
Abstract
The review considers the historical aspects of the formation and development of bioelectrochemistry and some issues of the analytical application of electrochemical methods (mainly voltammetry and related methods) to studies of the properties of biologically active compounds and biopolymers in the context of the interests of biochemistry and medicine. The importance of taking into account processes on the surface of electrodes generating an analytical signal is noted. The review deals with the principles of direct electrochemical analysis of biopolymers (proteins and nucleic acids) based on the electroactivity of amino acid residues, redox active sites, and nucleic base residues on unmodified electrodes. The electrochemical behavior of proteins, peptides, oligonucleotides, and single- and double-stranded nucleic acid molecules is discussed from the point of view of their spatial structures. Emphasis is placed on directions in the development of bioelectrochemistry.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
THE TERM BIOELECTROCHEMISTRY
The application of the methods and approaches of electrochemistry to studying biomolecules began to stand out as a separate field of knowledge about half a century ago, although the first works were published somewhat earlier. The interdisciplinary nature of this field was formed gradually, and the term bioelectrochemistry did not appear immediately. To date, it has no clear boundaries because its definition includes several concepts and cognitive approaches united by the prefix bio-. Nevertheless, the historical aspects of the formation and development of bioelectrochemistry in the format of the movement of ideas aimed at solving analytical problems are of interest.
In 1979, on the initiative of Giulio Milazzo, the international Bioelectrochemical Society was created and officially registered in France in 1981 (Bioelectrochemical Society, BES, http://www.bioelectrochemical-soc.org). It is interesting that a few years earlier Milazzo had been a participant in the VI Heyrovsky Discussion (1972, Liblice Castle near Prague) devoted to the problems of electroanalytical chemistry as applied to biomedicine. A series of symposiums of the Bioelectrochemical Society under the title Biannual Symposium Series of the Society on Bioelectrochemistry and Bioenergetics, started in 1971, is currently continuing, and the international journal Bioelectrochemistry and Bioenergetics, published since 1974, has changed its name to Bioelectrochemistry since 2000 (official site: https://www.journals.elsevier.com/bioelectrochemistry). The journal is devoted to electrochemical principles in biology and biological aspects of electrochemistry. In May 2021, the year of the 40th anniversary of the Society, the XXVI Bioelectrochemical Symposium was held for the first time in an online format (XXVI International Symposium on Bioelectrochemistry and Bioenergetics of the Bioelectrochemical Society, http://www.bes2021.org). It should be mentioned that the Czech tradition of holding the Heyrovsky Discussions has also been preserved (http://www.physchem.cz), and the conference was devoted to the electrochemistry of biologically active compounds, including biopolymers, almost half a century later.
The very history of the appearance of the term with the prefix bio- in the science of electrochemistry is of interest. Although biology and chemistry arose independently of each other, the connection between these sciences was realized only in the 19th century, when natural scientists began to purposefully conduct experiments with living organisms under the influence of various physical factors. Electrical experiments with the prefix bio- in their names were first carried out at the end of the 18th century. In 1771, the Italian anatomist and physiologist Luigi Galvani discovered and studied the phenomenon of muscle contraction in a dissected frog under the influence of an electric current [1]. Observing the contraction of muscles when they were connected by metal to nerves or the spinal cord, he drew attention to the fact that the muscle contracts when two different metals touch it simultaneously. Galvani explained these phenomena by the existence of “animal electricity,” due to which the muscles are charged like a Leyden jar. The phenomena discovered by Galvani referred to as “galvanism” for a long time. In the 1790s, the Italian physicist and physiologist Alessandro Volta, who was interested in animal electricity, conducted a series of experiments and showed that the phenomena observed by Galvani were associated with the presence of a closed circuit consisting of two dissimilar metals and a liquid. Volta considered the causes of galvanism to be physical and physiological actions to be one of the manifestations of this physical process. After conducting experiments with different pairs of electrodes, Volta found that the physiological irritation of nerves is the stronger, the further two metals are separated from each other in the following order: zinc, tin foil, tin, lead, iron, brass, etc., up to silver, mercury, and graphite. This famous Volta potential (activity) series was the core of the effect; the frog muscle was only a passive, albeit very sensitive, electrometer, and the active links were metals, from the contact of which their mutual electrization occurred. Conducting numerous comparative physiological experiments, Volta observed in animals a greater electrical excitability of nerves compared to muscles and smooth muscles of the intestines and stomach compared to skeletal ones. He discovered the electrical irritability of the organs of vision and taste in humans [1]. Currently, electrophysiology, whose father can rightfully be considered Galvani, deals with the study of galvanic effects in biology. As a tribute to history, the Bioelectrochemical Society traditionally presents the Luigi Galvani Prize every two years to a young scientist who has made the most significant contribution to the field of bioelectrochemistry (http://www.bioelectrochemical-soc.org/awards-LuigiGalvani.php).
The prefix bio- in the term bioelectrochemistry means biologically active substances, biopolymers (proteins and nucleic acids), whole cells, and living organisms. The first works devoted to the electrochemical behavior of proteins on a mercury electrode appeared in the early 1930s, that is, less than ten years after the discovery of polarography by Yaroslav Heyrovsky. The period of practical use of electrochemical methods, starting with polarography, in the analysis of biomedical objects covers a hundred years, whereas bioelectrochemistry itself as an interdisciplinary branch of science appeared relatively recently. At present, the term bioelectrochemistry includes a wide range of branches one way or another related to living systems and detection of electron transfer. Bioelectrochemistry studies the electrochemical regularities of biological processes (in particular, information transmission along nerve fibers, energy conversion, photosynthesis, and reception) and the impact of external electric and magnetic fields on biological systems [2]. The common stage of all these processes is the separation of charges (ions and electrons), which occurs in the course of a redox reaction or during the transport of ions through membranes. This results in the appearance of a membrane potential and ion concentration gradients between the interior of the cell and the environment. Free energy accumulated in the form of membrane potential or concentration gradients ensures the generation and transmission of nerve impulses, ATP synthesis, some types of mechanical movement, etc. According to Berezovchuk [3], bioelectrochemistry studies the structure and properties of the membranes of living cells, the mechanism of ion transfer through the membrane, the nature of the potential jump on the membrane of a living cell, and the mechanism of potential transfer along the nerve fiber. Knowledge of the cell membrane mechanism will make it possible to design various devices that work on the principle of a living cell. On the other hand, the processes of ion transport in living systems can be modeled using the knowledge of ionometry, a section of electrochemistry. Thus, bioelectrochemistry is a scientific direction the subject of which is the electrochemical aspects of the operation of living systems [4]. The term bioelectrochemistry is now also understood as the area of electrochemistry associated with the study of the electrochemical properties of biomolecules and the development of methods for their electrochemical determination in biosamples.
The appearance of biosensors gave a new breath to bioelectrochemistry: in 1962, Clark and Lyons [5] proposed the concept of enzymatic membrane electrodes. A solution of an enzyme (for example, glucose oxidase or urease) had to be placed between two membranes on the surface of an electrode that detected the loss of a substrate or the increase in the product of the enzymatic reaction. The pH and pO2 electrodes were used as sensor elements. The idea of Clark and Lyons [5] was to use the properties of enzymes in order to determine the analyte concentration in the course of cardiovascular operations and for the postoperative prognosis of patient’s condition. Thus, biosensors are chemical sensors in which the recognition system is based on a biochemical mechanism. According to the definition adopted in 1999 by the International Union of Pure and Applied Chemistry (IUPAC) [6], the chemical sensor is a device that transforms chemical information, from component concentration to composition, into an analytical signal. Chemical sensors usually consist of two main parts: a chemical (molecular) recognition system (receptor) and a physicochemical transducer. A biological recognition system transmits information, usually on the concentration of an analyte, from a biochemical receptor in the form of a chemical or physical output signal with certain sensitivity. According to the IUPAC recommendations [6], the term biosensor should be separated from analytical systems that include additional separation steps (such as high performance liquid chromatography), auxiliary devices and/or sample processing by introducing special reagents (for example, flow-injection analysis). Thus, the biosensor should be an analytical device that does not require additional reagents in addition to natural cosubstrates necessary for the determination of the analyte. At the same time, there is an alternative point of view: Turner [7] subdivided modern biosensors into (1) complex high-performance laboratory equipment capable of quickly, easily, and accurately detecting biological interactions and determining components of interest and (2) easy-to-use portable devices designed for a wide range of consumers for decentralized, in situ, or home analysis. The former are expensive, and the latter are large-scale produced and available. The families of biosensors are immunosensors, enzyme-based biosensors, and whole-cell biosensors. Biosensors have been applied to a wide range of analytical problems, including use in medicine, biomedical research, pharmacology, ecology, food industry, process industries, and security. The best-known example of an analytical device based on an electrochemical biosensor is a commercially available glucometer. The design and study of molecular and supramolecular structures with molecular bioreceptor and biomimetic properties for use in analytical devices is also a field of interest for bioelectrochemistry. Here, the focus is on the complementary intersection of molecular recognition, nanotechnology, molecular imprinting, and supramolecular chemistry to improve the analytical performance and reliability of devices. A field of bioelectronics that seeks to use biology in conjunction with electronics in a broader context, for example, covering biological fuel cells, bionics, and biomaterials for information processing and storage arose simultaneously with biosensorics. The key aspect here is the interface between biological materials and micro- and nanoelectronics.
Recent IUPAC recommendations [8] concerned the terminology of methods of bioanalytical chemistry, analysis, and study of biomacromolecules. They introduced clear definitions related to bioanalytical samples, enzymatic methods, immunoanalytical methods, methods used in genomics and nucleic acid analysis, proteomics, metabolomics, glycomics, lipidomics, and interactomics [8]. According to the IUPAC Recommendations 2018, the field of bioanalytical chemistry includes detection (identification, sequence decoding), characterization (polarity and charge, structure, and intermolecular interactions), and quantification and monitoring (stability, dynamics, fragmentation, degradation, metabolism, etc.). The term biosensor refers to a measuring tool (device, means) that does not require additional reagents and provides selective qualitative and/or quantitative analytical information using a biological recognition element, which is held in direct spatial contact with the transduction element (transducer) [8].
BIOELECTROCHEMISTRY: A HISTORICAL RETROSPECTIVE
The history of electrochemical methods of analysis can be traced from the middle of the 19th century [19]. The discovery of polarography by Heyrovsky in 1922 meant the emergence of a new field of electrochemistry, in which the polarograph became the main tool for recording polarization curves (polarograms) of a mercury drop electrode in test solutions. At first, inorganic compounds were, as a rule, the test materials, and the development of the theory of this method was associated with the interpretation of their polarographic behaviors. In 1925, Shikata [10] recorded a polarogram of the first organic compound, nitrobenzene, in the Heyrovsky laboratory in Prague. If we take into account that the nitro group is a part of the structure of many organic compounds with biological activity, we can accept this date as the beginning of analytical bioelectrochemistry. Thus, everything began with the polarography of low-molecular-weight organic compounds. From personal experience: in 1960, one of the authors of this review entered the graduate school of the Chemical Institute of the Kazan Branch of the USSR Academy of Sciences (Kazan) with a degree in polarography of organic compounds. The objects of investigation were semi- and thiosemicarbazones of aldehydes and ketones, of which a number of substances had antituberculosis activity. During his postgraduate studies, he completed an internship at the Polarographic Institute of the Czechoslovak Academy of Sciences initiated by Arbuzov and Kitaev. The internship took place under the patronage of Heyrovsky, who received the Nobel Prize in Chemistry “for his discovery and development of the polarographic methods of analysis” (1959). The atmosphere at the institute contributed to a deep acquaintance with the work in the field of polarography in biomedicine and pharmacy. Thus, at first, polarographic methods were successfully developed as effective means for studying the structure and reactivity of organic compounds in processes with the participation of electrons. Gradually, extensive information on the electrochemical properties of organic compounds in solutions became in demand in the development of methods for their determination in various objects.
It took almost half a century of research to show that not only mercury but also noble metals, their oxides, various carbon materials, pastes, and composites based on them can be used as materials for polarizable electrodes in voltammetry; so-called modified electrodes were also used in addition to conventional ones. The appearance of a stationary solid electrode meant a transition from cathodic (reduction) to anodic (oxidation) voltammetry, which turned out to be extremely promising for organic analysis. The dimensions of the electrodes themselves have decreased by tens and even hundreds of times, and unified manufacturing methods, for example, screen printing, have been used in order to achieve reproducibility of their characteristics. At the same time, research on hardware design, that is, on the improvement of devices for reliable detection of a useful signal in relation to noise (interference) was carried out based on the principles of microelectronics. A modern voltammograph (potentiostat) bears little resemblance to a set of rheostats and Heyrovsky batteries. As a rule, this is a small block or sometimes a board inserted into a computer or smartphone. For example, the PalmSens company (the Netherlands, official website: www.palmsens.com) successfully produces such mini devices, in particular, a Sensit Smart potentiostat compatible with a smartphone. Miniaturization touched equipment, electrodes, and electrochemical cells. This is due to the fact that electrodes have been used as signal converters in integrated devices such as thin layer flow cells, microfluidic cells, and labs on a chip. The miniaturization of such systems has made it possible to achieve outstanding success in reducing both the analyte concentrations determined and the volume of sample consumed. Thus, the prerequisites for the next qualitative leap and a transition to implantable devices that combine the functions of a diagnostician and a therapeutic agent have been created. In this case, the determined analyte amounts can be decreased to a level of pico- and femtograms in the sample.
Note that the development of devices to be used in electrochemical analysis, including mobile devices, is possible only on the basis of a deep understanding of both the problems of bioanalytical chemistry and the nature of processes occurring on the electrode surface. The analytical response to the test compound is formed in the course of a process occurring on the electrode, which often consists of several stages [11]. Usually, the actual mechanism of an electrode reaction remains outside the interests of the analyst. It is important that the response be repeatable within the required limits. Nevertheless, it is useful to have a general understanding of the mechanism. The mechanism is understood as the whole set of heterogeneous electron transfer reactions at the electrode/solution interface and homogeneous chemical reactions near the electrode and in the bulk of the solution. A study of the mechanism includes determining the sequence of these electrochemical (E) and chemical (C) stages, their rate constants, which make it possible to identify the rate-limiting stage of transfer, and the numbers of electrons and protons transferred per molecule participating in the reaction, the establishment of other fundamental electrochemical characteristics of individual stages, determination of the nature of the formed particles, etc. It should be kept in mind that each electrochemical and chemical process should include only one elementary act [11]. In most cases, the rate-limiting step of the process is one of the chemical steps preceding, subsequent, or parallel to electron transfer. To establish the nature of the reaction products formed at the electrode, electrolysis is carried out at a controlled potential, and then the products are identified using appropriate physicochemical methods.
It is necessary to note a global trend in electroanalytics in relation to biomedical objects. Of low-molecular-weight organic compounds, various flavonoids, including antioxidants, stay in fashion [12]. Advances are also observed in the field of biosensorics, in which both natural and artificial materials are used to organize a selective response, including synthetic receptors and nanozymes [13, 14]. We repeat by saying that we live in an age of major transformations in the fields of biomedicine and, hence, bioelectrochemistry. Here, areas with interdisciplinary interactions are of undoubted interest for bioelectroanalysts. These are the problems of early diagnosis of diseases, the functioning of cells, the control of the effectiveness and toxicity of drugs, the control of the concentrations of food dyes, etc.
Proteins. The beginning of protein electrochemistry [15] can be considered the work of Heyrovsky and Babicka [16] published in 1930. They observed a new wave appeared on the voltammogram at a potential of –1.6 V (0.2 V more positive than that of the reduction of \({\text{NH}}_{4}^{ + }\) ions in an ammonia buffer solution) when a protein was added to the polarographic cell. The wave was attributed to the process of electrochemical reduction of hydrogen on the surface of mercury catalyzed by the protein (Fig. 1). Then, in 1932, Herles and Vancura [17], two physicians studying the polarographic activity of various human biological fluids at the invitation of Heyrovsky, detected another less pronounced signal: the introduction of a protein into a sodium chloride solution caused an increase in current at a potential 0.3 V more positive than that of the reduction of sodium ions in supporting electrolyte. The signal was referred to as a pre-sodium wave (Fig. 1). Further studies showed that the processes occurring in a polarographic cell with the participation of a protein can be expressed by the following reaction equations [18]:
where P is the protein (catalyst), PH+ is the protonated form of the protein, and DH is the proton donor. The overvoltage of the catalytic hydrogen evolution in this case is described by the Tafel equation, which can be written as follows [18]:
where E is the electrode potential, V; I is the Faraday reduction current of hydrogen ions, A; a is a constant depending on the design of the working electrode; F is the Faraday constant (96 485 C mol–1); R is the universal gas constant (8.314 J mol–1 K–1 ); and T is the temperature, K. The protein is adsorbed on the surface of mercury due to the functional groups of amino acid residues, such as –NH2, =NH, –SH, and –COOH, and shifts the overvoltage of hydrogen evolution to the region of less negative potentials. In most cases, the detected reduction current is proportional to the concentration of the catalytic protein, and it is observed at ultralow protein amounts. The active use of the phenomenon of catalytic hydrogen evolution for the determination of proteins and peptides began only in the second half of the 1990s, when the group of Palecek proposed constant current chronopotentiometric stripping analysis (CPSA) [19]. The CPSA detects changes in the potential with time when a small direct current is applied to the working electrode. The method reduces the influence of background current and makes it possible to obtain a pronounced peak of hydrogen reduction in the (ΔE/Δt)–1–E coordinates (where E is the electrode potential, V and t is time, s) instead of a hard-to-detect wave. The signal detected by the CPSA was named peak H in honor of Heyrovsky (the founder of polarography), hydrogen evolution, and high sensitivity. Using CPSA on a mercury electrode, it is possible to detect proteins and peptides at nanomolar and subnanomolar concentrations. In recent years, a number of protein molecules have been tested by this method [19–22]. However, despite the known reaction mechanism, it was not revealed which amino acid residues make the main contribution to the recorded signal. It is believed that cysteine (Cys) residues play a key role in this case. In slightly acidic media, all peptides (with and without Cys residues) give an peak H. In weakly alkaline solutions, Cys-free peptides do not give a signal, while Cys-containing peptides show a clear peak under the same conditions. According to Palecek and Ostatna [22], almost any peptide and protein gives an peak H under appropriate conditions (Fig. 1).

Polarographic signals of proteins.
In 1933, Brdicka [23] observed a similar effect of catalytic hydrogen evolution when he tried to suppress the signal of cobalt in an ammonia buffer solution with blood serum: the signal was suppressed, but a new clearly pronounced double wave appeared at potentials more positive than that of the pre-sodium wave. This phenomenon was called the Brdicka catalytic reaction (Fig. 1). Of amino acids, only cystine (Cys–Cys) and Cys gave rise to this wave. The height of the new wave was hundreds of times greater than the reduction signal of the Cys–Cys SS groups, which indicated its catalytic nature. Because the height of the catalytic wave for Cys was two times lower than that for Cys–Cys at the same concentration, Brdicka [24] concluded that the SS groups of Cys–Cys were reduced to SH groups, and the SH groups, in turn, served as sources of hydrogen ions for the recorded signal. Using this method, Brdicka [25] analyzed more than 250 blood serum samples from cancer patients and healthy donors to reveal a significant difference between them. Thus, in the case of the blood serum of cancer patients, the height of a characteristic wave was always lower than that for the serum of healthy volunteers. Answering the question which of the groups, SH or SS, are responsible for the polarographic reaction, Brdicka [25] concluded that the observed effect was mainly related to the SS groups. A deeper study conducted with blood serum samples from 386 subjects (cancer patients, patients with other diseases, and healthy donors) revealed the influence of a number of factors (the age of patients and the type of tumor) on the Cys–Cys \( \rightleftarrows \) Cys equilibrium [26]. Since then, the polarographic study of proteins has been based mainly on catalytic waves obtained in the presence of cobalt [27–29]. The Brdicka catalytic reaction has found its application in bioanalysis for the determination of Cys-containing proteins, such as metallothioneins [30, 31] and phytochelatins [32]. It is believed that Cys- and/or Cys–Cys-containing proteins act as catalysts for the reaction, but its mechanism is still not fully understood.
In the 1950s–1960s, works on the structure of proteins, especially that of insulin (Sanger, Nobel Prize in Chemistry 1958) and the spatial structures of myoglobin and hemoglobin by X-ray diffraction analysis (Perutz and Kendrew, Nobel Prize in Chemistry 1962) brought research in the field of proteins and amino acids, including electrochemical studies, to a new level. In the 1960s, the polarographic reduction of proteins via Cys–Cys disulfide bonds (RSSR) was discovered [33, 34]. The overall equation of the processes occurring in this case on the surface of a mercury electrode can be written as follows [35, 36]:
According to the reaction mechanism proposed for the reduction of Cys–Cys [35], mercury(II) cysteinate is initially formed and then reduced (Eqs. (6) and (7), respectively):
Another mechanism was proposed for the reduction reaction of the oxidized form of glutathione (GSSG) (Eqs. (8) and (9)) [36]:
Note that the catalytic electroreduction of hydrogen in the presence of a protein required the reduction of the SS groups of Cys–Cys residues (Fig. 1), but reports of this kind have not been published. This contradiction can be explained by a significant difference in the heights of the two types of waves. The reduction currents of the SS groups of Cys–Cys proteins are about 0.1 μA, while the currents of the catalytic waves of hydrogen evolution are 10–100 μA. Thus, it was necessary to achieve a higher sensitivity of signal measurement in order to detect the reduction of SS bridges, as was first demonstrated by Cecil and Weitzman [33] in 1964 using insulin and a number of other proteins as examples. It is well known that the SH groups of Cys (RSH) after preliminary polarographic oxidation also form mercury(I) and mercury(II) cysteinates adsorbed on the electrode surface (Eqs. (10) and (11)) [37, 38]:
The concentration of protein molecules can also be determined by the reduction of the formed Hg–S bond (Eq. (7), Fig. 1). Later, in the 1970s, studies of the polarographic reduction of hemoproteins, proteins containing the Fe(II)/Fe(III) ion in the active center, revealed a direct electron transfer between the electrode and the prosthetic group to start the electrochemistry of cofactor-containing proteins [39–41].
In the 1970s, it was shown that the adsorption of protein molecules on the electrode surface is a key to detecting direct electron transfer between the electrode and the active site of a protein. The term direct electron transfer means the exchange of electrons between an enzyme (protein) cofactor and an electrode in the absence of any mediators [42]. Direct electron transfer has been described for a small number of enzymes and proteins, which are commonly referred to as redox active. In 1972, Betso et al. [39] described the reduction of the Fe(III) ion of cytochrome c (cyt c) heme in solution on mercury, platinum, and gold electrodes without irreversible protein denaturation. They concluded that protein adsorption on the electrode surface significantly affects the observed electrochemical signal, but it does not cause electrode contamination or loss of the electrode’s ability to transfer electrons [39]. In 1977, the reversible direct electron transfer between cyt c in solution and an electrode was detected [43, 44]:
Cyclic voltammetry in a range from 0.45 to –0.30 V (vs. Ag/AgCl) recorded a clear wave characteristic of diffusion-controlled direct electron transfer between cyt c and the electrode made of mixed oxides of indium and tin [43]. At the same time, the electron transfer between cyt c and the electrode surface of a gold disk electrode was detected only after the introduction of 4,4'-bipyridyl into the cell [44]. The presence of 4,4'-bipyridyl (which is nonelectroactive in this range of potentials) stimulated electron transfer between cyt c and the electrode. Eddowes and Hill [44] suggested that 4,4'-bipyridyl interacts with the protein and/or with the electrode to change its electrical double layer. More recently, Eddowes et al. [45] experimentally confirmed that 4,4'-bipyridyl forms an adsorption layer on the electrode surface, with which cyt c binds through lysine residues before electron transfer. Thus, 4,4'-bipyridyl was the first compound that served as a bridge between the protein and the electrode, orienting the protein on the electrode surface and stimulating direct electron transfer. More recent work has confirmed the importance of some kind of link between the active site of the enzyme and the electrode surface for efficient electron transfer. In particular, Rusling [46] observed a similar effect for hemoproteins using the films of surfactants.
Further studies in the electrochemistry of cofactor-containing proteins led to the immobilization of enzymes on the electrode surface and the discovery of a phenomenon of bioelectrocatalysis. The term bioelectrocatalysis is defined as the acceleration of electrochemical reactions with the use of biological catalysts [47]. Bioelectrocatalysis can also be called biocatalysis with the replacement of biological electron delivery systems by electrochemical systems. Direct bioelectrocatalysis assumes the absence of any freely diffusing or immobilized mediators; electrons freely pass between the active site of the enzyme and the electrode [47]. Pioneering works in the field of bioelectrocatalysis were carried out in the late 1970s–1980s by Soviet scientists led by Berezin. The first oxygen enzyme electrode was made by immobilizing laccase on a carbon black electrode [48]. Molecular oxygen was reduced at the active site of the enzyme, while electrons were supplied from the electrode material (Eq. (13)):
Laccase belongs to the blue copper oxidase family, which also includes bilirubin oxidase, ascorbate oxidase, and ceruloplasmin. Copper-containing oxidases have the ability to directly reduce oxygen to water (Eq. (13)) without the formation of highly reactive toxic oxygen intermediates, such as the superoxide radical anion \(\left( {{\text{O}}_{2}^{{- \bullet }}} \right),\) the hydroxyl radical (•OH), and hydrogen peroxide (H2O2). Hemoproteins are another large class of proteins containing redox-active cofactors. In 1979, Yaropolov et al. [49] published the first work describing bioelectrocatalysis with the participation of peroxidase hemoprotein immobilized on a soot electrode; five years later, Yaropolov et al. [50] discovered direct bioelectrocatalysis with the participation of another hemoprotein hydrogenase. Obviously, the principles of the electrochemistry of redox-active cofactor-containing proteins are based on biological electron-transfer reactions. Nature uses a relatively limited range of redox-active centers: heme, quinones, flavins, and iron-sulfur clusters. The understanding of biological principles that determine efficient electron transfer is important for the use of natural electron carriers (oxidoreductases, NADH-dependent dehydrogenases, and redox-active proteins) in biosensors, biofuel cells, and bioelectrosynthesis [51].
At the same time, the electrochemical oxidation of the sulfur-containing amino acids Cys and Cys–Cys on platinum and gold electrodes was demonstrated and studied in detail in the 1960s [52–54]. Two decades later, in 1980, Brabec and Mornstein [55] and Reynaud et al. [56] independently described the oxidation of proteins due to the amino acid residues of tyrosine (Tyr) and tryptophan (Trp) on electrodes made of carbon materials. The oxidation signal of ribonuclease, albumin, lysozyme, and insulin was observed on an impregnated graphite electrode at potentials of about 0.7–0.8 V (vs. saturated calomel electrode (SCE)) [55]. The electrooxidation signals of ribonuclease (RNase), albumin, and concanavalin A were recorded on a carbon-paste electrode at potentials of 0.8–0.9 V (vs. SCE), while no clear peaks due to oxidation of these proteins were obtained on gold, platinum, and glassy carbon electrodes [56]. Since then, the electrochemistry of proteins and peptides has been developed mainly with the use of solid electrodes.
Nucleic acids. E. Palecek (1930–2018, Brno, Czech Republic) stood at the origins of the electrochemistry of nucleic acids. He devoted his life to studying the properties of DNA and other biopolymers and left behind a huge number of works (books, reviews, and original articles). An active study of the electrochemical properties of nucleic acids began in the late 1950s, when DNA and RNA molecules were oxidized and reduced on a mercury electrode [57, 58]. In 1958, Palecek, a student of Heyrovsky, observed the reduction of adenine (Ade) and cytosine (Cyt) on a dropping mercury electrode, and the reduction of the latter proceeded not only in an acidic but also in a neutral medium; guanine (Gua) gave a characteristic anodic signal due to the oxidation of a DNA reduction product at high negative potentials [57, 59–61] (Fig. 2). Before that, it was believed that, among the components of nucleic acids, only Ade can be electrochemically reduced on mercury in strongly acidic media [62]. Curiously, in 1957, Berg [63] confirmed the electrochemical inertness of nucleic acids by determining protein traces in DNA and RNA samples in the presence of cobalt ions. In 1960, Miller [64, 65] was the first to demonstrate the ability of DNA to be adsorbed on the surface of a polarized mercury electrode. Palecek [57] recalled his work in those years:

Polarographic signals of nucleic bases.
Shortly after the publication of my article [60] on the oscillographic polarography of DNA from calf thymus and its degradation products in Nature (1960), I was invited by J. Marmur from Harvard University for an internship in his laboratory as a doctoral student. It took about two years before I was allowed to leave communist Czechoslovakia, but for me it was like a miracle. In 1962, the Polaroskop P 524 instrument for oscillographic polarography was manufactured only in Czechoslovakia. Marmur, who by this time had moved from Harvard to Brandeis University, advised me to bring this instrument with me. Thus, I sent the instrument by air cargo. Armed with a letter of recommendation from Heyrovsky, I traveled to the United States in November 1962 with no doubts about the suitability of my method for nucleic acid research. Unfortunately, the instrument, completely broken, arrived only nine months later. The prevailing opinion in the laboratory was that it had been carefully “searched” by secret services on both sides of the Iron Curtain.
In the United States, Palecek acquired necessary microbiological knowledge and practical experience in molecular biology. At that time, Marmur was one of the leading scientists in the field of DNA. Marmur was the first to discover the renaturation and hybridization of DNA, that is, its ability to restore a double strand from complementary single strands [66, 67]. This ability of DNA is being used today in a range of molecular biology techniques and in biotechnology, including electrochemical DNA sensors. Marmur also proposed a method for DNA extraction [68], which has been used with some modifications for several decades as a classic method for extracting DNA from bacteria and other organisms. After returning from the United States, Palecek continued his active work in the field of electroanalysis of nucleic acids. Palecek’s discovery laid the foundation for a further study of the electrochemical behavior of the molecules of nucleic acid and related compounds.
In the late 1970s, Cummings and Elving [69] performed the one-electron reduction of thymine (Thy, El/2 = –2.4 V) and uracil (Ura, El/2 = –2.3 V) on mercury [70] with the formation of the corresponding radical anions and proton transfer in a medium of dimethyl sulfoxide (Fig. 2). In this case, Ura and Thy anions formed insoluble salts with mercury, giving rise to the subsequent oxidation waves of reduction products (El/2 of from –0.1 to –0.3 V) [69, 70]. In contrast to Ura, in the case of Thy containing a methyl group in the 5-position, which stabilizes the free radical and creates a steric hindrance to dimerization, the resulting free radical was reduced [70]. It was shown that all nucleic bases and some other derivatives of purine and pyrimidine give anodic signals due to the formation of sparingly soluble compounds with electrode mercury, which was subsequently used for their cathodic inversion determination at nanomolar concentrations [71–73]. In the mid 1980s. Palecek’s group proposed an adsorption inversion analysis of nucleic acids, which made it possible to increase the sensitivity of determining the concentration of DNA by several orders of magnitude. It was found that DNA and RNA can be easily immobilized on the surface of mercury electrodes by simply immersing the electrode in a drop of a nucleic acid solution (3–10 μL) for a short time (30–180 s) [74, 75]. Due to strong adsorption, DNA and RNA formed a stable layer on the electrode surface; then, the electrode was washed, and voltammetric measurements were carried out in a new solution that no longer contained the analyte. This procedure made it possible to decrease the analyzed sample volume by two or three orders of magnitude, as compared to that in classical voltammetry. This technique was called adsorptive transfer stripping voltammetry (AdTSV). The combination of AdTSV with dc chronopotentiometry was particularly successful in the determination of nucleic acids using mercury electrodes [76].
In the first two decades, only mercury electrodes were used to determine the concentrations of nucleic acids. However, despite the unique advantages, mercury electrodes have a number of limitations, including mechanical instability and a narrow range of operating anode potentials, which is only rarely suitable for the oxidation of organic compounds. In the 1980s, Brabec [77, 78], who was a post-graduate student of Palecek and a coworker of Dryhurst, together with the latter introduced electrodes made of carbon materials into the electrochemistry of nucleic acids [79–83]. Considering Brabec’s contribution to the electrochemistry of proteins, he can safely be called the founder of the direct electrochemical analysis of biopolymers on solid electrodes. Surprisingly, the first publications on the electrochemical oxidation of high-molecular-weight nucleic acids on electrodes made of carbon materials appeared only in 1978 [79–81]. Already in the early 1960s. Smith and Elving [84, 85], who studied the behavior of the monomeric components of nucleic acids on a graphite electrode, showed that Gua and Ade are capable of being oxidized on this type of electrodes. More recently, Dryhurst and coauthors [86, 87] studied the oxidation of Ade and Gua on a pyrolytic graphite electrode in more detail and suggested possible mechanisms of electrode reactions. An important result of the study of the electrooxidation of low-molecular-weight components of nucleic acids was the discovery of the oxidation of Ade and Gua nucleosides at significantly higher positive potentials than the potentials characteristic of free bases [88, 89]. These studies also showed a significant difference between the peak potentials of guanosine and adenosine oxidation. In 1980, Brabec [82] published the quantitative regularities of the oxidation of DNA molecules from various natural objects on a pyrolytic graphite electrode found using differential pulsed voltammetry. The studied DNA molecules differed in the total concentrations of Gua and Cyt (Gua + Cyt) bases. All of the DNA samples exhibited peak G (at a potential of about 0.9 V vs. SCE, pH 6.4) corresponding to the oxidation of the Gua residue and peak A (about 1.2 V) corresponding to the oxidation of Ade in the voltammograms. It was noted that the potentials of the peaks G and A differed by 0.28 V, and they did not reflect the Gua + Cyt content of the DNA sample. However, it was found that the ratio of the heights of peaks A and G, denoted by K, was equal to the (Ade + Thy)/(Gua + Cyt) ratio of the DNA sample. This fact was used to develop a method for determining the Gua + Cyt content of DNA according to the formula
where K is the ratio of the heights of peaks A and G corresponding to the oxidation of Ade and Gua residues of DNA. It is interesting that the height ratio between peaks A and D was the same for native and thermally denatured DNAs. In the case of denatured DNA, the signal strengths noticeably increased compared to that of undenatured DNA [82].
The interest of electrochemists in nucleic acids was bolstered by the award of the Nobel Prize in Medicine in 1962 to Wilkins, Crick, and Watson for their discovery of the molecular structure of nucleic acids and their importance for the transmission of information in living systems. By the time of the first publications on the electrochemical activity of nucleic acids, the structure of DNA in the form of a double strand was described by Watson and Crick [90] simultaneously with Franklin and Gosling [91] in 1953. This happened almost a hundred years after the discovery of the DNA molecule by Miescher in 1869 [92]. Watson and Crick [90] proposed a system based on paired bases. The structure of DNA included two hydrogen bonds for the Ade and Tim pair and three hydrogen bonds for the Gua and Cyt pair. This nucleic acid molecule is now known as the B form of DNA stabilized by bound water, which fits perfectly into the minor groove. Altman gave the name to DNA in 1889, and its role in the transmission of genetic information was discovered by Griffith in 1923 [92]. In 1953, Zamenhof et al. [93] reported that the amount of Ade in DNA is always equal to the amount of Thy, and the amount of Gua is always equal to the amount of Cyt. This regularity was extremely important in the development of the double strand model. The amount of purines (Ade + Gua) is always equal to the amount of pyrimidines (Cyt + Thy), but the ratio (Gua + Cyt)/(Ade + Thm) varies from species to species.
Thus, we can conclude that, by the end of the 1980s, the foundation of the electrochemistry of proteins and nucleic acids was laid and the main types of analytical signals for the oxidation and reduction of these biomolecules on a mercury electrode and electrodes made of solid materials were published. However, despite the knowledge of the complex spatial organization of biopolymer molecules, the concept of a one-dimensional structure of proteins and nucleic acids when interpreting the results dominated in electrochemistry. Although many processes and phenomena, such as direct electron transfer or bioelectrocatalysis, had been discovered by this time, the nature of the analytical signals of biopolymers was not fully explained.
CURRENT CONCEPTS ON THE ELECTROCHEMICAL PROPERTIES OF PROTEINS AND NUCLEIC ACIDS
At present, electrochemists are coming to understand the need to expand their concepts of redox processes occurring on the surface of electrodes with the participation of biopolymers, such as proteins and nucleic acids, primarily taking into account their spatial (3D) structure [94]. For a long time, the electrochemical behavior of biopolymers was considered from the point of view of the total number of certain electroactive groups in the primary sequence of molecules based on the number of monomer units. However, recent studies indicated that not all potentially electroactive groups in a polypeptide or polynucleotide chain can simultaneously participate in electrochemical reactions [95]. The electrochemical signal decreases with an increase in the molecular weight of a biopolymer (its surface area) per electroactive group [95, 96]. With the development of computer technologies and bioinformatics methods that make it possible to model the spatial structure of macromolecules, it becomes obvious that the electrochemical signal of a biopolymer cannot be equal to the sum of the signals of its constituent monomers. On the one hand, this complicates the interpretation of an analytical signal and, on the other hand, opens up broad prospects for the application of electrochemistry to solve biochemical and medical problems. Based on the molecular structure, a scientist can use electrochemical analysis to detect changes such as the formation of bipolymer complexes [97], the aggregation [98] or degradation of biopolymers [99], and their posttranslational (PTM) [100] or postreplicative (PRM) modifications [101]. These changes in the structure of biopolymers occur in living systems both under normal conditions and in various pathologies [98–105]. Moreover, if earlier there was a clear division into electroactive and nonelectroactive substances, today it would be more correct to say that a biomolecule is nonelectroactive under the given experimental conditions. There is confidence that a search for suitable conditions will make it possible to discover new properties of substances, in particular, biopolymers, sugars, and lipids in the future. A deeper understanding of the relationship between the spatial organization of proteins and nucleic acids and their electrochemical behavior contributes to the development of fundamentally new (bio)sensor systems for both everyday use and biomedical research.
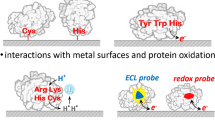
Proteins. To date, it is well known that proteins on solid electrodes are capable of producing an electrochemical signal (1) due to the oxidation of amino acid residues (in a range of potentials from 0.5 to 1.5 V) (2) and/or due to the reduction or oxidation of their prosthetic groups (for example, heme or flavin in a range from –0.5 to 0.5 V vs. Ag/AgCl) [47, 51, 106–112] (Fig. 3). Hasoň et al. [112] presented a detailed analysis of the electrochemical behavior of biomolecules, in particular, biopolymers, on electrodes made of carbon materials (graphite, graphene, carbon nanotubes, and boron-doped diamond) taking into account the effect of the electrode surface structure on the adsorption of molecules. It should be noted that, in contrast to routine graphite and glassy carbon electrodes, boron-doped diamond electrode has a wider operating range of positive potentials. However, as far as is known, no fundamentally new properties of protein and nucleic acid molecules have been discovered so far on electrodes made of boron-doped diamond [113] as well as on electrodes modified with graphene or carbon nanotubes. At the same time, recently, Suprun et al. [114] found that almost all proteinogenic amino acids can be oxidized on the surface of printed graphite electrodes at a potential of 0.95 V in amperometric flow-injection analysis. In some cases, a signal of the reduction of amino acid oxidation products (in particular, Trp) on a pencil graphite electrode [115]. Nevertheless, clear analytically significant signals of the reduction of proteinogenic amino acids and their residues have not yet been obtained. The Tyr, Trp, and Cys residues made the greatest contribution to the oxidation signal of a protein molecule at a potential of 0.6–0.8 V [109, 116–124], whereas a potential of about 1 V or higher (vs. Ag/AgCl, neutral medium) was required for the oxidation of His, Met, and Cys–Cys residues [118–124] (Fig. 4). Thus, it was found that the Aβ(1–42) peptide gave two clear peaks in a square wave voltammogram at potentials of 0.6 and 1.0 V and a wave in a potential range of 1.2–1.5 V, which were respectively assigned to the Tyr (Tyr-10), His (His-6, -13, and -14) and Met (Met-35) residues of the peptide [122]. On electrodes made of carbon materials and gold, the oxidation of proteins due to amino acid residues proceeded as an irreversible pH-dependent process [95, 117–120]. Note that the oxidation mechanism of amino acid residues in proteins and the oxidation reactions of most free amino acids remain not fully understood [109, 110, 125–127]. The products of electrochemical reactions should be identified using various physicochemical methods (in particular, NMR and IR spectroscopy). A natural question arises about the relationship between the structure of a protein and its electrochemical properties. One of the first attempts to answer this question was made by Brabec and Mornstein [55], who wrote: “Bovine serum albumin, also containing, similarly to lysozyme, tryptophan residues, yielded, however, only one peak at the potentials of the oxidation peak of tyrosine residues. This behaviour might be explained by the fact that bovine serum albumin contains approx. 10-times more tyrosine than tryptophan residues.” Thus, it can be seen that Brabec and Mornstein [55], who published this work in 1980, relied only on the primary structure of the protein, namely, on its amino acid sequence, and assumed that the number of electroactive residues, such as Tyr and Trp, determined the observed electrochemical behavior of proteins. It took three decades to understand that only amino acid residues located on the surface of the protein molecule, with electroactive groups oriented outward, are available for electrochemical oxidation on the electrode surface; therefore, they determine the signal of electrooxidation [95]. Proteins have a sufficiently rigid spatial structure, whereas the conformation of peptides is often characterized as a random coil. The electrochemistry of peptides can be considered as a separate area [128]: the signals of their amino acid residues are more pronounced and easier to interpret due to small molecular weights. A strong argument in favor of a close relationship between the signal intensity of electrooxidation and the protein structure is that the unfolding (denaturation) of protein leads to a significant increase in the strength of current [55, 120, 129]. On the contrary, the aggregation of proteins or peptides leads to a decrease in the electrooxidation current [130–134], which also suggests that the location of electroactive residues on the surface of the protein molecule rather than the total number of the residues is responsible for the observed electrochemical behavior. Interestingly, large peptide aggregates, such as those of β-amyloid, which is involved in the pathogenesis of Alzheimer’s disease, are unable to produce any significant oxidation signal: monomers and small oligomers make the main contribution to the overall sample signal in the course of peptide aggregation [130]. In fact, this makes electrochemistry a unique method for the monitoring of peptide aggregation by detecting a decrease in the oxidation current caused by the depletion of a pool of peptide monomers/oligomers when they were included in aggregates [131–134]. It was also found that the electrooxidation signal is sensitive to substitutions of individual amino acids in the polypeptide chain, as occurs in the case of β-amyloid mutants [122, 123, 135] and genetically modified variants of acetylcholinesterase [117]. In the latter case, some substitutions of electroactive residues for conditionally nonelectroactive ones led to an increase in the electrooxidation signal, indicating the significance of conformational changes associated with substitutions for the availability of electroactive residues for oxidation on the electrode surface [95]. Thus, conformational changes can also affect the intensity of electrooxidation signals along with the concentration of a protein or peptide [55, 120, 122, 136].

Oxidation and reduction potentials of proteins and nucleic acids on solid electrodes.

Potentials and sites of the amino acids tyrosine, tryptophan, cysteine, histidine, methionine, and cystine oxidation on solid electrodes.
Returning to active centers, note that the detection of the redox activity of proteins bearing prosthetic groups requires a special modification of the electrode surface, which allows one to connect the active center and the electrode [46, 47, 108]. In fact, this also confirms the hypothesis on the preservation of the spatial structure of a redox-active protein under the conditions of an electrochemical experiment on the electrode surface [46]. As a rule, electrode reactions involving the active sites of proteins proceed in a thin layer on the electrode surface, and they are characterized by a linear dependence of the peak current on the potential scan rate [46, 47, 106–108]. The protein cyt c is perhaps the only exception to this rule, and it gives a reversible signal of the Fe(III)/Fe(II) heme ion from a solution drop even on an unmodified electrode [39, 43]. In the case of redox-active proteins, their oriented immobilization on the electrode surface becomes of decisive importance for achieving efficient electron transfer and bioelectrocatalysis [47, 137]. It is possible to achieve an ordered arrangement of enzyme molecules on the electrode surface, that is, to orient them for observing the most efficient bioelectrocatalysis, with the use of conductive polymers or conductive nanomaterials. In addition, mutant forms of enzymes with specially introduced amino acid substituents are widely used. Direct electron transfer from the electrode to the active center of an enzyme (protein) makes it possible to determine the concentration of redox-active proteins by recording the peak current of the reduction or oxidation of its active centers [138–140], while the enzymatic activity of a protein can be initiated by applying an appropriate potential [137, 141]. However, the search for a suitable matrix for the immobilization of redox-active proteins is not an easy task, and it is often impossible to achieve efficient coupling of a biochemical enzymatic reaction with an electrode reaction. In some cases, direct electron transfer between the active site of the enzyme (protein) and the electrode is unattainable by definition due to glycosylation or the large size of a protein globule. The relationship between the structure and functions of a protein enzyme can be determined by studying the mechanisms of direct bioelectrocatalysis [47], which is important for understanding biochemical processes and for developing biosensor systems. The targeted introduction of amino acid residues, such as His and Cys, peptides, or biotin fragments using genetic engineering technologies has made it possible to develop redox-active enzymes with desired electrochemical properties including the possibility of their targeted immobilization [137, 142]. It is well known that some amino acid substitutions (mutations) can have a noticeable effect on the enzymatic activity of proteins. Thus, the study of the electrochemical and electrocatalytic activity of hemoproteins from the cytochrome P450 (CYP) family, namely human CYP 2C9 and its two polymorphic variants, which are present in approximately 35% of the Caucasian population, immobilized on the surface of a glassy carbon electrode showed a lower catalytic activity for the forms CYP 2C9*3. (ks = 3 ± 1 min–1) and CYP 2C9*2 (ks = 12 ± 2 min–1), as compared with wild-type CYP 2C9 (ks = 18 ± 1 min–1) in the presence of S-warfarin, a marker substrate of CYP 2C9 [143]. The developed bioelectrochemical method makes it possible to determine therapeutically significant differences in the metabolism of drugs associated with polymorphism, which is responsible for adverse drug reactions, in particular, in a significant part of the population of the Caucasus. With the use of single amino acid substitutions in the enzyme sequence, it is also possible to solve the problem of increasing the rate of direct electron transfer in electrochemical biosensors, as shown by the example of tobacco mosaic peroxidase [144]. Olloqui-Sariego et al. [144] observed a significant increase in the rate of direct electron transfer between the graphite electrode and tobacco mosaic peroxidase after replacing the Leu residue located near the heme pocket on the enzyme surface with Trp [144]. An increase in the rate of direct electron transfer between the redox active center of a protein and the electrode and an increase in the sensitivity of the determination of an enzyme substrate can be achieved by changing the spatial structure of the protein, that is, the directed unfolding (denaturation) of a polypeptide chain, as found with the examples of hemoglobin [145] and cyt c [146] with respect to hydrogen peroxide and the nitrite ion. Note that many PTM have been found for hemoproteins [102]. The question of whether these PTM affect the ability of the Fe(III)/(II) ion of the protein heme to donate or accept electrons can be answered using electrochemistry. For example, Gómez-Mingot et al. [147] found that horse skeletal muscle myoglobin with electrochemically nitrated Tyr-103 gives a lower and less pronounced reduction peak than that of native myoglobin in cyclic voltammograms. Unfortunately, despite the importance of the effect of PTM on the structure and functions of redox-active hemoproteins, such as cyt c [148] or cytochrome P450 2B1 [149], changes in the intensity of direct electron transfer and bioelectrocatalysis of these proteins as a result of PTM are currently poorly understood.
Modern technologies make it possible to obtain the three-dimensional structure of a protein molecule with the localization of all amino acid residues. Currently, the Protein Data Base (PDB, https:// www.rcsb.org) contains many 3D models of structures, including protein complexes. Of course, the results of computer simulation do not necessarily predict the true localization of the electroactive groups of amino acid residues under the conditions of an electrochemical experiment, and it should be borne in mind that their location can be influenced to some extent by (1) the composition of an electrolyte (solvent), (2) the electric field of the electrode, and (3) the degree of rigidity of the protein molecule as such. Another important aspect of protein electrooxidation is that some amino acid residues in the protein sequence can be modified with different functional groups due to the phenomenon of posttranslational modification [150]. These new functional groups can generate their own electrochemical signals or influence (suppress or enhance) the original signal of amino acid residues [100, 151]. Currently, both amino acid sequences and expected PTM can be retrieved for the vast majority of proteins in a comprehensive database known as the Universal Protein Resource (UniProt, https://www.uniprot.org). Thus, the electrochemical behavior of almost any protein molecule at the electrode can be predicted using the Protein Data Base and the Universal Protein Resource, and this makes it possible to simplify the interpretation of this behavior solely on the basis of the protein amino acid sequence (for example, see [118, 152]). Taking into account the spatial structure of a protein for interpreting its electrochemical signals allows a deeper understanding of the real processes occurring on the electrode surface [153].
Nucleic acids. The residues of nitrogen bases (Gua, Ade, Thy, Cyt, and Ura) are known to be involved in the reduction and oxidation of nucleic acids on solid electrodes [79, 82, 94, 96, 97, 112, 154–161]. However, among various types of electrodes, only the pyrolytic graphite electrode made it possible to detect both the reduction and oxidation of nitrogen base residues in nucleosides [162] and oligonucleotides [159] due to a wide working potential window from –2 to 2 V (vs. Ag/AgCl; acetate buffer solution, pH 5). In addition, both the signals of the oxidation of reduction products and the signals of the reduction of oxidation products of nitrogen bases in nucleosides and oligonucleotides were also recorded on pyrolytic graphite [159, 162] (Fig. 3). The reduction or oxidation reactions of nucleic bases or their residues require sufficiently high negative (approximately from –1.5 to ‒2.0 V) or positive (approximately from 1.0 to 1.5 V) potentials, respectively [79, 82, 86, 87, 154, 155, 158, 161–173]. The signals of oxidation or reduction of Gua and Ade bases and their residues in nucleic acid molecules can be easily obtained under various experimental conditions and on various types of solid electrodes, whereas Thy, Cyt, and Ura require higher positive or negative potentials and more stringent experimental conditions; as a consequence, the attribution of their signals is often questionable (Fig. 5). Thus, the oxidation of mononucleotides on carbon electrodes manifested itself as anodic peaks at potentials of 0.9–1.0 V for guanosine monophosphate, 1.2–1.3 V for adenosine monophosphate, 1.4–1.5 V for thymidine monophosphate, and 1.5–1.6 V for cytidine monophosphate (vs. Ag/AgCl; phosphate buffer solution, pH 7.4) [163, 174]. Interestingly, the addition of sugar and phosphate groups to a nitrogen base (with the formation of a nucleoside or nucleotide) shifted the maximum oxidation potential of molecules to more positive values, and vice versa [86, 87, 164, 165]. However, the oxidation and reduction processes of nucleic bases in DNA molecules occurred at potentials different from the potentials characteristic of the corresponding free nucleosides or nucleotides, which indicated that the oxidation processes of base residues in monomeric and polymeric molecules proceed according to different mechanisms and give different products [159, 162]. The mechanisms and products of the electrochemical oxidation or reduction of free nitrogen bases and their nucleosides and nucleotides are well studied [55, 86, 87, 159, 162, 164–168] (Fig. 5), but there are no similar detailed studies on base residues in polymeric DNA or RNA molecules; therefore, any conclusions based on a knowledge of monomers are rather speculative [162]. Both oxidation and reduction of nucleic bases can involve the formation of intermediate products in the form of radicals and their chemical reactions, which are sensitive to the location of active fragments [162]. Many factors, such as the sequence and structure of nucleic acids, the analyte concentration, the electrode surface properties, and the pH and composition of the supporting electrolyte, can affect the mechanisms of electrode reactions of nucleic bases [162]. Note that, to date, signals due to the reduction of the base residues of high-molecular-weight DNA or RNA from natural sources on solid electrodes have not been detected. At the same time, oxidation and reduction signals were obtained for all base residues in short synthetic oligonucleotides of a certain sequence with the predominance of the oxidation signals of Gua and Ade residues [159, 161, 173–176]. However, it should be noted that, unfortunately, most studies did not provide any information on the purity of oligonucleotides or the additional purification of obtained synthetic preparations and on the conformation of single-stranded DNA (ssDNA) molecules.

Reaction schemes of the primary oxidation of guanine and adenine on carbon electrodes [83].
As for high-molecular-weight double-stranded DNA (dsDNA) from natural sources, its oxidation on solid electrodes due to base residues, which was first demonstrated in the pioneering works of Brabec [82, 154, 155], is still controversial [112, 158, 169, 175]. The dsDNA molecules show only two oxidation peaks at about 0.7–0.9 and 1.0–1.2 V (vs. Ag/AgCl, neutral medium) in voltammograms recorded on electrodes made of carbon materials, and these peaks were attributed to Gua and Ade residues, respectively [79, 82, 83, 155, 172]. It was hypothesized that the electroactive groups of these nitrogen bases, which are not involved in the formation of hydrogen bonds in the DNA double strand, remain free for electrooxidation on the electrode surface [82, 154, 155]. It was also found that the peak currents of the oxidation of native dsDNA are noticeably lower than the signals of denatured DNA [82, 95, 154, 155], and they regularly decrease with an increase in the logarithm of the molecular weight of DNA in both native and denatured biopolymers [96]. To explain the observed differences in the electrochemical behaviors of native and denatured DNA, it has been suggested that DNA is adsorbed on the electrode surface, and the flexible polymer chain of denatured DNA can fit better or repeat the uneven electrode surface than a more rigid molecule of native dsDNA [95, 154, 155 ]. As a result, the number of DNA segments in contact with or in close proximity to the electrode surface so that they can be electrooxidized in denatured DNA is higher than that in native dsDNA. In fact, this is a vivid example of considering the DNA molecule as a one-dimensional structure. Indeed, this hypothesis was partly confirmed by the conclusions that preliminary electrode polarization at positive potentials (from 0.2 to 1.3 V) for several minutes was required for the reliable detection of oxidation signals from both native and denatured DNA and even from small oligonucleotides on carbon and gold electrodes [96, 154, 155, 169–171, 174]. This was explained by more efficient adsorption of negatively charged biomolecules on positively charged electrodes. For example, it was found that, after the prepolarization of an electrode made of highly ordered pyrolytic graphite at 0.4 V (vs. Ag/AgCl) for 15 min, dsDNA from calf thymus exhibited pronounced oxidation peaks of Gua and Ade residues, which cannot be obtained without prepolarization [171]. However, Brabec and Dryhurst [79] failed to obtain the oxidation signal of Gua residues of native dsDNA from calf thymus even after prepolarization. Moreover, Wang et al. [177], using amperometric flow-injection analysis at a potential of 1 V, demonstrated that native dsDNA from calf thymus produced higher oxidation currents on carbon-paste electrodes than those of denatured DNA. It is now increasingly recognized that the nitrogen bases hidden inside the DNA double strand may be difficult to access or not available at all for electrode reactions [157, 178]. Thus, the results of experiments on the electrochemical oxidation of dsDNA so far raise more questions than answers.
One of the explanations for the observed discrepancies in the electrochemical behavior of DNA can be degradation, especially, with consideration for the well-known dependence of the electrooxidation current on the size of DNA [96]. Degradation can lead to the formation of low-molecular-weight DNA fragments and/or, possibly, free monomers, which can significantly affect the result of an electrochemical experiment. Indeed, DNA degradation was demonstrated for dsDNA from calf thymus immobilized on the surface of a gold electrode at a potential of 0.5 V (vs. Ag/AgCl) [179]. The electrode prepolarization procedure can potentially contribute to this degradation. The presence of metal ions (or their complexes) in the sample in combination with the application of a negative potential to the electrode can destroy DNA molecules, as shown for DNA and Cu(II)-bipyridyl complexes at –0.6 V [180]. Nevertheless, whether DNA degrades on electrodes made of carbon materials during an electrochemical experiment is currently not completely known [155].
Although the use of dsDNA from natural sources in research is extremely attractive due to the availability and low cost of commercial preparations, these preparations suffer from molecular size heterogeneity and the potential presence of both ssDNA and dsDNA, as rightly pointed out by Brabec and Koudelka [96] in 1980. Unfortunately, the biochemical characterization of DNA samples used in many electrochemical studies was not given [171, 172, 177, 181]; because of this, it is difficult to compare the results obtained by different authors and interpret them. Obviously, polymerase chain reaction (PCR) amplicons, which are ideal dsDNA fragments of a given length, which can vary from tens to hundreds of base pairs, are a more suitable model for studying the electrooxidation of dsDNA. Their application would allow a better understanding of the mechanisms underlying the electrooxidation of dsDNA in an electrochemical experiment. Recently, Suprun et al. [175] found that the electrooxidation of DNA of natural origin on printed graphite electrodes proceeds predominantly by the oxidation of Gua and Ade residues of ssDNA molecules, which was expressed by two signals at potentials of about 0.75 and 1.05 V (phosphate buffer solution, pH 7.4), respectively. For ssDNA, it was found that the oxidation reactions of both residues are controlled by diffusion. In dsDNA molecules, even as short as 24 nucleotides long, the residues of these bases seem to be inaccessible for oxidation on carbon electrodes. It can be concluded that electrooxidation signals for a particular heterogeneous DNA preparation of natural origin are produced predominantly by low-molecular-weight ssDNA fragments [175]. This electrochemical behavior of DNA is in good agreement with the results obtained for protein molecules, in which electroactive amino acid residues hidden inside a protein globule [95] or a peptide aggregate [128, 130, 132] are inaccessible for electrode reactions.
Although the direct electrochemistry of nucleic acids is characterized by high oxidation (1 to 2 V) and reduction (–1 to –2 V) potentials and relatively low detectable currents, electrochemical (bio)sensor systems have been developed based on a decrease in the electrooxidation signals upon the formation of a double strand [99, 178, 182]. In order to improve the analytical characteristics, electrodes modified with various materials are widely used for electrocatalysis with respect to nucleic acids and their monomers and, as a result, for increasing the sensitivity of the analysis [156]. An alternative strategy proposed by the group of Hocek and Fojta [183, 184] is based on direct electrochemical probing of DNA with modified bases. A palette of electrochemically active groups, which can be introduced into DNA sequences by polymerase incorporation of chemically modified nucleotides, was developed [183, 184]. This approach made it possible to significantly increase the sensitivity and selectivity of the electrochemical detection of nucleic acids on various electrodes. Moreover, the multipotential redox coding of nucleic acids became possible. This approach can have a wide range of analytical applications, including the detection of DNA mutations, damage, hybridization, and amplification and the analysis of DNA–protein interactions. The electrochemical properties of nucleic acids can potentially be used to monitor various PRM of DNA and RNA [101, 185]. Like amino acid residues in proteins, a large number of modified nucleosides were identified in the DNA and RNA of living organisms, viruses, mitochondria, and chloroplasts as a result of normal and pathogenic enzymatic or nonenzymatic processes [185]. Various electrochemical strategies were developed to detect DNA or RNA as potential cancer biomarkers, for example, methylated DNA of gene promoters, circulating tumor DNA, viral nucleic acids, or short noncoding RNA molecules, in particular microRNA [101]. In conclusion, it should be noted that the selectivity of the electrochemical determination of substances is primarily determined by sample preparation, separation, and preconcentration methods. In particular, the analysis of biosamples for the presence of viral or bacterial DNA is preceded by a targeted amplification step implemented using PCR or isothermal amplification [186], which makes it possible to detect up to one nucleic acid molecule of a given sequence in a sample.
Nanopore sequencing of biopolymers. In modern bioelectrochemistry, one more promising direction based on a conductivity change during the passage of a biomolecule through a nanosized channel, a so-called nanopore—nanopore sequencing—can be recognized. A readable conductometric signal is due to the structure peculiarities of biopolymers. Nanopore sequencing has been developing since the early 1990s; recently, it enabled Long [187] to perform successful DNA sequencing. This method of detecting the electrochemical signal of a biomolecule opens up possibilities for studying population heterogeneities and the conformational dynamics of systems ranging from individual DNA to individual proteins [188]. Nanopores, which ensure the placement of individual objects of analysis in a limited space, transform the behavior of a single molecule into a detected electrochemical signal with a high signal-to-noise ratio. A large amount of research was devoted to the electrochemical detection of various objects from nucleic acids, peptides, proteins, and biomolecular complexes to organic low- and high-molecular-weight molecules with the use of nanopores. Due to the successive retention of a part of a molecule in a nanopore, new signal reading mechanisms shed light on the relationship between the structure of a single molecule and its conductometric activity. Thus, Li et al. [189] used the T232K/K238Q mutant aerolysin nanopore with enhanced electrostatic interaction at the T232K region and a high repulsive barrier at the K238Q region to study the phosphorylation of a 9-mer Tau protein peptide, which is involved in the pathogenesis of Alzheimer’s disease. The signal produced by a sensor based on T232K/K238Q aerolysin made it possible to identify the characteristic distribution of unphosphorylated Tau peptide, pS262-Tau peptide, pT263-Tau peptide, and pS262/pT263-Tau peptide with almost 100% accuracy (where p refers to a phosphorylated amino acid). The excellent sensitivity of this protein nanopore was due to the extremely low translocation rate, which increased the duration of signal reading to tens or hundreds of milliseconds for a peptide of nine amino acid residues [189].
Oxford Nanopore Technologies currently produces a series of DNA sequencing devices under the brand Oxford Nanopore (https://nanoporetech.com). In Russia, SkyGen is the exclusive distributor of Oxford Nanopore Technologies products (https://www.skygen.com). SkyGen regularly holds conferences and seminars and provides scientific support to users of Oxford Nanopore sequencers. The Oxford Nanopore devices detect a change in the ion current at the moment of passage of a biomolecule through a nanopore. Information on current changes is used to identify the analyzed biomolecule. Specially designed and patented pore-forming proteins are used to create pores in the membranes of instruments made in accordance with the Oxford Nanopore technology. Pore-forming proteins are widespread in nature. For example, the protein α-hemolysin and similar protein pores naturally occur in cell membranes where they act as channels for transporting ions or molecules in and out of cells. The protein α-hemolysin is a heptamer with a pore with an inner diameter of 1 nm. The sizes of many biomolecules, including DNA, lie in the same range. The pores are very stable. Note that nanopore sequencing technology would not be feasible in practice without the bioinformatic processing of received signals. The company has also developed and patented electronics that allow parallel recording of signals from several nanopores and collecting and analyzing data in real time. The simplest version of an instrument based on this technology is MinION Oxford Nanopore Technologies, which makes it possible to read entire DNA sequence corresponding to tens of kilobases and limited only by the intrinsic length of the test molecules [190]. Oxford Nanopore is looking for new nanopore solutions with properties that can improve instrument performance. Protein nanopores are sufficiently strong, easily reproducible, inexpensive, and easily modifiable. However, it is likely that the future generations of nanopore-based sensor devices will use nanopores made from synthetic materials. At present, solid-state nanopores do not have the chemical specificity of protein.
Thus, the electrochemistry of biopolymers is based on the measurement of the reduction or oxidation signals of redox-active centers of proteins or/and the oxidation of their amino acid residues and the oxidation or reduction of nitrogen bases in DNA or RNA (Fig. 3). It was found that almost all proteinogenic amino acids are prone to specific electrochemical oxidation (at a potential of 0.5 to 1.5 V); at the same time, all nucleotides that make up DNA and RNA are capable of both being irreversibly oxidized (at a potential of 1 to 2 V) and reduced (at a potential of –1 to –2 V) due to nitrogen base residues on the electrodes from carbon materials (graphite and glassy carbon). Electrochemical reactions of free amino acids and nucleic bases and their residues in proteins and nucleic acids should be systematically studied in order to identify reaction products and establish mechanisms. In addition, the electrochemistry of biopolymers currently requires the characterization of series of protein and nucleic acid molecules with known spatial structures, monomer sequences, and molecular weights in order to identify general structure–property relationships. The use of preparations without additional biochemical characterization and, if necessary, additional purification in electrochemical studies makes it difficult to interpret and compare the experimental results of different scientific groups. It is necessary to consider the electrochemical behavior of proteins and nucleic acids from the point of view of their spatial structure. The modern level of technology allows one to model the spatial structure of a single biomolecule and to detect ultralow currents with a high signal-to-noise ratio. With the use of electrochemical approaches, the nucleotide sequence of DNA molecules can be currently determined, and the determination of the primary structure of proteins is not far off.
* * *
In conclusion, we compare the two considered areas of biopolymer electrochemistry: proteins and nucleic acids. The first studies on the electroactivity of nucleic acids appeared about 30 years later than the pioneering works in the polarography of proteins; by now, they experience rapid growth and are catching up and even ahead of them. Interestingly, the same scientists, Palecek, Brabec, Fojta, Wang, Oliveira-Brett, and others, made a significant contribution to the development of these two parallel areas—the electrochemistry of DNA and proteins. Those who started with the study of the electrochemistry of DNA then moved on to proteins and vice versa. One can only admire the fortitude of Palecek, who remained faithful to the mercury electrode until the end of his days in studying the behavior of various classes of biopolymers. The end of the 1970s and the beginning of the 1980s, when Brabec published the results of his studies on the electrooxidation of DNA molecules and proteins on electrodes made of carbon materials, can be considered the time of the foundation of the electrochemical analysis of biopolymers on solid electrodes. This review summarizes data on the presently known intrinsic electrochemical properties of biopolymers that manifest themselves on unmodified electrodes. Knowing the fundamental properties, researchers, depending on the specific analytical problem, can always choose a system where these properties manifest themselves in a desired way. The development of the electrochemistry of proteins and nucleic acids expands the scope of electroanalysis in general—not only for quantitative determination but also for qualitative analysis of changes in the structure of molecules. By in vitro modeling complex biological systems and processes, scientists can explore the influence of various external and internal factors, such as pH or mutations. Thus, electrochemistry provides special information on about the properties of biomolecules, which, in combination with other physicochemical methods, allows one to look deeper into the molecular mechanisms of processes in living organisms.
REFERENCES
Kudryavtsev, P.S., Kurs istorii fiziki (History of Physics), Moscow: Prosveshchenie, 1982.
Khimicheskaya entsiklopediya (Chemical Encyclopedia), Knunyants, I.L., Ed., Moscow: Sov. Enthiklopediya, 1988.
Berezovchuk, A.V., Fizicheskaya khimiya: konspekt lektsii (Physical Chemistry: Lecture Notes), Moscow: Eksmo, 2015.
Chizmadzhev, Yu.A., Sorosov. Obrazovat. Zh., 2000, vol. 6, no. 3, p. 23.
Clark, L.C. and Lyons, C., Ann. N. Y. Acad. Sci., 1962, vol. 102, no. 1, p. 29.
Theavenot, D.R., Toth, K., Durst, R.A., and Wilson, G.S., Pure Appl. Chem., 1999, vol. 71, no. 12, p. 2333.
Turner, A.P.F., Chem. Soc. Rev., 2013, vol. 42, no. 8, p. 3184.
Labuda, J., Bowater, R.P., Fojta, M., Gauglitz, G., Glatz, Z., Hapala, I., Havlis, J., Kilar, F., Kilar, A., Malinovska, L., Siren, H.M.M., Skladal, P., Torta, F., Valachovic, M., Wimmerova, M., Zdrahal, Z., and Hibbert, D.B., Pure Appl. Chem., 2018, vol. 90, no. 7, p. 1121.
Lubert, K.H. and Kalcher, K., Electroanalysis, 2010, vol. 22, nos. 17–18, p. 1937.
Shikata, M., Trans. Faraday Soc., 1925, vol. 21, no. 8, p. 42.
Kitaev, Yu.P., Troepol’skaya, T.V., and Budnikov, H.C., Promezhutochnye produkty v elektrokhimicheskikh reaktsiyakh (Intermediates in Electrochemical Reactions), Moscow: Nauka, 1982.
Ziyatdinova, G.K. and Budnikov, H.C., in Farmatsevticheskii analiz (Pharmaceutical Analysis), vol. 16 of Problemy analiticheskoi khimii (Problems of Analytical Chemistry), Budnikov, H.C. and Garmonov, S.Yu., Eds., Moscow: Argamak-Media, 2013, p. 230.
Zavolskova, M.D., Nikitina, V.N., Maksimova, E.D., Karyakina, E.E., and Karyakin, A.A., Anal. Chem., 2019, vol. 91, no. 12, p. 7495.
Komkova, M.A., Zarochintsev, A.A., Karyakina, E.E., and Karyakin, A.A., J. Electroanal. Chem., 2020, vol. 872, 114048.
Suprun, E.V., Doctoral (Biol.) Dissertation, Moscow: Res. Inst. Biomed. Chem., 2017.
Heyrovsky, J. and Babicka, J., Collect. Czech. Chem. Commun., 1930, vol. 2, p. 370.
Herles, F. and Vancura, A., Bull. Int. Acad. Sci. Boheme, 1932, vol. 33, p. 119.
Bobrowski, A. and Zarebski, J., Electroanalysis, 2000, vol. 12, no. 15, p. 1177.
Tomschik, M., Havran, L., Fojta, M., and Paleček, E., Electroanalysis, 1998, vol. 10, no. 6, p. 403.
Paleček, E., Ostatna, V., Masarik, M., Bertoncini, C.W., and Jovin, T.M., Analyst, 2008, vol. 133, no. 1, p. 76.
Zivanovic, M., Aleksic, M., Ostatna, V., Doneux, T., and Paleček, E., Electroanalysis, 2010, vol. 22, nos. 17–18, p. 2064.
Paleček, E. and Ostatna, V., Electroanalysis, 2007, vol. 19, no. 23, p. 2383.
Brdicka, R., Collect. Czech. Chem. Commun., 1933, vol. 5, p. 112.
Brdicka, R., Collect. Czech. Chem. Commun., 1933, vol. 5, p. 148.
Brdicka, R., Nature, 1937, vol. 139, no. 3528, p. 1020.
Walker, I.C. and Reimann, S.P., Am. J. Cancer, 1939, vol. 37, no. 4, p. 585.
Millar, G.J., Biochem. J., 1953, vol. 53, no. 3, p. 385.
Millar, G.J., Biochem. J., 1953, vol. 53, no. 3, p. 393.
Kalous, V. and Pavlicek, Z., Biochim. Biophys. Acta, 1962, vol. 57, no. 1, p. 44.
Kizek, R., Trnkova, L., and Paleček, E., Anal. Chem., 2001, vol. 73, no. 20, p. 4801.
Raspor, B., Paic, M., and Erk, M., Talanta, 2001, vol. 55, no. 1, p. 109.
Fojta, M., Fojtova, M., Havran, L., Pivonkova, H., Dorcak, V., and Sestakova, I., Anal. Chim. Acta, 2006, vol. 558, nos. 1–2, p. 171.
Cecil, R. and Weitzman, P.D.J., Biochem. J., 1964, vol. 93, no. 1, p. 1.
Zikan, J. and Kalous, V., Collect. Czech. Chem. Commun., 1967, vol. 32, no. 1, p. 246.
Kolthoff, I.M., Stricks, W., and Tanaka, N., J. Am. Chem. Soc., 1955, vol. 77, no. 18, p. 4739.
Stricks, W. and Kolthoff, I.M., J. Am. Chem. Soc., 1952, vol. 74, no. 18, p. 4646.
Stankovich, M.T. and Bard, A.J., J. Electroanal. Chem., 1977, vol. 75, no. 2, p. 487.
Ralph, T.R., J. Electroanal. Chem., 1994, vol. 375, no. 1, p. 1.
Betso, S.R., Klapper, M.H., and Anderson, L.B., J. Am. Chem. Soc., 1972, vol. 94, no. 23, p. 8197.
Scheller, F., Janchen, M., Lampe, J., Prumke, H.J., Blanck, J., and Paleček, E., Biochim. Biophys. Acta, 1975, vol. 412, no. 1, p. 157.
Scheller, F., Bioelectrochem. Bioenerg., 1977, vol. 4, no. 4, p. 490.
Ludwig, R., in Encyclopedia of Applied Electrochemistry, Kreysa, G., Ota, K., and Savinell, R.F., Eds., New York: Springer, 2014, p. 330.
Yeh, P. and Kuwana, T., Chem. Lett., 1977, vol. 6, no. 10, p. 1145.
Eddowes, M.J. and Hill, H.A.O., J. Chem. Soc., Chem. Commun., 1977, no. 21, p. 771.
Eddowes, M.J., Hill, H.A.O., and Uosaki, K., J. Electroanal. Chem., 1980, vol. 116, p. 527.
Rusling, J.F., Microporous Mater., 1994, vol. 3, nos. 1–2, p. 1.
Karyakin, A.A., Bioelectrochemistry, 2012, vol. 88, p. 70.
Berezin, I.V., Bogdanovskaya, V.A., Varfolomeev, S.D., Tarasevich, M.R., and Yaropolov, A.I., Dokl. Akad. Nauk SSSR, 1978, vol. 240, p. 615.
Yaropolov, A.I., Malovik, V., Varfolomeev, S.D., and Berezin, I.V., Dokl. Akad. Nauk SSSR, 1979, vol. 249, no. 6, p. 1399.
Yaropolov, A.I., Karyakin, A.A., Varfolomeyev, S.D., and Berezin, I.V., Bioelectrochem. Bioenerg., 1984, vol. 12, nos. 3–4, p. 267.
Bioelectrochemistry: Fundamentals, Experimental Techniques, and Applications, Bartlett, P.N., Ed., Chichester: Wiley, 2008.
Koryta, J. and Pradac, J., J. Electroanal. Chem., 1968, vol. 17, nos. 1–2, p. 167.
Koryta, J. and Pradac, J., J. Electroanal. Chem., 1968, vol. 17, nos. 1–2, p. 177.
Koryta, J. and Pradac, J., J. Electroanal. Chem., 1968, vol. 17, nos. 1–2, p. 185.
Brabec, V. and Mornstein, V., Biochim. Biophys. Acta, Protein Struct., 1980, vol. 625, no. 1, p. 43.
Reynaud, J.A., Malfoy, B., and Bere, A., J. Electroanal. Chem. Interfacial Electrochem., 1980, vol. 116, p. 595.
Paleček, E., Talanta, 2002, vol. 56, no. 5, p. 809.
Paleček, E., Fojta, M., Jelen, F., and Vetterl, V., in The Encyclopedia of Electrochemistry, Bard, A.J. and Stratmann, M., Eds., Weinhei: Wiley, 2002, vol. 9, ch. 12, p. 365.
Paleček, E., Naturwissenschaften, 1958, vol. 45, no. 8, p. 186.
Paleček, E., Nature, 1960, vol. 188, p. 656.
Paleček, E., Collect. Czech. Chem. Commun., 1960, vol. 25, no. 9, p. 2283.
Bendich, A., in The Nucleic Acids, Chargaff, E. and Davidson, J.N., Eds., New York: Academic, 1955, vol. 1, p. 119.
Berg, H., Biochem. Z., 1957, vol. 329, no. 3, p. 274.
Miller, I.R., J. Mol. Biol., 1961, vol. 3, no. 3, p. 229.
Miller, I.R., J. Mol. Biol., 1961, vol. 3, p. 357.
Marmur, J. and Lane, D., Proc. Natl. Acad. Sci. U. S.A., 1960, vol. 46, no. 4, p. 453.
Marmur, J., Rownd, R., and Schildkraut, C.L., in Progress in Nucleic Acid Research, Davidson, J.N. and Cohn, W.E., Eds., London: Academic, 1963, vol. 1, p. 231.
Marmur, J., J. Mol. Biol., 1961, vol. 3, no. 2, p. 208.
Cummings, T.E. and Elving, P.J., J. Electroanal. Chem., 1979, vol. 102, p. 237.
Cummings, T.E. and Elving, P.J., J. Electroanal. Chem., 1978, vol. 94, p. 123.
Paleček, E., Anal. Biochem., 1980, vol. 108, no. 1, p. 129.
Paleček, E., Jelen, F., Hung, M.A., and Lasovsky, J., Bioelectrochem. Bioenerg., 1981, vol. 8, no. 6, p. 621.
Paleček, E., Anal. Chim. Acta, 1985, vol. 174, p. 103.
Paleček, E., Anal. Biochem., 1988, vol. 170, no. 2, p. 421.
Teijeiro, C., Nejedly, K., and Paleček, E., J. Biomol. Struct. Dyn., 1993, vol. 11, no. 2, p. 313.
Paleček, E., Tomschik, M., Stankova, V., and Havran, L., Electroanalysis, 1997, vol. 9, no. 13, p. 990.
Brabec, V. and Paleček, E., J. Electroanal. Chem., 1970, vol. 27, no. 1, p. 145.
Brabec, V., Biophys. Chem., 1980, vol. 11, no. 1, p. 1.
Brabec, V. and Dryhurst, G., J. Electroanal. Chem., 1978, vol. 89, no. 1, p. 161.
Brabec, V. and Dryhurst, G., Stud. Biophys., 1978, vol. 67, p. 23.
Brabec, V. and Dryhurst, G., J. Electroanal. Chem., 1978, vol. 91, no. 2, p. 219.
Brabec, V., J. Electroanal. Chem., 1980, vol. 116, p. 69.
Brabec, V., J. Electroanal. Chem., 1981, vol. 128, p. 437.
Smith, D.L. and Elving, P.J., Anal. Chem., 1962, vol. 34, no. 8, p. 930.
Elving, P.J. and Smith, D.L., Anal. Chem., 1960, vol. 32, no. 13, p. 1849.
Dryhurst, G. and Elving, P.J., J. Electrochem. Soc., 1968, vol. 115, no. 10, p. 1014.
Dryhurst, G. and Pace, G.F., J. Electrochem. Soc., 1970, vol. 117, no. 10, p. 1259.
Dryhurst, G., Anal. Chim. Acta, 1971, vol. 57, no. 1, p. 137.
Dryhurst, G., Talanta, 1972, vol. 19, no. 6, p. 769.
Watson, J.D. and Crick, F.H.C., Nature, 1953, vol. 171, p. 737.
Franklin, R.E. and Gosling, R.G., Nature, 1953, vol. 17, p. 156.
Wilson, W.D., in Nucleic Acids in Chemistry and Biology, Blackburn, G.M. and Gait, M.J., Eds., New York: Oxford Univ. Press, 1996.
Zamenhof, S., Brawerman, G., and Chargaff, E., Biochim. Biophys. Acta, 1952, vol. 9, p. 402.
Suprun, E.V., Electrochem. Commun., 2021, vol. 125, 106983.
Suprun, E.V., Zharkova, M.S., Morozevich, G.E., Veselovsky, A.V., Shumyantseva, V.V., and Archakov, A.I., Electroanalysis, 2013, vol. 25, p. 2109.
Brabec, V. and Koudelka, J., J. Electroanal. Chem. Interfacial Electrochem., 1980, vol. 116, p. 793.
Chiorcea-Paquim, A.M. and Oliveira-Brett, A.M., Electrochim. Acta, 2014, vol. 126, p. 162.
Veloso, A. and Kerman, K., Anal. Bioanal. Chem., 2013, vol. 405, p. 5725.
Fojta, M., Daňhel, A., Havran, L., and Vyskočil, V., TrAC, Trends Anal. Chem., 2016, vol. 79, p. 160.
Suprun, E.V., TrAC, Trends Anal. Chem., 2019, vol. 116, p. 44.
Bartosik, M. and Jirakova, L., Curr. Opin. Electrochem., 2019, vol. 14, p. 96.
Lin, Y.W., Arch. Biochem. Biophys., 2018, vol. 641, p. 1.
Soskic, V., Groebe, K., and Schrattenholz, A., Exp. Gerontol., 2008, vol. 43, p. 247.
Pacher, P., Beckman, J.S., and Liaudet, L., Physiol. Rev., 2007, vol. 87, p. 315.
Humphrey, S.J., James, D.E., and Mann, M., Trends Endocrinol. Metab., 2015, vol. 26, p. 676.
Scheller, F.W., Wollenberger, U., Lei, C., Jin, W., Ge, B., Lehmann, C., Lisdat, F., and Fridman, V., Rev. Mol. Biotechnol., 2002, vol. 82, p. 411.
Ferapontova, E.E., Shleev, S., Ruzgas, T., Stoica, L., Christenson, A., Tkac, J., Yaropolov, A.I., and Gorton, L., Perspect. Bioanal., 2005, vol. 1, p. 517.
Scheller, F.W., Bistolas, N., Liu, S., Janchen, M., Katterle, M., and Wollenberger, U., Adv. Colloid Interface Sci., 2005, vol. 116, p. 111.
Paleček, E., Tkáč, J., Bartosik, M., Bertók, T., Ostatná, V., and Paleček, J., Chem. Rev., 2015, vol. 115, p. 2045.
Suprun, E.V., Shumyantseva, V.V., and Archakov, A.I., Electrochim. Acta, 2014, vol. 140, p. 72.
Vacek, J., Zatloukalova, M., and Novak, D., Curr. Opin. Electrochem., 2018, vol. 12, p. 73.
Hasoň, S., Daňhel, A., Schwarzová–Pecková, K., and Fojta, M., in Nanotechnology and Biosensors, Nikolelis, D.P. and Nikoleli, G.P., Eds., Amsterdam: Elsevier, 2018, p. 51.
Baluchová, S., Daňhel, A., Dejmková, H., Ostatná, V., Fojta, M., and Schwarzová-Pecková, K., Anal. Chim. Acta, 2019, vol. 1077, p. 30.
Suprun, E.V., Karpova, E.V., Radko, S.P., and Karyakin, A.A., Electrochim. Acta, 2020, vol. 331.
Özcan, A. and Şahin, Y., Biosens. Bioelectron., 2012, vol. 31, p. 26.
Vestergaard, M., Kerman, K., Saito, M., Nagatani, N., Takamura, Y., and Tamiya, E., J. Am. Chem. Soc., 2005, vol. 127, p. 11892.
Somji, M., Dounin, V., Muench, S.B., Schulze, H., Bachmann, T.T., and Kerman, K., Bioelectrochemistry, 2012, vol. 88, p. 110.
Enache, T.A. and Oliveira-Brett, A.M., Bioelectrochemistry, 2013, vol. 89, p. 11.
Topal, B.D., Özkan, S.A., and Uslu, B., J. Electroanal. Chem., 2014, vol. 719, p. 14.
Chiku, M., Nakamura, J., Fujishima, A., and Einaga, Y., Anal. Chem., 2008, vol. 80, p. 5783.
Dorcak, V., Ostatna, V., and Paleček, E., Electrochem. Commun., 2013, vol. 31, p. 80.
Suprun, E.V., Khmeleva, S.A., Radko, S.P., Kozin, S.A., Archakov, A.I., and Shumyantseva, V.V., Electrochem. Commun., 2016, vol. 65, p. 53.
Suprun, E.V., Radko, S.P., Khmeleva, S.A., Mitkevich, V.A., Archakov, A.I., Makarov, A.A., and Shumyantseva, V.V., Electrochem. Commun., 2017, vol. 75, p. 33.
Enache, T.A. and Oliveira-Brett, A.M., Bioelectrochemistry, 2017, vol. 114, p. 13.
Nguyen, N.T., Wrona, M.Z., and Dryhurst, G., J. Electroanal. Chem. Interfacial Electrochem., 1986, vol. 199, p. 101.
Enache, T.A. and Oliveira-Brett, A.M., J. Electroanal. Chem., 2011, vol. 655, p. 9.
Enache, T.A. and Oliveira-Brett, A.M., Electroanalysis, 2011, vol. 23, p. 1337.
Sek, S., Vacek, J., and Dorcak, V., Curr. Opin. Electrochem., 2019, vol. 14, p. 166.
Ostatna, V., Cernocka, H., Kurzatkowska, K., and Paleček, E., Anal. Chim. Acta, 2012, vol. 735 P, p. 31.
Suprun, E.V., Khmeleva, S.A., Kiseleva, Y.Y., Radko, S.P., Archakov, A.I., and Shumyantseva, V.V., Electroanalysis, 2016, vol. 28, p. 1977.
Suprun, E.V., Radko, S.P., Andreev, E.A., Khmeleva, S.A., Kozin, S.A., Makarov, A.A., Archakov, A.I., and Shumyantseva, V.V., J. Electroanal. Chem., 2017, vol. 791, p. 152.
Suprun, E.V., Radko, S.P., Farafonova, T.E., Mitkevich, V.A., Makarov, A.A., Archakov, A.I., and Shumyantseva, V.V., Electroanalysis, 2017, vol. 29, p. 2906.
Suprun, E.V., Radko, S.P., Kozin, S.A., Mitkevich, V.A., and Makarov, A.A., J. Electroanal. Chem., 2018, vol. 830-831, p. 34.
Suprun, E.V., Radko, S.P., Kozin, S.A., Mitkevich, V.A., and Makarov, A.A., J. Electroanal. Chem., 2020, vol. 861, 113938.
Suprun, E.V., Khmeleva, S.A., Radko, S.P., Archakov, A.I., and Shumyantseva, V.V., Electrochim. Acta, 2016, vol. 187, p. 677.
Chikae, M., Fukuda, T., Kerman, K., Idegami, K., Miura, Y., and Tamiya, E., Bioelectrochemistry, 2008, vol. 74, p. 118.
Ferapontova, E.E., Grigorenko, V.G., Egorov, A.M., Borchers, T., Ruzgas, T., and Gorton, L., J. Electroanal. Chem., 2001, vol. 509, p. 19.
Ashe, D., Alleyne, T., and Iwuoha, E., Biotechnol. A-ppl. Biochem., 2007, vol. 46, p. 185.
Suprun, E.V., Shilovskaya, A.L., Lisitsa, A.V., Bulko, T.V., Shumyantseva, V.V., and Archakov, A.I., Electroanalysis, 2011, vol. 23, p. 1051.
Pandiaraj, M., Sethy, N.K., Bhargava, K., Kameswararao, V., and Karunakaran, C., Biosens. Bioelectron., 2014, vol. 54, p. 115.
Shumyantseva, V.V., Kuzikov, A.V., Masamrekh, R.A., Bulko, T.V., and Archakov, A.I., Biosens. Bioelectron., 2018, vol. 121, p. 192.
Gilardi, G., Fantuzzi, A., and Sadeghi, S.J., Curr. Opin. Struct. Biol., 2001, vol. 11, p. 491.
Panicco, P., Castrignano, S., Sadeghi, S.J., Di Nardo, G., and Gilardi, G., Bioelectrochemistry, 2021, vol. 138, 107729.
Olloqui-Sariego, J.L., Zakharova, G.S., Polozni-kov, A.A., Calvente, J.J., Hushpulian, D.M., Gorton, L., and Andreu, R., Electrochim. Acta, 2020, vol. 351, 136465.
Wu, H., Wang, X., Qiao, M., Zhang, H., Jin, X., and Fan, S., Sens. Actuators, B, 2015, vol. 221, p. 694.
Lancellotti, L., Borsari, M., Bellei, M., Bonifacio, A., Bortolotti, C.A., di Rocco, G., Ranieri, A., Sola, M., and Battistuzzi, G., Electrochim. Acta, 2020, vol. 363, 137237.
Gomez-Mingot, M., Alcaraz, L.A., Heptinstall, J., Donaire, A., Piccioli, M., Montiel, V., and Iniesta, J., Arch. Biochem. Biophys., 2013, vol. 529, p. 26.
Díaz-Moreno, I., García-Heredia, J.M., Díaz-Quintana, A., Teixeira, M., and Miguel, A., Biochim. Biophys. Acta, 2011, vol. 1807, p. 1616.
Roberts, E.S., Lin, H.L., Crowley, J.R., Vuletich, J.L., Osawa, Y., and Hollenberg, P.F., Chem. Res. Toxicol., 1998, vol. 11, p. 1067.
Walsh, C.T., Garneau-Tsodikova, S., and Gatto, G.J., Jr., Angew. Chem., Int. Ed. Engl., 2005, vol. 44, p. 7342.
Suprun, E.V., Radko, S.P., Farafonova, T.E., Kozin, S.A., Makarov, A.A., Archakov, A.I., and Shumyantseva, V.V., Electrochim. Acta, 2017, vol. 258, p. 1182.
Fernandes, I.P. and Oliveira-Brett, A.M., Bioelectrochemistry, 2020, vol. 133, 107451.
Suprun, E.V., Zharkova, M.S., Veselovsky, A.V., Archakov, A.I., and Shumyantseva, V.V., Russ. J. Electrochem., 2017, vol. 53, p. 97.
Brabec, V., Biopolymers, 1979, vol. 18, p. 2397.
Brabec, V., Biophys. Chem., 1979, vol. 9, p. 289.
de-los-Santos-Álvarez, P., Lobo-Castañón, M.J., Miranda-Ordieres, A.J., and Tunon-Blanco, P., Electroanalysis, 2004, vol. 16, p. 1193.
Oliveira-Brett, A.M., in Electrochemical DNA Assays, Bioelectrochemistry: Fundamentals, Experimental Techniques, and Applications, Bartlett, P.N., Ed., Chichester: Wiley, 2008, p. 411.
Ferapontova, E.E., Annu. Rev. Anal. Chem., 2018, vol. 11, p. 197.
Špaček, J., Daňhel, A., Hasoň, S., and Fojta, M., Electrochem. Commun., 2017, vol. 82, p. 34.
Pontinha, A.D., Chiorcea-Paquim, A.M., Eritja, R., and Oliveira-Brett, A.M., Anal. Chem., 2014, vol. 86, p. 5851.
Vidláková, P., Pivoňková, H., Kejnovská, I., Trnko-vá, L., Vorlíčková, M., Fojta, M., and Havran, L., Anal. Bioanal. Chem., 2015, vol. 407, p. 5817.
Špaček, J., Fojta, M., and Wang, J., Electroanalysis, 2019, vol. 3, p. 2057.
Oliveira-Brett, A.M., Piedade, J.A.P., Silva, L.D., and Diculescu, V.C., Anal. Biochem., 2004, vol. 332, p. 321.
Goncalves, L.M., Batchelor-McAuley, C., Barros, A.A., and Compton, R.G., J. Phys. Chem. C, 2010, vol. 114, p. 14213.
Li, Q., Batchelor-McAuley, C., and Compton, R.G., J. Phys. Chem. B, 2010, vol. 114, p. 7423.
Oliveira-Brett, A.M. and Matysik, F.M., J. Electroanal. Chem., 1997, vol. 429, p. 95.
Ferapontova, E.E., Electrochim. Acta, 2004, vol. 49, p. 1751.
Goyal, R.N., Singh, U.P., and Abdullah, A.A., Indian J. Chem., 2003, vol. 42A, p. 42.
Ferapontova, E.E. and Dominguez, E., Electroanalysis, 2003, vol. 15, p. 629.
Brett, C.M., Oliveira-Brett, A.M., and Serrano, S.H., J. Electroanal. Chem., 1994, vol. 366, p. 225.
Wu, L., Zhou, J., Luo, J., and Lin, Z., Electrochim. Acta, 2000, vol. 45, p. 2923.
Yao, T., Wasa, T., and Musha, S., Bull. Chem. Soc. Jpn., 1978, vol. 51, p. 1235.
Brotons, A., Mas, L.A., Metters, J.P., Banks, C.E., and Iniesta, J., Analyst, 2013, vol. 138, p. 5239.
Stempkowska, I., Ligaj, M., Jasnowska, J., Langer, J., and Filipiak, M., Bioelectrochemistry, 2007, vol. 70, p. 488.
Suprun, E.V., Kutdusova, G.R., Khmeleva, S.A., and Radko, S.P., Electrochem. Commun., 2021, vol. 124, 106947.
Shumyantseva, V.V., Bulko, T.V., Tikhonova, E.G., Sanzhakov, M.A., Kuzikov, A.V., Masamrekh, R.A., Pergushov, D.V., Schacher, F.H., and Sigolaeva, L.V., Bioelectrochemistry, 2021, vol. 140, 107736.
Wang, J., Chen, L., and Chicharro, M., Anal. Chim. Acta, 1996, vol. 319, p. 347.
Diculescu, V.C., Chiorcea-Paquim, A.M., and Oliveira-Brett, A.M., TrAC, Trends Anal. Chem., 2016, vol. 79, p. 23.
Ferapontova, E.E. and Shipovskov, S.V., Biochemistry (Moscow), 2003, vol. 68, p. 99.
Yang, Z.S., Wang, Y.L., and Zhang, Y.Z., Electrochem. Commun., 2004, vol. 6, p. 158.
Honeychurch, K.C., O’Donovan, M.R., and Hart, J.P., Biosens. Bioelectron., 2007, vol. 22, p. 2057.
Ferapontova, E.E., Curr. Opin. Electrochem., 2017, vol. 5, p. 218.
Hocek, M. and Fojta, M., Chem. Soc. Rev., 2011, vol. 40, p. 5802.
Fojta, M., in Biosensors for Security and Bioterrorism Applications, Nikolelis, D.P., and Nikoleli, G.-P., Eds., New York: Springer, 2016, p. 309.
Oberacher, H., Erb, R., Plattner, S., and Chervet, J.P., TrAC, Trends Anal. Chem., 2015, vol. 70, p. 100.
Zhao, Y., Chen, F., Li, Q., Wang, L., and Fan, C., Chem. Rev., 2015, vol. 115, p. 12491.
Long, Y.-T., Small Methods, 2020, vol. 4, 2000695.
Lin, Y., Ying, Y.-L., and Long, Y.-T., Nanopore confinement for electrochemical sensing at the single-molecule level, Curr. Opin. Electrochem., 2018, vol. 7, p. 172.
Li, S., Wu, X.Y., Li, M.Y., Liu, S.C., Ying, Y.L., and Long, Y.T., Small Methods, 2020, vol. 4, no. 11, 2000014.
Laver, T., Harrison, J., O’Neill, P.A., Moore, K., Farbos, A., Paszkiewicz, K., and Studholme, D.J., Biomol. Detect. Quantif., 2015, vol. 3, p. 1.
ACKNOWLEDGMENTS
This work was carried out within the framework of the Program of Fundamental Scientific Research in the Russian Federation for a long-term period (2021–2030).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of interest.
Additional information
Dedicated to the 100th anniversary of the discovery of polarography by J. Heyrovsky
Translated by V. Makhlyarchuk
Rights and permissions
About this article

Cite this article
Suprun, E.V., Budnikov, H.C. Bioelectrochemistry as a Field of Analysis: Historical Aspects and Current Status. J Anal Chem 77, 643–663 (2022). https://doi.org/10.1134/S1061934822060168
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1061934822060168