
Abstract
Specific features of chromatographic determination of xanthophyll esters are studied using an example of marigold flower lutein diesters under reversed-phase HPLC conditions. The developed two-column method made it possible to establish that, in samples with a low solubility of carotenoids in the used solvent and on a chromatography column in using a mobile phase with a low solubility of carotenoids, the precipitation of diesters is possible, which detrimentally affects the accuracy of the chromatographic determination. A critical factor in this case isì temperature: the storage of samples (solutions) in a refrigerator is not always advisable, because freezing of the main components is possible. It was shown that the use of a mobile phase containing from 0 to 10 vol % acetonitrile in acetone at a temperature not lower than 20°C is acceptable for the separation of all-trans-lutein diesters from cis-derivatives of lutein and zeaxanthin derivatives on “monomeric” C18-phases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
According to the type of functional groups, carotenoids are divided into carotenes, including hydrocarbons and their epoxides, and xanthophylls, containing OH groups (or keto groups). Xanthophylls in natural sources are most often esterified with several different higher fatty acids [1]. This significantly complicates the separation of individual carotenoids by reversed-phase HPLC [2]; therefore, the common practice is the saponification of xanthophyll esters before their subsequent determination [3, 4].
Among the economically important sources of xanthophyll esters, plants of the genus Tagetes (marigolds) with orange flower petals can be distinguished [5, 6]. Xanthophylls (mainly lutein) of this plant are used in pharmacy to prevent age-related macular degeneration, in bird feeding, as a food additive, and as dyes for the food industry [5]. The uniqueness of marigold flower petals lies in the biosynthesis of lutein diesters in high concentrations. Several examples of using reversed-phase HPLC [6–11] for the control of xanthophylls in dried marigold flower petals have been described, although a more detailed study of marigold carotenoids was presented in [12]. Additional information on the composition of lutein diesters was obtained using the “polymeric” C30 stationary phase instead of the “monomeric” C18 [13, 14]. However, a number of issues related to the chromatographic determination of xanthophyll diesters remain unexplored. This became especially important after the publication of [15], in which it was found that there may be a discrepancy between the results of HPLC determinations of carotenoids on a number of commercial chromatography columns and the amount of the injected sample, the reasons for which remain unclear.
The discrepancies noted in [15] stimulated the present study of the chromatographic behavior of xanthophyll esters under reversed-phase chromatography conditions with the assessment of the degree of the discovery (the ratio of the amount of the found substance to the amount of the substance added to the column) of carotenoids using the two-column method developed in this work.
EXPERIMENTAL
We used extracts of marigold (Tagetes erecta), ornamental physalis (Physalis alkekengi), and rowan berries (Sorbus aucuparia) grown in Belgorod in 2022. Marigold petals, physalis calyx, blister-like inflated with fruits, and rowan berries were dried without access to direct sunlight. Extraction was carried out by rubbing the plant material under a layer of an extractant (acetone) in a porcelain mortar until the last portion of the extractant became discolored; all portions of the extract were mixed in a measuring vessel and the volume was brought to the mark with acetone.
To determine the concentration of xanthophyll diesters, a Shimadzu UV-VIS spectrophotometer with quartz cells was used. After an appropriate dilution of the filtrate with acetone, it was passed through a packed filter with a pore diameter of 0.2 μm. The molar absorption coefficient of 144 500 L mol–1 cm–1 at 456 nm was used for calculations [16]. It was found that the linear dependence between the concentration of carotenoids and the absorbance of solutions was preserved, at least in the range of absorbances from 0.1 to 1.1.
For chromatographic separation, we used extracts diluted with acetone to a concentration of no more than 1.5 × 10–4 M with respect to the main xanthophyll. Such extracts can be stored at a temperature not lower than 20°C for up to 3 days in a dark place. Refrigeration should be avoided.
For separation, we used chromatography columns 4.6 × 250 mm Kromasil 100-5C18, 4.6 × 250 mm Kromasil 150-5C18, 4.6 × 100 mm Kromasil 100-5C18 and a Kromasil C18 guard column using an Agilent 1200 Infinity chromatograph with a diode array detector. Chromatograms were recorded at 445 nm, stored and processed using the Agilent ChemStation software.
RESULTS AND DISCUSSION
In determining xanthophyll esters in extracts of marigold petals, it is desirable to separate lutein and zeaxanthin derivatives, because not only lutein derivatives, but also zeaxanthin derivatives are necessary for the prevention of age-related macular degeneration [17]. The determination of zeaxanthin diesters (ZE) against the background of lutein diesters (LE) is not an easy task, because LE can be present not only as all-trans-lutein derivatives, but also as a mixture of cis isomers, later eluting on “monomeric” C18-stationary phases (as well as ZE). However, according to our experience, in freshly prepared samples, the concentration of cis isomers is noticeably lower than the concentration of ZE, which is confirmed by the electronic absorption spectra of the corresponding chromatographic peaks. Therefore, for an approximate assessment of the level of the accumulation of zeaxanthin derivatives, one can use the area of peaks eluting immediately after the peaks corresponding to all-trans LE isomers. At that, it is necessary to separate zeaxanthin derivatives from lutein derivatives, one of radicals in the structure of which belongs to unsaturated acids [12]. It is known that the retention of triacylglycerols decreases when the palmitic acid radical is replaced by the oleic acid radical [18]. By analogy, the alkyl octadecenoate ester of the all-trans isomer of lutein will elute before the corresponding alkyl palmitate.
To control the separation of complex mixtures for a given “monomeric” stationary C18 phase, one can use a change in the type of organic components of the mobile phase, their concentration, or temperature. In [19], by changing the components of the mobile phase, it was possible to avoid the superposition of the peaks of diesters of capsanthin, bearing one carbonyl group in the structure, and capsorubin, bearing two carbonyl groups in the molecule.
Effect of the type of mobile phase components. Both the extractant and the mobile phase for separating carotenoids should dissolve these compounds well. In acetonitrile and methanol (component 1, C1), as in the most commonly used mobile phase components, the solubilities of β-carotene are equal and rather low, 10 mg/L [16]. In this regard, the mobile phase should contain a solvent with a higher solubility of carotenoids in it as the main component (C2), and the number of such solvents from the list commonly used in HPLC is limited [16]. Figure 1 shows chromatograms of marigold extract in four types of eluents containing acetonitrile (C1) and four different components (C2), the concentrations of which (based on preliminary studies) were selected so that the retentions of basic diesters were close for all four components. One can see that the same order of elution of lutein diesters was observed in all chromatographic systems. The retention of LE increased with an increase in the number of carbon atoms (ΣNC) in two fatty acid radicals, because the affinity of the stationary C18 phase to them increased. After each all-trans-lutein derivative (for example, 5 in Fig. 1), there appeared a mixture (5a) containing all-trans-zeaxanthin and the sum of cis-lutein isomers with a similar ΣNC. They were followed, if possible, by the diester of all-trans-lutein (6b), in which the palmitoyl radical was replaced by an octadecenyl radical (presumably an oleic acid radical) with ΣN(C + 2).

Separation of marigold flower extract carotenoids in four types of eluents: (a) 70 vol % acetone and 30 vol % acetonitrile; (b) 52 vol % propanol-2 and 48 vol % acetonitrile; (c) 49 vol % ethyl acetate and 51 vol % acetonitrile; (d) 34 vol % methyl tert-butyl ether and 56 vol % acetonitrile, 0.8 mL/min. Column: 150 × 4.6 mm Kromasil 100-5C18, 30°C. Substances: (1) lutein, (2) and (3) two regioisomers of lutein myristate and palmitate; lutein diesters: (4) laurate-myristate, (5) dimyristate, (6) myristate-palmitate, (7) dipalmitate, (8) palmitate-stearate, (9) distearate; the letter a denotes the corresponding derivatives of zeaxanthin, the letter b denotes esters containing octadecenoic acid radicals.
The same elution order for four different additions to acetonitrile to the mobile phase is not surprising, because lutein and zeaxanthin derivatives are isomers with the same set of functional groups. Therefore, for the case under consideration, acetone, isopropanol, ethyl acetate, and methyl tert-butyl ether had approximately equal selectivities.
Effect of the concentration of an organic modifier in the mobile phase. As the concentration of C2 in the mobile phase increased, the retention of analytes decreased with an increase the number of dispersion interactions (or the number of carbon and hydrogen atoms) between the sorbate and the components of the mobile phase. In this case, the degree of separation of pairs of diesters of types 5a and 6b will decrease, which is confirmed experimentally (Fig. 2).

Separation of marigold flower extract carotenoids in four mobile phases of acetone–acetonitrile composition containing: (a) 100 vol % acetone; (b) 95 vol % acetone; (c) 90 vol % acetone; (d) 85 vol % acetone; (e) 80 vol % acetone, 0.8 mL/min. Column: 250 × 4.6 mm Kromasil 100-5C18 with pre-column Kromasil C18, 30°C. Lutein esters: (1) laurate-myristate, (2) dimyristate, (3) myristate-palmitate, (4) dipalmitate, (5) palmitate-stearate, (6) distearate.
The data in Fig. 2 indicate that the lower the concentration of C2 in the mobile phase, the better the separation of the mixture components. However, a decrease in the concentration of C2 is associated with another problem, i.e., with a decrease in the solubility of carotenoids in the mobile phase.
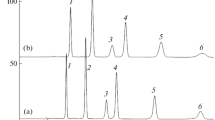
Effect of sample solvent on the shape of the chromatogram. It was shown in [20] that, in the separation of anthocyanins, the concentration of acetonitrile in the sample solvent can lead to artifacts distorting the chromatogram. To test the effect of sample solvent on the separation of carotenoids, two samples were prepared with equal concentrations of carotenoids using acetone (S1) as a solvent in the first sample, and a mixture of acetone (70 vol %) with acetonitrile (S2) in the second sample. The chromatograms of both samples recorded in the day of sample preparation were almost identical, which indicated a possibility of using samples in a solvent with a higher dissolving power, which is convenient for sample preparation. However, in the second day, the differences were significant (Fig. 3). In the S2 chromatogram, peak areas decreased (relative to peak areas for S1) with an increase in the retention of diesters, i.e., with a decrease in their solubility in the sample solvent: from ~80% for lutein laurate-myristate and dimyristate to ~50, ~40, and ~30% for lutein dipalmitate, palmitate-stearate and distearate, respectively. The reason for this phenomenon became clear after the discovery of a precipitate on the bottom of the container containing the second sample.

Separation of carotenoids of two samples of the extract of marigold flowers in the eluent composition 49 vol % ethyl acetate and 51 vol % acetonitrile for samples S1 (A1) and S2 (A2). Column 150 × 4.6 mm Kromasil 100-5С18, 30°С, 0.8 mL/min. Peak numbering is as in Fig. 1.
Therefore, the most important reason for the appearance of artifacts in the chromatography of carotenoids may be their insufficient solubility both in the sample solvent and in the mobile phase.
A precipitate forms at the insufficient solubility of carotenoids in the sample solvent. When solutions are stored at low temperatures, solubility decreases; i.e., one should carefully store sample solutions in a refrigerator.
The insufficient solubility of carotenoids in the mobile phase, according to our experience, can also be detected by a change in pressure values during chromatography: with the spontaneous crystallization of components during chromatography, a gradual increase in pressure with jumps was observed. In studying the solubility of anthocyanins [21], the formation of precipitates was observed even in eluents with high eluting capacities. The fact is that in chromatography, the sum of substances (e.g., diesters) is separated into individual components, and the probability of crystallization increases; the melting point also increases.
Two-column method of the control of chromatographic processes. A specific feature of the chromatographic determination of a mixture of substances, even in the presence of standard samples, is that we do not know beforehand the shape of the chromatogram (in terms of peak areas). The problem is easily solved using the following method. We record the chromatogram several times (e.g., three times) using column No. 1 and find the average values of peak areas and their sums. Then we attach column No. 2 in series and again record the chromatogram three times. The processing of chromatogram areas in both cases makes it possible to estimate the losses or their absence on column No. 2 in comparison with the first one. Then one can swap the columns and evaluate the properties of the first column.
In a number of studies on the proposed method, it was found that, with a relatively weak elution strength of the mobile phase, the degree of discovery of carotenoids can be significantly below 100% (Table 1). However, in going to extraction with 100% acetone, the loss of carotenoids within the experimental error (of the order 1–1.5%) cannot be detected (Table 2). Moreover, a certain increase in the areas of peaks in Table 2 may be due to errors in determining the areas of the incompletely separated peaks (Rs < 1), which are more accurately determined in chromatograms with the better separation of the components. In addition, as was mentioned above, a stable pressure line at the column inlet provides an evidence for the absence of artifacts. Therefore, the reason for a decrease in the degree of discovery of carotenoids may be the formation of a precipitate either in the sample (when stored at low temperature) or in the column during chromatography. It can be added that there is another reason for a decrease in the discovery of analytes in chromatography, i.e., the diffusion of analyte molecules into gallery pores [22]. This effect ismost probably also present, but it cannot be detected because of the nontransparent walls of the column. However, according to our experience, in solid-phase extraction of carotenoids on DIAPAK C18 cartridges, the complete washing of the cartridge from the yellow color (in the back extraction of carotenoids) is very problematic.
Conditions for the determination of lutein diesters and the results of the determination of carotenoids of marigold flowers. It was experimentally found that, at 19–20°С, a precipitate did not form in samples with concentrations of lutein diesters lower than 0.0003 M dissolved in acetone, but in storage in a household refrigerator, a precipitate appeared after two days at a concentration of 2.8 × 10–4 M.
It was found that eluent composition of 10 vol. % acetonitrile and 90 vol. % acetone could also be used to separate xanthophyll diesters without problems with precipitation in separating at a temperature not lower than 20°C on a 250 × 4.6 mm Kromasil 100-5C18 column protected by a Kromasil C18 guard pre-column, at a mobile phase rate of 0.5 mL/min or on two columns 100 × 4.6 mm Kromasil 100-5C18 and 250 × 4.6 mm Kromasil 100-5C18 connected in series using the same mobile phase at the same temperature; moreover, under these conditions, there was no need in reducing the flow rate of the mobile phase (below 1 mL/min) (Fig. 4).

Separation of carotenoids of marigold flower extract in the eluent composition 10 vol % acetonitrile and 90 vol % acetone. (a) column 250 × 4.6 mm Kromasil 100-5С18 with pre-column Kromasil С18, 20°С, 0.5 mL/min. (b) and (c): columns 100 × 4.6 mm Kromasil 100-5C18 and 250 × 4.6 mm Kromasil 100-5C18, 20°C, 1.0 mL/min. Designation of substances 1–9 as in Fig. 1, (10) lutein dilaurate. Additional letters denote all-trans-lutein derivatives with one more unsaturated acid radical.
The type of fatty acid radicals in lutein diesters is usually identified by mass spectrometry [7, 12], but for diesters of marigold flowers, one can use information available from the literature [12] on the formation of esters mainly by myristic and palmitic acids with a smaller contribution of stearic and lauric acids. Then it is sufficient to use an extract of ornamental physalis, containing zeaxanthin dipalmitate as the main component [23], to determine lutein dipalmitate, followed by assignment of compounds eluted earlier and later. The incremental approach [12, 24] ensures the detection of lutein dilaurate in a group of closely eluted compounds. The chromatogram reveals another group of peaks with electronic absorption spectra corresponding entirely to trans-lutein, and according to the results of monitoring the position of convergence points [24] of diesters formed with the participation of another monounsaturated acid; peaks of this group of substances are indicated by the addition of letter c.
Table 3 presents the results of determining the molar fraction of the types of lutein diesters by peak areas in five samples (of different geni) of marigold flowers. The presented data indicate the high reliability of determining the fatty acid composition of diesters under the conditions used. Other compounds were also found in the chromatogram, including lutein itself (peak 1), the concentration of which in the extract was in the range 0.1–1.0%, lutein monoesters (peaks 2 and 3), 2.0–2.6%; a mixture of diesters of zeaxanthin and cis isomers of lutein 7.7–9.8%; therefore, diesters of all-trans lutein accounted for 84.1–84.8%.
REFERENCES
Mariutti, L.R.B. and Mercadante, A.Z., Arch. Biochem. Biophys., 2018, vol. 648, p. 36.
Mercadante, A.Z., Rodrigues, D.B., Petry, F.C., and Mariutti, L.R.B., Food Res. Int., 2017, vol. 99, p. 830.
Sarkar, C.R., Bhagawati, B., Das, L., and Goswami, B.C., Ann. Biol. Res., 2012, vol. 3, p. 1461.
Abdala, A.F., Gallardo, A.P., Olvera, L.G., and Silva, E.M.E., Bioresour. Bioprocess., 2017, vol. 4, p. 5.
Raut, S. and Thaneshwari, T., Ecol. Environ. Conserv., 2022, vol. 28, special issue, p. S315.
Piccaglia, R., Marotti, M., and Grandi, S., Ind. Crops Prod., 1998, vol. 8, p. 45; Gregory, G.K., Chen, T.-S., and Philip, T., J. Food Sci., 1986, vol. 51, p. 1093; Rivas, J.D.L., J. Chromatogr. A, 1989, vol. 464, p. 442.
Tsao, R., Yang, R., Young, J.C., Zhu, H., and Manolis, T., J. Chromatogr. A, 2004, vol. 1045, p. 65.
Sowbhagya, H.B., Sampathu, S.R., and Krishnamurthy, N., Food Rev. Int., 2004, vol. 20, p. 33.
Jiang, X.-Y., Chen, L.-S., and Zhou, C.-S., J. Cent. South Univ. Technol., 2005, vol. 12, p. 306.
Hayashi, T., Oka, H., Ito, Y., Goto, T., Ozeki, N., Itakura, Y., Matsumoto, H., Ohno, H., Yoshida, K., Miyazawa, T., and Nagase, H., J. Liq. Chromatogr. Relat. Technol., 2005, vol. 27, p. 335.
Vechpanich, J. and Shotipruk, A., Sep. Sci. Technol., 2010, vol. 46, p. 265.
Lapshova, M.S., Deineka, V.I., Deineka, L.A., Blinova, I.P., and Tret’yakov, M.Yu., J. Anal. Chem., 2013, vol. 68, no. 11, p. 1014.
Abdel-Aal, E.-S.M., Rabalski, I., and Blackwell, B.A., J. Agric. Food Chem., 2007, vol. 55, p. 4965.
Deineka, V.I., Lapshova, M.S., Zakharenko, E.V., and Deineka, L.A., Russ. J. Phys. Chem. A, 2013, vol. 87, no. 11, p. 1912.
Epler, K.S., Sander, L.C., Ziegler, R.G., Wise, S.A., and Craft, N.E., J. Chromatogr. A, 1992, vol. 595, p. 89.
Craft, N.E. and Soares, J.H., Jr., J. Agric. Food Chem., 1992, vol. 40, p. 431.
Mrowicka, M., Mrowicki, J., Kucharska, E., and Majsterek, I., Nutrients, 2022, vol. 14, p. 827.
Turtygin, A.V., Deineka, V.I., and Deineka, L.A., J. Anal. Chem., 2013, vol. 68, no. 6, p. 558.
Deineka, V.I., Burzhinskaya, T.G., and Deineka, L.A., Anal. Kontrol’, 2019, no. 4, p. 501.
Deineka, V.I., Sidorov, A.N., Deineka, L.A., and Tynyanaya, I.I., Sorbtsionnye Khromatogr. Protsessy, 2016, vol. 16, no. 3, p. 384.
Deineka, V.I., Deineka, L.A., Sidorov, A.N., Kostenko, M.O., and Blinova, I.PRuss. J. Phys. Chem. A, 2016, vol. 90, p. 861.
Deineka, V.I., Deineka, L.A., Sidorov, A.N., Saenko, I.I., and Kostenko, O.M., Sorbtsionnye Khromatogr. Protsessy, 2016, vol. 16, no. 5, p. 624.
Weller, P. and Breithaupt, D.E., J. Agric. Food Chem., 2003, vol. 51, p. 7044.
Deineka, V.I. and Deineka, L.A., Sorbtsionnye Khromatogr. Protsessy, 2006, vol. 6, no. 3, p. 366.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of interest.
Additional information
Translated by V. Kudrinskaya
Rights and permissions
About this article

Cite this article
Deineka, V.I., Burzhinskaya, T.G., Blinova, I.P. et al. Specific Features of the Determination of Xanthophyll Esters under Reversed-Phase HPLC Conditions. J Anal Chem 78, 759–765 (2023). https://doi.org/10.1134/S1061934823060023
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1061934823060023