
Abstract
A possibility of replacing acetonitrile for methanol in mobile phases containing an organic modifier, 10 vol. % of formic acid (pH regulator), and water in the separation of anthocyanins by reversed-phase HPLC on a C18 stationary phase (Symmetry®C18) is studied. It is found that in replacing acetonitrile for methanol, it is necessary to use two components of the mobile phase, containing only methanol and only formic acid, as on the joint presence of these substances, a prolonged retention drift is observed, presumably because of the acylation of methanol. It is found that the replacement of acetonitrile for methanol results in small changes in the selectivity of the separation of anthocyanins with identical substituents but different aglycones and with different carbohydrate substituents for one and the same aglycone. There are also no significant differences in the separation efficiency (by the number of theoretical plates) of anthocyanins, which makes it possible to reject expensive acetonitrile in favor of methanol. The developed procedure was applied to determine anthocyanins in cranberry fruits of various degrees of ripeness (by color). The specific features of the anthocyanin composition of cranberry fruits are determined and the need in controlling the composition of the sample solvent for excluding artifact peaks is shown.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
In reversed-phase HPLC (RP HPLC) for the elution of analytes, it is necessary to use aqueous-organic mixtures with pH-adjusting additives, which must meet a number of requirements for physical properties, including limitations on viscosity, volatility, and, if possible, the use of spectrophotometric detection in the required wavelength region. To determine anthocyanins in RP HPLC, the elution strength of the mobile phase is most often controlled by changing the concentration of acetonitrile. Recently, there has been a trend to the refuse of this solvent, which is toxic to humans and the environment, and the cost of which has increased significantly in recent years [1]. In this respect, methanol, being toxic to humans, is less hazardous to the environment [1, 2], and belongs to inexpensive solvents. Acetonitrile is sufficiently chemically inert to be used without problems in a mixture with water and various acids, including formic acid, the addition of which (up to 10 vol %) is necessary to convert anthocyanins into the flavilic form [3]. Unlike acetonitrile, methanol can be esterified with carboxylic acids to form significantly more hydrophobic esters; therefore, the retention times of analytes can be reduced using mixtures of methanol with formic acid. However, the presence of a large amount of water in the mobile phases, acceptable for the determination of anthocyanins, shifts the equilibrium of the esterification reaction towards the starting acid and alcohol. It should be noted that the published data described the use of mixtures of methanol, formic acid, and water for the separation of anthocyanins in one component of the mobile phase, while there were no indications of problems with the stability of the retention times of analytes in the isocratic mode [4, 5], or gradient elution modes [6–10] under the conditions of reversed-phase HPLC.
The aim of this work was to assess a possibility of replacing acetonitrile for methanol in the mobile phases containing formic acid for the separation of anthocyanins and to reveal a change in the separation selectivity and efficiency (in terms of the number of theoretical plates) at such a replacement in RP HPLC.
EXPERIMENTAL
Extracts of anthocyanins from cercis leaves [11], red currant [12], and cherry [13] were used. The extracts were prepared by infusing a plant material in a 0.1 M aqueous solution of HCl and separated from the plant material by filtration through a paper filter. Partial purification was carried out by solid-phase extraction on DIAPAK C18 concentrating nozzles (cartridges) (BioKhimMak ST, Moscow).
Anthocyanins were separated on an Agilent 1200 Infinity chromatograph with a diode array detector. A chromatographic column 150 × 4.6 mm Symmetry®C18 with a particle size of 3.5 nm at a column thermostat temperature of 40°C was used. Chromatograms were recorded and processed using the ChemStation software.
The mobile phases were prepared using methanol of analytical reagent grade and acetonitrile of HPLC Gradient grade (Fisher Chemical, Germany), formic acid of high-purity grade (85%, China), and distilled water.
The following abbreviations were used to denote anthocyanins: anthocyanidins Dp (delphinidin), Cy (cyanidin), Pt (petunidin), Pn (peonidin), and Mv (malvidin); glycosides Glu (glucoside), Sopho (2"-glucosyl glucoside), Sam (2"-xylosyl glucoside), Rut (6"-ramnosyl glucoside or rutinoside), XRut (2"-xylosyl rutinoside), GRut (2"-glucosyl rutinoside), with substitution in position 3 of the aglycone, see Scheme 1.

Scheme 1 . Structure of anthocyanin aglycones (anthocyanidins).
The following procedure for the determination of anthocyanins in cranberry fruits was used:
(1) Anthocyanins were extracted by the infusion of cranberry fruits, divided into three groups by the color intensity. For this, 100 mL of a 0.1 M aqueous solution of HCl were added to 7–8 g of fruits, and the mixture was allowed to stand overnight for infusion. In the next day, the extract was separated from the plant material and filtered through a paper filter. A single extraction was sufficient for the almost complete extraction of anthocyanins (more than 98%).
(2) The obtained extracts were used for the spectrophotometric determination of anthocyanins in terms of Cy3Glu [14], but without excluding the absorption of polymeric anthocyanins at pH 4.5. The presence of polymeric anthocyanins was assessed by the chromatographic method [15]. Using this method, the contribution of polymeric anthocyanins can be found from the area of the diffuse peak starting in the dead volume (time) range; as a result, no more than 2% of polymer forms were obtained.
(3) The next step was sample preparation for the determination of the species composition of anthocyanins by reversed-phase HPLC. The extracts obtained in step 1 were adsorbed on a DIAPAK C 18 concentrating cartridge activated by passing 5 mL of acetone and conditioned by passing 10–15 mL of a 0.1 M aqueous solution of HCl. Anthocyanins were back extracted from a cartridge with 3 mL of methanol containing 10 vol % of formic acid. The back extract was diluted with a solution of formic acid (10 vol. %) in water in a ratio of 1 : 5 for subsequent chromatography.
(4) Next, the anthocyanins were separated under the conditions of RP HPLC. Solution A, containing 30 vol % of methanol in water, and solution B, containing 20 vol % of formic acid in water, were prepared. For elution, a gradient pump (two-channel) was used, but with a constant ratio of solutions A and B (1 : 1) at a mobile phase flow rate of 0.8 mL/min. The chromatographic column and separation temperature are indicated above. Chromatograms were recorded three times. Chromatograms were recorded, stored, and processed using the Agilent ChemStation software. The difference in the retention times of the main components were no more than 2 s, and the discrepancy between the results of determining the proportion of anthocyanins did not exceed 0.5%.
RESULTS AND DISCUSSION
A possibility of the esterification reaction was indirectly confirmed in the first experiments. In adjusting the steady-state conditions of the chromatographic system in an eluent containing 12 vol % of methanol and 10 vol % of formic acid in water, we could not reach for the retention time drift to stop even after 3 h (Fig. 1), although usually 20–30 min was sufficient to stabilize the retention of anthocyanins. In this case, a decrease in the retention times of all sample components predicted above was observed.

Drift of the retention of anthocyanins in the mobile phase CH3OH–HCOOH–H2O (12 : 10 : 78, vol.). The time interval between the injection of samples A–F is 25–30 min. Anthocyanins: (1) Dp3Glu; (2) Cy3Glu; (3) Pt3-Glu; (4) Pn3Glu; and (5) Mv3Glu. Column 4.6 × 150 mm Symmetry C18, 3.5 μm. Temperature 40°С, mobile phase flow rate 0.8 mL/min.
The reduced concentration of formic acid used in a number of studies (less than 10 vol %) also led to a drift of retention times, but more slowly (Fig. 2). It should be noted that no indications of a drift of retention times were found in the literature in working with similar mixtures in mobile phase compositions. The solution to the problem was to mix two solutions, one of which contained only methanol in addition to water, and the second was a solution of formic acid in water. This approach was used in some works on the determination of anthocyanins in gradient modes [11, 12]. We have experimentally found that this method allowed the chromatographic system to be brought to a steady-state mode in the usual time (20–30 min), but, unfortunately, single-channel isocratic pumps in this case became unsuitable. This method was used later in the study of a possibility of replacing acetonitrile for methanol.

Drift of retention times of anthocyanins in eluents based on methanol and formic acid. Anthocyanins: (1) and (1a) Cy3Glu, (2) Pn3Glu. Mobile phases for (1) and (2) 10 vol % HCOOH and 12 vol % CH3OH in water, for (1a) 5 vol % HCOOH and 13 vol % CH3OH in water.
Comparison of the selectivity of anthocyanin separation. To compare the selectivity of the separation of 3‑glucosides of five of the six main anthocyanidins in replacing acetonitrile for methanol, the most effective method to date is a comparison of separation maps in the version of relative retention, proposed in [16] (Fig. 3).

Map of the separation of anthocyanins for the system of mobile phases I: methanol –10 vol % formic acid–water in comparison with the anthocyanin separation map for system II: acetonitrile–10 vol % formic acid–water. The dotted lines and the number with letter a indicate the relative retention in system II. Anthocyanins: (1) Dp3Glu, (2) Cy3Glu; (3) Pt3Glu; (4) Pn3Glu, and (5) Mv3Glu.
It turned out that replacing acetonitrile for methanol as a component with a higher viscosity resulted in a slight increase in pressure (only by 10–15%) for the compositions of mobile phases used in this work. At that, a more compact elution occurred in the order unchanged for both organic modifiers of the mobile phase:
Such compactness can be used to reduce the time required for the separation of all or some of the components of natural mixtures of anthocyanins, when there was no sense in increasing the elution time of the most strongly retained component.
The addition of an OH-group to ring B in the transition from Cy3Glu to Dp3Glu led, when both types of organic modifiers of the mobile phase were used, to a logical decrease in retention because of an increase in the relative hydrophilicity of the adsorbate. Therefore, the trend lines in the separation map for Dp3Glu were located below the trend line for Cy3Glu. However, this increase in hydrophilicity for the mobile phases based on methanol turned out to be somewhat smaller than for the mobile phases based on acetonitrile. With the addition of methoxy groups, the trend lines were always located higher, which could be interpreted as an increase in the lipophilicity of anthocyanins. In this case, the trend lines for Mv3Glu and Pn3Glu for the mobile phases containing methanol were located below similar trend lines for the mobile phases based on acetonitrile. These two factors resulted in a decrease in the range of retention times of 3-glucosides (and, consequently, any other glycosides of the same type) of five of the six main natural anthocyanidins.
A rigorous explanation of the changes found for one composition of the mobile phase is problematic, because in reality each adsorbate had its own balance of hydrophilic and hydrophobic properties, i.e. its retention should be described by at least a two-parameter equation. The equations of relative retention [17] are just two-parameter:
where the logarithm of the retention factor of sorbate i is compared with the logarithm of the retention factor of the sorbate selected the reference substance (Cy3-Glu). In this equation, parameter a1 can be considered the relative characteristic of the lipophilicity of the sorbate (with respect to the reference substance). The parameter a0 is not an unambiguous characteristic of hydrophilicity; to refine it, it is necessary to determine the abscissas of the convergence points [18]. However, for anthocyanins, there are no homologues necessary to find the coordinates of such points. However, comparing the slopes of the trend lines in Fig. 3, one can find that, in fact, the relative lipophilicity of anthocyanins on the addition of methoxy groups increases more strongly for mobile phases containing methanol.
For the trend lines in Fig. 3, there is one more feature useful for confirming their correctness. Thus, in the approximation to the right, i.e. for long retention times (or for small amounts of methanol or acetonitrile), the lines for the same substance must intersect at the point at which the concentration of the organic modifier (except for formic acid) becomes zero. Indeed, analyzing the equations of the relative retention, we found that the abscissas of the intersection points for the pairs of trend lines for the substances Dp3G, Pn3G, and Mv3G were close to each other: 1.99, 2.09, and 2.02, which was satisfactory for the lines plotted only from three experimental points for each dependence.
In studying the effect of replacing acetonitrile for methanol on the separation of different types of glycosides of the same anthocyanidin (cyanidin), the trend lines for identical compounds were similar (Table 1). At that, the slope of the trend line for diglycosides was about 0.1 greater than that for 3-glycosides of cyanidin with a noticeable increase for 3-rutinoside because of the presence of a methyl group in the carbohydrate substituent; for 3-triglycosides, the slope increased by another 0.15, as in using acetonitrile as a modifier. Thus, the replacement of the solvent virtually did not result in a change in the selectivity of anthocyanin separation with the complication of the structure of the carbohydrate substituent, and the parameter a1, as in using acetonitrile [19], remained a unique indicator of the number of monoses in the carbohydrate substituent, which is convenient for the preliminary identification of anthocyanins without using mass-spectrometry detection.
Comparison of efficiencies. To study the efficiency of chromatographic systems, an analysis of experimental data using the dynamic approach according to the van Deemter equation [20] is usually used. In our case, such an analysis made it possible to establish that the minimum height of the theoretical plate was achieved at an irrationally low flow rate of the mobile phase (less than 0.20 mL/min) (Fig. 4). In this regard, for chromatography, it was necessary to find a compromise between the degree of separation (Rs) and the time of chromatography, i.e. the degree of separation can be increased, if necessary, by reducing the flow rate of the mobile phase. Data analysis of Fig. 4 shows that the efficiency of chromatographic systems even slightly increases on replacing acetonitrile for methanol.

Van Deemter’s plot for Cy3Glu in two mobile phases. Mobile phases: (1) 16 vol % CH3OH and 10 vol % HCOOH in water; (2) 8.8 vol % CH3CN and 10 vol % HCOOH in water.
Determination of anthocyanins in cranberry fruits. The relative proportion of various types of anthocyanins in a cranberry fruit extract can be determined at one wavelength without introducing corrections [15]. This is due to the fact that the electronic absorption spectra of all six main 3-glycosides virtually do not differ.
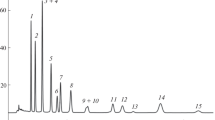
The separation of anthocyanins in cranberry fruits is illustrated in Fig. 5, and Table 2 summarizes the results of the determination of anthocyanins and their changes as the cranberries ripen. In addition to the six main anthocyanins, which were found in a number of studies [21–25], several minor components were also noticeable in the chromatogram, the fraction of which in the sum of anthocyanins by peak areas did not exceed 3–5%. The structure of these anthocyanins was not determined in this work, but an analysis of the trend line parameters suggests that they (except for component a in Fig. 5) are cyanidin monoglycosides, and their acylation with some aliphatic acids is not excluded. Acylation with aliphatic acids (in our experience) has little effect on the slope of the trend lines, but retention in this case strongly depends on the position of acylation of the carbohydrate substituent. Considering that in the sample preparation and storage, the acylation of anthocyanins with formic acid is not excluded, the version in which the sample was prepared by replacing formic acid with orthophosphoric acid was investigated. It turned out that such a replacement in no way affected the type of the chromatogram, i.e., minor peaks were not artifacts, but a consequence of peculiarities of the biosynthesis of anthocyanins in cranberry fruits. However, as the fruits ripen (or depending on color intensity), changes in the anthocyanin composition were observed. An increase in the fraction of Cy3Glu and Pn3Glu was especially noticeable.

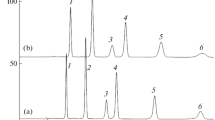
Separation of red cranberry anthocyanins in the mobile phase 15 vol % CH3OH and 10 vol % HCOOH in water. Sample solvents—mixtures of methanol, formic acid, and water in the ratios: A—20 : 10 : 70; B—50 : 10 : 40, vol Detection at 215 nm. Compounds: (1) Cy3Gala; (2) Cy3Glu; (3) Cy3Ara; (4) Pn3Gala; (5) Pn3Glu; and (6) Pn3Ara; a, b, c, d, e—not identified.
In one of works on the HPLC of anthocyanins performed in our laboratory [26], it was shown that, with incorrect sample preparation, artifact peaks could appear in the chromatogram. They could occur when the sample was dissolved in a solvent with a higher elution capacity than the mobile phase used. In using methanol, the assessment of the effect of solvent composition on the quality of the resulting chromatogram was especially important, as there were many publications in the literature in which it was recommended to dissolve the sample in methanol in the end of sample preparation for subsequent chromatography. Samples were prepared, dissolved in methanol-based solvents (with 10 vol % of formic acid) with equal concentrations of anthocyanins, but different concentrations of methanol (from 50 to 10 vol %). In the subsequent chromatography, it was found that, even at a methanol concentration of 50% in the sample, the peaks of anthocyanins with shorter retention times split and the peaks of anthocyanins with longer retention times were significantly broadened (Fig. 5b). Artifacts were absent only when the sample was dissolved in a mixture close in composition to the mobile phase used; at that, a change in methanol concentration only by ±50% was conditionally permissible. Therefore, it was not surprising that in [27], in which a sample dissolved in methanol was chromatographed, bifurcated peaks of substances with the shortest retention times appeared in the chromatogram.
CONCLUSIONS
Thus, methanol is an important alternative to the environmentally less favorable acetonitrile for the determination of anthocyanins under the conditions of reversed-phase HPLC with 10 vol % of formic acid as a pH regulator of the mobile phase.
REFERENCES
Bordonaba, J.G., Crespo, P., and Terry, L.A., Food Chem., 2011, vol. 129, p. 1265.
Yabre, M., Ferey, L., Some, I.T., and Gaudin, K., Molecules, 2018, vol. 23, p. 1065.
Pina, F., Oliveira, J., and de Freitas, V., Tetrahedron, 2015, vol. 71, p. 3107.
Mozetic, B., Trebse, P., and Hribar, J., Food Technol. Biotechnol., 2002, vol. 40, p. 207.
Koh, K., Youn, J.E., and Kim, H.-S., J. Food Sci. Technol., 2014, vol. 51, p. 377.
Flamini, R. and Tomasi, D., Vitis, 2000, vol. 39, p. 79.
Luczkiewicz, M. and Cisowski, W., Chromatographia, 1998, vol. 48, p. 360.
Valls, J., Millan, S., Marti, M.P., Borras, E., and Arola, L., J. Chromatogr. A, 2009, vol. 1216, p. 7143.
Revilla, I., Pérez-Magariño, S., González-Sanjosé, M.L., and Beltrán, S., J. Chromatogr. A, 1999, vol. 847, p. 83.
Comandini, P., Blanda, G., Cardinali, A., Cerretani, L., Bendini, A., and Fiorenza, M., J. Sep. Sci., 2008, vol. 31, p. 3257.
Doronin, A.G., Deineka, V.I., Deineka, L.A., Tretiakov, M.Y., Tokhtar, V.K., and Chulkov, A.N., Adv. Biol. Sci. Res., 2019, vol. 7, p. 90.
Deineka, V.I., Chulkov, A.N., Deineka, L.A., and Sorokopudov, V.N., Sorbtsionnye Khromatogr. Protsessy, 2015, vol. 15, no. 4, p. 280.
Deineka, L.A., Chulkov, A.N., Deineka, V.I., Sorokopudov, V.N., and Shevchenko, C.M., Nauchn. Ved. Belgorod. Gos. Univ., Ser. Estestv. Nauki, 2011, vol. 9(104), no. 15/1, p. 364.
Giusti, M.M. and Wrolstad, R.E., Characterization and measurement of anthocyanins by UV-visible spectroscopy, Curr. Protoc. Food Anal. Chem., 2001, no. 1, p. F1.2.1.
Deineka, V.I., Oleinits, E.Yu., Kul’chenko, Ya.Yu., Blinova, I.P., and Deineka, L.A., J. Anal. Chem., 2020, vol. 75, p. 1443.
Deineka, V.I., Russ. J. Phys. Chem A, 2006, vol. 80, p. 429.
Deineka, V.I., Russ. J. Phys. Chem A, 2006, vol. 80, p. 425.
Deineka, V.I., Deineka, L.A., and Turtygin, A.V., Sorbtsionnye Khromatogr. Protsessy, 2008, vol. 8, no. 3, p. 465.
Deineka, V.I. and Grigor’ev, A.M., J. Anal. Chem., 2004, vol. 59, p. 270.
Katz, E., Ogan, K.L., and Scott, R.P.W., J. Chromatogr. A, 1983, vol. 270, p. 51.
Brown, P.N., Paul, R., and Shipley, P.R., J. AOAC Int., 2011, vol. 94, p. 459.
Vorsa, N. and Polashock, J.J., J. Am. Soc. Hortic. Sci., 2005, vol. 130, p. 711.
Jurikova, T., Skrovankova, S., Mlcek, J., Balla, S., and Snopek, L., Molecules, 2019, vol. 24, p. 24.
Cesoniene, L., Daubaras, R., Jasutiene, I., Miliauskiene, I., and Zych, M., J. Appl. Bot. Food Qual., 2015, vol. 88, p. 295.
Huopalahti, R., Jrvenp, E.P., and Katina, K., J. Liq. Chromatogr. Relat. Technol., 2000, vol. 23, p. 2695.
Deineka, V.I., Sidorov, A.N., Deineka, L.A., and Tynyanaya, I.I., Sorbtsionnye Khromatogr. Protsessy, 2016, vol. 16, no. 3, p. 384.
Andrade, P.B., Carvalho, A.R.F., Seabra, R.M., and Ferreira, M.A., J. Agric. Food Chem., 1998, vol. 46, p. 968.
Funding
This work was supported by a grant from the Russian Foundation for Basic Research “Postgraduates,” project no. 20-33-90031/20.
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated by V. Kudrinskaya
Rights and permissions
About this article

Cite this article
Deineka, V.I., Oleinits, E.Y., Blinova, I.P. et al. Selectivity Control of the Separation of Anthocyanins: Replacing Acetonitrile for Methanol in the Mobile Phase. J Anal Chem 76, 939–945 (2021). https://doi.org/10.1134/S1061934821060022
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1061934821060022