to the analyzed soil samples; the best results were obtained when 5 g of a sample was moistened with 10 mL of water [103, 112].
The choice of an extraction solvent plays a critical role in achieving the maximum recovery of organic compounds during the extraction step. Depending on the nature of the analytes and the task at hand, the solvent must selectively extract the analytes or groups of analytes and be easily separated from water. In addition, in choosing a solvent, its compatibility with the subsequent chromatographic determination, cost, safety, and environmental friendliness are taken into account. In the original QuEChERS method, acetonitrile was chosen as a solvent and, as further studies showed, acetonitrile turned out to be the best extraction solvent not only for the multicomponent extraction of pesticides with a wide range of polarities from fruits and vegetables [98, 100, 111], but also for other food products [98, 107, 108, 110], soils [103, 112], medicinal substances from food products [103, 112], and many other organic compounds from natural and biological samples [106, 107, 110, 111]. In addition to acetonitrile, other solvents are used to extract lipophilic compounds from fat matrices, such as methanol, acetone, ethyl acetate, a mixture of acetone with hexane, or other organic solvents [100, 106, 111]. Thus, in the review [100] it was noted that, for the extraction of 12 PAHs from ham or 33 PAHs from fish, satisfactory results were obtained using ethyl acetate or a mixture of acetone, ethyl acetate, and isooctane, respectively; for the extraction of antibiotics from fish, a mixture of acetonitrile with methanol was used [106], and ethyl acetate and a mixture of acetone with hexane were used to extract persistent organic pollutants [111]. In addition to the nature of the solvent, its volume also affects the efficiency of analyte extraction. The review [106] noted that, in most cases, the ideal solvent/sample ratio is 1 : 1, too small volume results in the incomplete recovery of the target analytes, and excessive volume increases the cost of an analysis.
Salting out agents play an important role at the extraction/separation step. During the development of the QuEChERS methodology, a wide variety of salts and their mixtures were tested as salting out agents: MgSO4, NaCl, Na2SO4, ammonium chloride and formate, and sodium acetate and citrate [100, 106, 112]. In a number of works listed in the review [106] it was noted that the best inorganic salt promoting liquid–liquid phase separation was MgSO4, whose dehydration ability is approximately four times higher than that of anhydrous Na2SO4. A mixture of MgSO4 and NaCl in a ratio of 4 : 1 is most often used as an effective salting out agent in the extraction of pesticides and most other organic analytes [106]. A mixture of MgSO4, NaCl, and sodium acetate in a ratio of 4 : 1 : 1 provided the best separation efficiency while simultaneously isolating 42 pesticides and 23 other organic compounds belonging to different classes from soils [112]. However, these salts tend to be deposited as particulate matter on the surfaces of the mass spectrometer ion source and possibly inside the analyzer or in the gas chromatograph inlet tubing, resulting in the loss of instrument performance and requiring longer maintenance. Therefore, in some cases they are replaced with ammonium chloride or formate [100].
Traditional sorbents that are most often used at the purification stage include the PSA amino sorbent (a mixture of primary and secondary amines), C18 and graphitized carbon black. The PSA sorbent is commonly used to remove sugars, organics, and fatty acids. Silica C18 is effective for purifying the extract from various fats, sterols, humic acids, and other non-polar compounds, and graphitized soot is used to remove pigments (carotenoids and chlorophyll) [103, 106, 111]. Depending on the nature of the analyzed matrices and analytes, the above sorbents can be used together at the purification stage; MgSO4 is often added to them [101]. The quantities of traditional sorbents and their combinations used at the purification stage in the official methods of AOAC and CEN 15662 were given in the review [101]. These sorbents are increasingly used not only for the purification of extracts of vegetables, fruits [98, 105, 109], other food products [101, 108], and soils [103, 112] in determining pesticides in them, but also for the purification of food product extracts in the determination of medicinal substances, PAHs, and other organic compounds [101, 102, 106, 111]. In addition, new, alternative sorbents have been developed and used for the sample purification of complex matrices. Let’s give a few examples. For the selective removal of chlorophyll from green plant extracts, instead of graphitized soot, the main disadvantage of which is a decrease in the recovery of analytes with a planar structure, it was proposed to use the ChloroFiltr® sorbent. This sorbent has been tested in the determination of hundreds of pesticides and herbicides and has shown a reduction in the chlorophyll content of more than 82% without a loss of analytes [100, 101]. Other new commercially available sorbents are Z-Sep and Z-Sep Plus, developed by Supelco (United States). Z-Sep is a sorbent based on zirconia-modified silica, and the Z-Sep Plus sorbent includes zirconia and C18. These sorbents remove more fats and pigments from the extracts than traditional PSA and C18, and also provide higher analyte recovery and better reproducibility [101, 110]. As an alternative to Z-sorbents for improved fat removal, Agilent (United States) has developed and produced an innovative material, EMR-Lipid (enhanced matrix removal) [107, 110, 111]. The action of EMR-Lipid is based on a unique combination of exclusion and hydrophobic interactions. According to the manufacturer, EMR-Lipid selectively removes major classes of lipids from extracts of fat-containing samples, such as avocado and animal tissue, without a loss of pesticides, veterinary drugs, or PAH.
In addition to the sorbents listed above, which are included in QuEChERS kits produced by different companies, a number of studies were devoted to assessing a possibility of using new nanostructured sorbents, such as multi-walled carbon nanotubes and their derivatives, magnetic nanoparticles, metal-organic frameworks, covalent organic frameworks, graphene oxide, and some others. A complete list of nanostructured sorbents that have found application in the QuEChERS method can be found in one of the latest reviews [114], which examines the strengths and weaknesses of each nanomaterial, and also discusses the problems that can be encountered in using them.
An analysis of review works carried out as part of this review indicates that the QuEChERS method has found wide application to chemical analysis as an effective method for the sample preparation of samples of varied composition and complexity with the subsequent determination of organic compounds by chromatographic methods. Separate reviews were devoted to the application of the QuEChERS method for the extraction of pesticides from fruits and vegetables [98, 105, 109], food products [101, 108], and soils [103, 112]; antibiotics and other medicinal substances from food [101, 102, 106]; PAHs and other persistent organic pollutants from food [101, 111]; mycotoxins from food products [116]. Reviews are devoted to sample preparation of environmental samples, food products and biological samples using the QuEChERS method for determining various organic compounds in them [99, 107, 108]. The features of combining sample preparation using the QuEChERS method with the subsequent determination of pesticides with various traditional chromatographic detectors for GC and HPLC were discussed in the review [104]. In the review [102], using an example of medicinal substances, features of a combination of sample preparation using the QuEChERS method with the subsequent determination of compounds by GC-MS were discussed. The reviews cited above contain tables that indicate the analyzed samples and analytes, sample weight, extractants, salting out agents, sorbents and their quantities, as well as the recoveries of analytes and analytical ranges.
Several reviews have compared the QuEChERS method with other solid sample preparation methods discussed above [99, 103, 110, 112]. The most comprehensive such comparison is given in a review [110], where the table lists analytes and samples for which QuEChERS is an effective high-throughput alternative capable of providing similar or better analytical performance, including lower matrix effects, without the need for special devices. You can also find examples where the QuEChERS method demonstrated worse analytical performance compared to other sample preparation methods.
Another significant advantage of the method is that it allows one to simultaneously extract a very large number of analytes belonging to different classes with high efficiency. Thus, the review [110], as an example, discussed a possibility of the simultaneous extraction of 243 pesticides from cardamom and 137 veterinary drugs and their metabolites from fish. The review [112] provides a link to an original work indicating a possibility of the simultaneous extraction of 225 pesticides from soils along with 80 other organic compounds. It is important to note that the QuEChERS method meets most of requirements for green analytical methods, because it reduces the consumption of toxic solvents and reagents and produces much less wastes.
The review [113], written by one of the developers of the QuEChERS method, described and explained the changes made to this method led to the development of the QuEChERSER method, with the terms “Efficient and Robust” added to the standard abbreviation. The author noted that the QuEChERSER method should be considered a “mega method” covering a wider range of polar and non-polar analytes in different sample types. It was noted that the emergence of QuEChERSER stimulated the development of chromatographic analytical equipment with mass spectrometric detection. Obviously, the most effective way to increase the efficiency of an analytical laboratory is to reduce the number of methods required for analyzing an identical list of substances. For example, four separate methods are typically used to monitor pesticides, environmental contaminants, veterinary drugs, and mycotoxins in relevant foods, but with QuEChERSER, an identical sample can be prepared using the same method to detect contaminants in all types of food products. The review includes a table comparing changes made to QuEChERSER versus QuEChERS during sample preparation, extraction, and purification. It was noted that great attention should be paid to grinding the samples, which should be carried out using liquid nitrogen. As modern analytical instruments are capable of providing limits of detection lower by several orders of magnitude compared to those at the time of QuEChERS development, the QuEChERSER developers recommend, on the one hand, to reduce the mass of the analyzed samples to 1–5 g, and on the other hand, to increase the volume of the extractant. For example, to extract residues of veterinary drugs from 2 g of food products of animal origin, it was recommended to use 10 mL of a mixture of acetonitrile with water in a ratio of 4 : 1 (by volume) as an extractant. Moreover, this solvent-to-sample ratio (5 mL/g) ensures the quantitative extraction of not only veterinary drugs, but also of pesticides, mycotoxins, and other organic pollutants from various matrices in one step. Information about other changes can be obtained both from the text and from the informative table provided in this overview. In addition, the review discussed modern approaches to automating sample preparation using the QuEC-hERSER method.
CONCLUSIONS
Thus, from the two parts of the literature review on methods for extracting organic compounds from solid samples, the following conclusions can be drawn. Solid sample preparation, including liquid–liquid extraction, is an inevitable and one of the most important steps in any analysis. This stage largely determines the accuracy of an analysis as a whole, so it should be treated with the maximum attention. The most commonly used methods for extracting organic compounds from solid matrices are shaking liquid–liquid extraction, Soxhlet extraction, ultrasound-assisted extraction (UAE), microwave-assisted extraction (MAE), pressurized liquid extraction (PLE), subcritical water extraction (SWE), supercritical fluid extraction (SFE), matrix solid-phase dispersion (MSPD), and the QuEChERS method. As a diagram, Fig. 5 shows the total number of reviews published on each of these methods over the period from the inception of the method to the present time, and the number of reviews published over the past five years. There is a clear trend towards the rapid development of simple, rapid, cost-effective, and user-friendly sample preparation methods, such as UAE and QuEChERS, with 7 and 10 reviews published in the last five years, respectively. The QuEChERS method has become particularly popular around the world because it meets the needs of modern laboratories, including the reduced use of solvents and materials, as well as the reduced time and cost of an analysis. Over the past five years, as seen in the reviews on pressurized liquid extraction and matrix solid-phase dispersion, many interesting papers have been published indicating that these methods, which combine analyte extraction with the subsequent purification of the extracts, continue to evolve. This is largely due to the emergence of new sorbents, including nanostructured ones, “green” solvents, and miniaturized schemes that, in the case of MSPD, combine extraction with other sample preparation options. As for microwave-assisted extraction, subcritical water extraction, and supercritical fluid extraction, their use in chemical analysis has noticeably decreased, but the number of works devoted to the use of these methods for extracting biologically active substances from plants has increased.

Number of reviews on methods for isolating organic compounds from solid samples published over the entire period and over the last five years. Abbreviations: UAE is ultrasound-assisted extraction, MAE is microwave-assisted extraction, PLE is pressurized liquid extraction, SWE is subcritical water extraction, SFE is supercritical fluid extraction, MSPD is matrix solid-phase dispersion.
REFERENCES
Majors, R.E., LC GC Eur., 2007, vol. 20, p. 71.
Ridgway, K., Lalljie, S.P.D., and Smith, R.M., J. Chromatogr. A, 2007, vol. 1153, p. 36. https://doi.org/10.1016/j.chroma.2007.01.134
Brinco Silva, J., Mateus, E.P., Ribeiro, A.B., Guedes, P., and Gomes, M., Microchem. J., 2023, vol. 195. https://doi.org/10.1016/j.microc.2023.109465
Qin, H., Liu, H., Liu, Y., Di, S., Bao, Y., Zhai, Y., and Zhu, S., TrAC, Trends Anal. Chem., 2023, vol. 164, p. 117112. https://doi.org/10.1016/j.trac.2023.117112
Picot-Allain, C., Mahomoodally, M.F., Ak, G., and Zengin, G., Curr. Opin. Food Sci., 2021, vol. 40, p. 144. https://doi.org/10.1016/j.cofs.2021.02.009
Armenta, S., Garrigues, S., Esteve-Turrillas, F.A., and de la Guardia, M., TrAC, Trends Anal. Chem., 2019, vol. 116, p. 248. https://doi.org/10.1016/j.trac.2019.03.016
Aly, A.A. and Górecki, T., Molecules, 2020, vol. 25, p. 1719. https://doi.org/10.3390/molecules25071719
Syrgabek, Y., Alimzhanova, M., García-Encina, P.A., Jiménez, J.J., and López-Serna, R., Trends Environ. Anal. Chem., 2023, vol. 39. https://doi.org/10.1016/j.teac.2023.e00206
Usman, M., Nakagawa, M., and Cheng, S., Processes, 2023, vol. 11, p. 3444. https://doi.org/10.3390/pr11123444
Moreda-Piñeiro, J. and Moreda-Piñeiro, A., TrAC, Trends Anal. Chem., 2019, vol. 118, p. 1. https://doi.org/10.1016/j.trac.2019.05.026
Moreda-Piñeiro, J. and Moreda-Piñeiro, A. TrAC, Trends Anal. Chem., 2023, vol. 169, p. 117411 https://doi.org/10.1016/j.trac.2023.117411
Maciel-Silva, F.W., Lachos-Perez, D., Buller, L.S., Sganzerla, W.G., Pérez, M., Rostagno, M.A., and Forster-Carneiro, T., Molecules, 2022, vol. 27, p. 6272. https://doi.org/10.3390/molecules27196272
Cetinkaya, A., Kaya, S.I., and Ozkan, S.A., TrAC, Trends Anal. Chem., 2023, vol. 168, p. 117330. https://doi.org/10.1016/j.trac.2023.117330
Dmitrienko, S.G., Apyari, V.V., Tolmacheva, V.V., Gorbunova, M.V., Furletov, A.A., and Zolotov, Yu.A., J. Anal. Chem., 2024, vol. 79, no. 8 (in press).
Mendiola, J.A., Herrero, M., Cifuentes, A., and Ibañez, E., J. Chromatogr. A, 2007, vol. 1152, p. 234. https://doi.org/10.1016/j.chroma.2007.02.046
Herrero, M., Castro-Puyana, M., Mendiola, J.A., and Ibañez, E., TrAC, Trends Anal. Chem., 2013, vol. 43, p. 67. https://doi.org/10.1016/j.trac.2012.12.008
Amador-Luna, V.M., Montero, L., and Herrero, M., TrAC, Trends Anal. Chem., 2023, vol. 169, p. 117410.
Gallego, R., Bueno, M., and Herrero, M., TrAC, Trends Anal. Chem., 2019, vol. 116, p. 198. https://doi.org/10.1016/j.trac.2019.04.030
Yousefi, M., Rahimi-Nasrabadi, M., Pourmortazavi, S.M., Wysokowski, M., Jesionowski, T., Ehrlich, H., and Mirsadeghi, S., TrAC, Trends Anal. Chem., 2019, vol. 118, p. 182. https://doi.org/10.1016/j.trac.2019.05.038
Uwineza, P.A. and Waskiewicz, A., Molecules, 2020, vol. 25, p. 3847.
Dias, A.L.B., de Aguiar, A.C., and Rostagno, M.A., Ultrason. Sonochem., 2021, vol. 74, p. 105584. https://doi.org/10.1016/j.ultsonch.2021.105584
Arumugham, T., Rambabu, K., Hasan, S.W., Show, P.L., Rinklebe, J., and Banat, F., Chemosphere, 2021, vol. 271, p. 29525. https://doi.org/10.1016/j.chemosphere.2020
Qamar, S., Torres, Y.J.M., Parekh, H.S., and Falconer, J.R., J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2021, vol. 1167, p. 122581. https://doi.org/10.1016/j.jchromb.2021.12258
López-Hortas, L., Rodríguez, P., Díaz-Reinoso, B., Gaspar, M.C., de Sousa, H.C., Braga, M.E.M., and Dominguez, H., J. Supercrit. Fluids, 2022, vol. 188, p. 105652. https://doi.org/10.1016/j.supflu.2022.105652
Fraguela-Meissimilly, H., Bastias-Monte, J.M., Vergara, C., Ortiz-Viedma, J., Lemus-Mondaca, R., Flores, M., Toledo-Merma, P., Alcazar-Alay, S., and Gallon-Bedoya, M., Molecules, 2023, vol. 28, p. 4421. https://doi.org/10.3390/molecules28114421
Bjorklund, E., Nilsson, T., and Bowadt, S., TrAC, Trends Anal. Chem., 2000, vol. 9, p. 434. https://doi.org/10.1016/S0165-9936(00)00002-9
Giergielewicz-Możajska, H., Dąbrowski, Ł., and Namieśnik, J., Crit. Rev. Anal. Chem., 2001, vol. 31, p. 149. https://doi.org/10.1080/20014091076712
Ramos, L., Kristenson, E.M., and Brinkman, U.A.T., J. Chromatogr. A, 2002, vol. 975, p. 3. https://doi.org/10.1016/s0021-9673(02)01336-5
Luque-Garcia, J.L. and Luque de Castro, M.D., TrAC, Trends Anal. Chem., 2004, vol. 23, p. 102.
Carabias-Martinez, R., Rodríguez-Gonzalo, E., Revilla-Ruiz, P., and Hernández-Méndez, J., J. Chromatogr. A, 2005, vol. 1089, p. 1. https://doi.org/10.1016/j.chroma.2005.06.072
Schantz, M.M., Anal. Bioanal. Chem., 2006, vol. 386, p. 1043. https://doi.org/10.1007/s00216-006-0648-2
Bjorklund, E., Sporring, S., Wiberg, K., Haglund, P., and von Holst, C., TrAC, Trends Anal. Chem., 2006, vol. 25, p. 318.
Nieto, A., Borrull, F., Pocurull, E., and Marce, R.M., TrAC, Trends Anal. Chem., 2010, vol. 29, p. 752. https://doi.org/10.1016/j.trac.2010.03.014
Runnqvist, H., Bak, S.A., Hansen, M., Styrishave, B., Halling-Sorensen, B., and Björklund, E., J. Chromatogr. A, 2010, vol. 1217, p. 2447. https://doi.org/10.1016/j.chroma.2010.02.046
Mustafa, A. and Turner, C., Anal. Chim. Acta, 2011, vol. 703, p. 8. https://doi.org/10.1016/j.aca.2011.07.018
Sun, H., Ge, X., Lv, Y., and Wang, A., J. Chromatogr. A, 2012, vol. 1237, p. 1. https://doi.org/10.1016/j.chroma.2012.03.003
Carro, A.M., González, P., and Lorenzo, R.A., J. Chromatogr. A, 2013, vol. 1296, p. 214. https://doi.org/10.1016/j.chroma.2013.04.068
Subedi, B., Aguilar, L., Robinson, E.M., Hageman, K.J., Björklund, E., Sheesley, R.J., and Usenko, S., TrAC, Trends Anal. Chem., 2015, vol. 68, p. 119. https://doi.org/10.1016/j.trac.2015.02.011
Vazquez-Roig, P. and Picó, Y., TrAC, Trends Anal. Chem., 2015, vol. 71, p. 55. https://doi.org/10.1016/j.trac.2015.04.014
Hoff, R.B. and Pizzolato, T.M., TrAC, Trends Anal. Chem., 2018, vol. 109, p. 83. https://doi.org/10.1016/j.trac.2018.10.002
Andreu, V. and Picó, Y., TrAC, Trends Anal. Chem., 2019, vol. 118, p. 709. https://doi.org/10.1016/j.trac.2019.06.038
Cao, Y., Liu, W., Gong, X., Yu, J., Tu, P., Li, J., and Song, Y., J. Pharm. Biomed. Anal., 2021, vol. 205, p. 114332. https://doi.org/10.1016/j.jpba.2021.114332
Fontanals, N., Pocurull, E., Borrull, F., and Marcé, R.M., Trends Environ. Anal. Chem., 2021, vol. 29, p. e00111. https://doi.org/10.1016/j.teac.2020.e00111
Barp, L., Višnjevec, A.M., and Moret, S., Foods, 2023, vol. 12, p. 2017. https://doi.org/10.3390/foods12102017
Smith, R.M., J. Chromatogr. A, 2002, vol. 975, p. 31. https://doi.org/10.1016/S0021-9673(02)01225-6
Weingartner, H. and Franck, E.U., Angew. Chem., Int. Ed. Engl., 2005, vol. 44, p. 2672. https://doi.org/10.1002/anie.200462468
Smith, R.M., Anal. Bioanal. Chem., 2006, vol. 385, p. 419. https://doi.org/10.1007/s00216-006-0437-y
Ong, E.S., Cheong, J.S.H., and Goh, D., J. Chromatogr. A, 2006, vol. 1112, p. 92. https://doi.org/10.1016/j.chroma.2005.12.052
Kronholm, J., Hartonen, K., and Riekkola, M.-L., TrAC, Trends Anal. Chem., 2007, vol. 26, p. 396. https://doi.org/10.1016/j.trac.2007.03.004
Teo, C.C., Tan, S.N., Yong, J.W.H., Hew, C.S., and Ong, E.S., J. Chromatogr. A, 2010, vol. 1217, p. 2484. https://doi.org/10.1016/j.chroma.2009.12.050
Plaza, M. and Turner, C., TrAC, Trends Anal. Chem., 2015, vol. 71, p. 39. https://doi.org/10.1016/j.trac.2015.02.022
Gbashi, S., Adebo, O.A., Piater, L., Madala, N.E., and Njobeh, P.B., Sep. Purif. Technol., 2016, vol. 46, p. 21. https://doi.org/10.1080/15422119.2016.1170035
Castro-Puyana, M., Marina, M.L., and Plaza, M., Curr. Opin. Green Sustainable Chem., 2017, vol. 5, p. 31. https://doi.org/10.1016/j.cogsc.2017.03.00
Borisova, D.R., Statkus, M.A., Tsizin, G.I., and Zolotov, Yu.A., J. Anal. Chem., 2017, vol. 72, no. 8, p. 823. https://doi.org/10.1134/S1061934817080044
Zhang, J., Wen, C., Zhang, H., Duan, Y., and Ma, H., Trends Food Sci. Technol., 2019, vol. 95, p. 183. https://doi.org/10.1016/j.tifs.2019.11.018
Essien, S.O., Young, B., and Baroutian, S., Trends Food Sci. Technol., 2020, vol. 97, p. 156. https://doi.org/10.1016/j.tifs.2020.01.014
Cheng, Y., Xue, F., Yu, S., Du, S., and Yang, Y., Molecules, 2021, vol. 26, p. 1. https://doi.org/10.3390/molecules26134004
Hawthorne, S.B., Anal. Chem., 1990, vol. 62, p. 633. https://doi.org/10.1021/ac00210a722
Janda, V., J. Chromatogr. A, 1993, vol. 642, p. 283.
Camel, V., Tambuté, A., and Caude, M., J. Chromatogr. A, 1993, vol. 642, p. 263.
Bowadt, S. and Hawthorne, S.B., J. Chromatogr. A, 1995, vol. 703, p. 549. https://doi.org/10.1016/0021-9673(95)00051-n
Lehotay, S.J., J. Chromatogr. A, 1997, vol. 785, p. 289. https://doi.org/10.1016/s0021-9673(97)00461-5
Motohashi, N., Nagashima, H., and Párkányi, C., J. Biochem. Biophys. Methods, 2000, vol. 43, p. 313. https://doi.org/10.1016/s0165-022x(00)00052-x
Radcliffe, C., Maguire, K., and Lockwood, B., J. Biochem. Biophys. Methods, 2000, vol. 43, p. 261. https://doi.org/10.1016/s0165-022x(00)00058-0
Lang, Q. and Wai, C.M., Talanta, 2001, vol. 53, p. 771. https://doi.org/10.1016/S0039-9140(00)00557-9
Turner, C., Eskilsson, C.S., and Bjórklund, E., J. Chromatogr. A, 2002, vol. 947, p. 1. https://doi.org/10.1016/s0021-9673(01)01592-8
Zougagh, M., Valcárcel, M., and Ríos, A., TrAC, Trends Anal. Chem., 2004, vol. 23, p. 399. https://doi.org/10.1016/s0165-9936(04)00524-2
Anitescu, G. and Tavlarides, L.L., J. Supercrit. Fluids, 2006, vol. 38, p. 167. https://doi.org/10.1016/j.supflu.2006.03.024
Abbas, K.A., Mohamed, A., Abdulamir, A.S., and Abas, H.A., Am. J. Biochem. Biotechnol., 2008, vol. 4, p. 345. https://doi.org/10.3844/ajbbsp.2008.345.353
Sunarso, J. and Ismadji, S., J. Hazard. Mater., 2009, vol. 161, p. 1. https://doi.org/10.1016/j.jhazmat.2008.03.069
Sapkale, G., Patil, S., Surwase, U., and Bhatbhage, P., Int. J. Chem. Sci., 2010, vol. 8, p. 729.
Herrero, M., Mendiola, J.A., Cifuentes, A., and Ibanez, E., J. Chromatogr. A, 2010, vol. 1217, p. 2495. https://doi.org/10.1016/j.chroma.2009.12.019
Machida, H., Takesue, M., and Smith, R.L., J. Supercrit. Fluids, 2011, vol. 60, p. 2. https://doi.org/10.1016/j.supflu.2011.04.016
Xu, L., Zhan, X., Zeng, Z., Chen, R., Li, H., Xie, T., and Wang, S., Afr. J. Pharm. Pharmacol., 2011, vol. 5, p. 1196. https://doi.org/10.5897/AJPP11.228
Huang, Z., Shi, X., and Jiang, W., J. Chromatogr. A, 2012, vol. 1250, p. 2. https://doi.org/10.1016/j.chroma.2012.04.032
Pokrovskii, O., Analitika, 2013, vol. 6, p. 23.
Pourmortazavi, S.M., Rahimi-Nasrabadi, M., and Hajimirsadeghic, S.S., Curr. Anal. Chem., 2014, vol. 10, p. 3.
Zoccali, M., Donato, P., and Mondello, L., TrAC, Trends Anal. Chem., 2019, vol. 116, p. 158. https://doi.org/10.1016/j.trac.2019.04.028
Barker, S.A., Long, A.R., and Short, C.R., J. Chromatogr. A, 1989, vol. 475, p. 353.
Walker, C.C., Lott, H.M., and Barker, S.A., J. Chromatogr. A, 1993, vol. 642, p. 225.
Barker, S.A., J. Chromatogr. A, 2000, vol. 880, p. 63. https://doi.org/10.1016/s0021-9673(99)01290-x
Barker, S.A., J. Chromatogr. A, 2000, vol. 885, p. 115.
Karasová, G., Brandšteterová, E., and Lachová, M., Czech J. Food Sci., 2003, vol. 21, p. 219.
Kristenson, E.M., Brinkman, U.A.Th., and Ramos, L., TrAC, Trends Anal. Chem., 2006, vol. 25, p. 96. https://doi.org/10.1016/j.trac.2005.05.011
Barker, S.A., J. Biochem. Biophys. Methods, 2007, vol. 70, p. 151. https://doi.org/10.1016/j.jbbm.2006.06.005
Bogialli, S. and Di Corcia, A., J. Biochem. Biophys. Methods, 2007, vol. 70, p. 163. https://doi.org/10.1016/j.jbbm.2006.07.007
García-López, M., Canosa, P., and Rodríguez, I., Anal. Bioanal. Chem., 2008, vol. 391, p. 963. https://doi.org/10.1007/s00216-008-1898-y
Moreda-Piñeiro, J., Alonso-Rodríguez, E., López-Mahía, P., Muniategui-Lorenzo, S., Prada-Rod-ríguez, D., Romarís-Hortas, V., Míguez-Framil, M., Moreda-Piñeiro, A., and Bermejo-Barrera, P., TrAC, Trends Anal. Chem., 2009, vol. 28, p. 110. https://doi.org/10.1016/j.trac.2008.09.016
Capriotti, A.L., Cavaliere, C., Giansanti, P., Gubbiotti, R., Samperi, R., and Lagana, A., J. Chromatogr. A, 2010, vol. 1217, p. 2521. https://doi.org/10.1016/j.chroma.2010.01.030
Capriotti, A.L., Cavaliere, C., Lagana, A., Piovesana, S., and Samperi, R., TrAC, Trends Anal. Chem., 2013, vol. 43, p. 53.
Capriotti, A.L., Cavaliere, C., Foglia, P., Samperi, R., Stampachiacchiere, S., Ventura, S., and Lagana, A., TrAC, Trends Anal. Chem., 2015, vol. 71, p. 186. https://doi.org/10.1016/j.trac.2015.03.012
Tu, X. and Chen, W., Molecules, 2018, vol. 23, p. 2767. https://doi.org/10.3390/molecules23112767
Ramos, L., TrAC, Trends Anal. Chem., 2019, vol. 118, p. 751. https://doi.org/10.1016/j.trac.2019.07.006
Wianowska, D. and Gil, M., TrAC, Trends Anal. Chem., 2019, vol. 112, p. 29. https://doi.org/10.1016/j.trac.2018.12.028
El-Deen, A.K., Sep. Purif. Rev., 2023, vol. 53, p. 1. https://doi.org/10.1080/15422119.2023.2172734
Anastassiades, M., Lehotay, S., Stajnbaher, D., and Schenck, F., J. AOAC Int., 2003, vol. 86, p. 412.
Lehotay, S.J., Anastassiades, M., and Majors, R.E., LC GC Eur., 2010, vol. 23, p. 418.
Wilkowska, A. and Biziuk, M., Food Chem., 2011, vol. 125, p. 803. https://doi.org/10.1016/j.foodchem.2010.09.094
Bruzzoniti, M.C., Checchini, L., De Carlo, R.M., Orlandini, S., Rivoira, L., and Del Bubba, M., Anal. Bioanal. Chem., 2014, vol. 406, p. 4089. https://doi.org/10.1007/s00216-014-7798-4
González Curbelo, M.Á., Socas-Rodríguez, B., Herrera-Herrera, A., González-Sálamo, J., Hernández-Borges, J., and Rodríguez-Delgado, M.Á., TrAC, Trends Anal. Chem., 2015, vol. 71, p. 169.
Rejczak, T. and Tuzimski, T., Open Chem., 2015, vol. 13, p. 980. https://doi.org/10.1515/chem-2015-0109
Schmidt, M.L. and Snow, N.H., TrAC, Trends Anal. Chem., 2016, vol. 75, p. 49. https://doi.org/10.1016/j.trac.2015.07.012
Pszczolinska, K. and Michel, M., J. AOAC Int., 2016, vol. 99, p. 1403. https://doi.org/10.5740/jaoacint.16-0274
Rahman, M.M., Abd, El-Aty A.M., Kim, S.-W., Shin, S.C., Shin, H.-C., and Shim, J.-H., J. Sep. Sci., 2016, vol. 40, p. 203. https://doi.org/10.1002/jssc.201600889
Lawal, A., Wong, R.C.S., Tan, G.H., Abdulra’uf, L.B., and Alsharif, A.M.A., J. Chromatogr. Sci., 2018, vol. 56, p. 656. https://doi.org/10.1093/chromsci/bmy032
Zhang, C., Deng, Y., Zheng, J., Zhang, Y., Yang, L., Liao, C., Su, L., Zhou, Y., Gong, D., Chen, L., and Luo, A., TrAC, Trends Anal. Chem., 2019, vol. 118, p. 517. https://doi.org/10.1016/j.trac.2019.06.01
Santana-Mayor, Á., Socas-Rodríguez, B., Herrera-Herrera, A.V., and Rodriguez-Delgado, M.Á., TrAC, Trends Anal. Chem., 2019, vol. 116, p. 214. https://doi.org/10.1016/j.trac.2019.04.018
Musarurwa, H., Chimuka, L., Pakade, V.E., and Tavengwa, N.T., J. Food Compos. Anal., 2019, vol. 84, p. 103314. https://doi.org/10.1016/j.jfca.2019.103314
Alcântara, D.B., Fernandes, T.S.M., Nascimento, H.O., Lopes, A.F., Menezes, M.G.G., Lima, A.C.A., Carvalho, T.V., Grinberg, P., Milhome, M.A.L., Oliveira, A.H.B., Becker, H., Zocolo, G.J., and Nascimento, R.F., Food Chem., 2019, vol. 298, p. 124958. https://doi.org/10.1016/j.foodchem.2019.124958
Perestrelo, R., Silva, P., Porto-Figueira, P., Pereira, J.A.M., Silva, C., Medina, S., and Camara, J.S., Anal. Chim. Acta, 2019, vol. 1070, p. 1. https://doi.org/10.1016/j.aca.2019.02.036
Kim, L., Lee, D., Cho, H.-K., and Choi, S.-D., Trends Environ. Anal. Chem., 2019, vol. 22, p. e00063. https://doi.org/10.1016/j.teac.2019.e00063
González-Curbelo, M.Á., Varela-Martínez, D.A., and Riaño-Herrera, D.A., Molecules, 2022, vol. 27, p. 4323. https://doi.org/10.3390/molecules27134323
Lehotay, S.S., LC GC North Am., 2022, vol. 40, p. 13.
Zhou, Q., Yu, C., Meng, L., Ji, W., Liu, S., Pan, C., Lan, T., Wang, L., and Qu, B., Crit. Rev. Food Sci. Nutr., 2023, p. 1. https://doi.org/10.1080/10408398.2023.2225613
Santana-Mayor, A., Rodríguez-Ramos, R., Herrera-Herrera, A.V., Socas-Rodríguez, B., and Rodríguez-Delgado, M.A., TrAC, Trends Anal. Chem., 2023, vol. 169, p. 117375.
Sadighara, P., Basaran, B., Afshar, A., and Nazmara, S., Microchem. J., 2024, vol. 197, p. 109711.
ACKNOWLEDGMENTS
The authors express their gratitude to the Interdisciplinary Scientific and Educational School of Moscow University “The Future of the Planet and Global Environmental Changes.”
Funding
The work was carried out within the framework of a state assignment, topic no. AAAA-A21-121011990021-7.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors of this work declare that they have no conflicts of interest.
Additional information
Translated by V. Kudrinskaya
Publisher’s Note.
Pleiades Publishing remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Dmitrienko, S.G., Apyari, V.V., Tolmacheva, V.V. et al. Methods for Extraction of Organic Compounds from Solid Samples: 2. Sub- and Supercritical Extraction. Matrix Solid-Phase Dispersion. QuEChERS Method. Review of Reviews. J Anal Chem 79, 1167–1187 (2024). https://doi.org/10.1134/S1061934824700540
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1061934824700540