
Abstract
The last couple of decades have witnessed the emergence of the wide and diverse field of bio-electrochemistry which nowadays provides enough research to fill several international meetings per year. As part of this research, topics such as the electrochemical analysis of biological samples, including electrochemical biosensors, and the characterization of redox properties of proteins and enzymes first come to mind. Indeed, these areas of biological electrochemistry have blossomed ever since the first pioneering studies on electrochemical biosensors in the 1980s [1]. The field has moved on considerably since then, of course, and various aspects of modern biological electrochemistry have recently formed part of a special issue of ChemPhysChem [2].
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
7.1 Introduction
The last couple of decades have witnessed the emergence of the wide and diverse field of bio-electrochemistry which nowadays provides enough research to fill several international meetings per year. As part of this research, topics such as the electrochemical analysis of biological samples, including electrochemical biosensors, and the characterization of redox properties of proteins and enzymes first come to mind. Indeed, these areas of biological electrochemistry have blossomed ever since the first pioneering studies on electrochemical biosensors in the 1980s [1]. The field has moved on considerably since then, of course, and various aspects of modern biological electrochemistry have recently formed part of a special issue of ChemPhysChem [2].
Today, bio-electrochemistry is moving into various directions, as illustrated in Fig. 7.1, including the development of electrochemical biosensors which do no longer require the “help” of enzymes to detect specific molecules in complex solutions [3]. Related bioanalytical systems have also been developed which enable the electrochemical detection of specific analytes in (partially purified) biological samples, including N-acetylcysteine, cysteine, and disulfides in urine (see also Sect. 5) [4, 5]. The development of such devices has recently been the subject of several expert reviews [6, 7]. Sensors also form the topic of some of the other chapters of this book.

The figure illustrates the widespread applications of traditional and modern electrochemical methods in key areas of biochemical and biological research. Some of these applications are discussed as part of this chapter. Please note that the figure as well as the text can only provide a—necessarily incomplete—selection of applications and do not reflect the full breadth of modern-day bio-electrochemistry
Besides biosensors, bio-electrochemistry also has a long tradition among bioinorganic chemists involved in the study of (redox-active) metalloproteins and enzymes [8]. Here, direct electrochemistry has been at the forefront of developments, often in conjunction with “intelligent” electrodes, such as (modified) film electrodes [9]. While most of these investigations have focused on metal-based biomolecules, more recently, the electrochemistry of non-metal, cysteine-containing proteins and enzymes has also been considered, in part for the characterization of these proteins, but also in part for rapid electrochemical detection [10–13].
Nonetheless, there are also some lesser known and often still emerging fields of bio-electrochemistry which catch the interest of researchers active in the field of biological chemistry or biochemistry. As part of this chapter, we will therefore consider a selection of electrochemical methods, which have already proven to be rather useful as part of biological chemical research, yet often still require further development to become widely accessible and applicable. Here, we will take the perspective of a biological redox chemist, with her/his own specific research interests, topics, and also problems.
Before we start, however, we first need to consider why electrochemical methods are particularly interesting and at the same time also well suited for the analysis of biological materials, from unusual natural products to complex biological systems, such as single cells. This question is not entirely unreasonable, since at first sight, electrochemistry, apart from a few basic applications, is hardly an integral part of Biology, Pharmacy, or Medicine. Nonetheless, we need to realize that electrochemistry is rather diverse and provides a whole arsenal of different methods and techniques which in one aspect or another may be very useful for the rapid and reliable analysis of biological materials. Such electrochemical methods are particularly valuable in bioanalysis because they are often readily available, widely applicable, extraordinarily informative and sensitive, yet at the same time also highly reliable and reproducible, and hence often considerably more robust when compared to alternative approaches, such as spectroscopy. Importantly, electrochemical methods, such as (Cyclic) Voltammetry (CV) or differential pulse polarography (DPP), are also highly selective and therefore enable the analysis of specific substances and processes in complex biological mixtures.
Within this context, the electrochemical glucose sensor may come to mind: This device is more or less selective for glucose, can be operated in whole blood, and is able to reliably and reproducibly sense even small changes in blood glucose concentrations despite the presence of a wealth of other blood components. Importantly, such a sensor is simple, small, and also rather cost-effective when compared to possible alternatives [14–16].
In the following sections of this chapter, we will focus on four rather interesting and powerful applications of electrochemistry in Biology and drug development: (1) electrochemical methods to monitor the proliferation of cells in real time and without the need for aliquots and staining; (2) electrochemical techniques to analyze cellular processes, such as metabolism, intra- and extracellular levels of reactive oxygen species (ROS), reactive nitrogen species (RNS), and oxidative stress (OS) at the single-cell level; (3) electrochemistry of sulfur and selenium proteins, which are at the center of important cellular regulatory and signaling processes (such as the cellular “thiolstat”); and last but not the least (4) electrochemistry of chalcogen-containing natural products and synthetic substances, with the aim to characterize such compounds and to derive at certain “chemical structure-electrochemical potential-biological activity relationships”.
Together, these four applications will highlight the considerable practical potential of electrochemical methods in biochemical and pharmaceutical research, especially in the context of research problems which are difficult to address or to resolve by other, more conventional methods. Nonetheless, these examples represent just a small section of a wider, thriving, and continuously growing area of bio-electrochemical research and applications.
7.2 The Chalcogen-Specific Electrochemical Tool Kit
Several of the techniques considered as part of this chapter are linked to the redox chemistry of chalcogens, i.e., oxygen, sulfur, selenium, and tellurium. Since the methods available for the analysis of such redox systems are often not widely known or straightforward, we will briefly consider them as part of the emerging electrochemical “toolkit” which can be employed for the detection and characterization/analysis of these systems in vitro and possibly also in vivo.
In stark contrast to the well-established electrochemistry of metal-based redox systems, which include various metalloproteins and enzymes, the analysis of chalcogen-based redox systems is still far from trivial and sometimes even controversial [17–19]. This is rather disappointing, as there is a strong interest among biological chemists to identify and subsequently quantify ROS and RNS inside and outside living cells [20]. Such ROS include, for instance, the superoxide radical anion (O2 •−), hydrogen peroxide (H2O2), and the hydroxyl radical (•OH); nitric oxide (•NO) and peroxynitrite (ONOO−) are among the most prominent RNS [21]. Table 7.1 provides a brief overview of the most common ROS and RNS, some of which will be discussed later on in the context of electrochemical detection. From a biochemical point of view, these reactive species are rather important as they play a pivotal role in cell signaling as well as host defense against bacteria and microbes. At the same time, the intracellular concentrations of various ROS and RNS increase during OS and may subsequently cause cell death via apoptosis. Modulating levels of ROS and RNS can therefore also serve a therapeutic purpose, for instance in the context of treating cancer. This has led to the emerging strategy of redox modulation which may also benefit from the input of electrochemistry and will be discussed in Sect. 7 [22].
Not surprisingly, methods enabling the determination of ROS and RNS concentrations in cells have received considerable attention. These detection techniques also include electrochemical methods. ROS, such as H2O2, peroxynitrite (ONOO−), nitric oxide •NO, and nitrite NO2 −, can be detected fairly easily by a coulometric method using a platinized carbon-based electrode [20, 24].
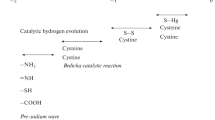
While most ROS and •NO therefore can be detected, characterized, and quantified by fairly standard and straightforward electrochemical methods, the electrochemistry of their cellular targets/reaction partners, e.g., sulfur- or selenium-based redox systems, is considerably more complex [17]. This certainly applies to most sulfur- (i.e., cysteine- and methionine-)-based proteins and enzymes, whose redox properties have only sporadically been analyzed by electrochemical methods (see Sect. 5). Yet it also applies to rather simple, small sulfur-containing molecules, such as disulfides and polysulfanes (see Sect. 6). Here, the “electrochemical tool kit” available to date is rather limited, crude, and prone to error: Traditionally, such sulfur-containing substances have been analyzed with the assistance of a metal-based electrode, such as a gold or mercury electrode [25–27]. These electrodes adsorb the sulfur-containing materials rather well, and therefore allow detection and basic characterization. Indeed, some flow-through detectors used in conjunction with chromatography (often FPLC and HPLC) are based on electrochemical detection of the thiol/disulfide redox couple, either in small molecules or in native, modified, or tagged proteins, such as metallothionein (MT) [4, 5, 11]. A schematic view of such an analytical setup is shown in Fig. 7.2.

Schematic view of an electrochemical method to monitor the presence of thiol-containing materials (including proteins). The example provided illustrates the use of CV in conjunction with a dropping mercury electrode to monitor the elution of the cysteine-rich protein metallothionein (MT). This method has been used, for instance, for small thiol-containing compounds by Stenken et al. (using a gold-amalgam instead of a “pure” Hg electrode) and for MT proteins by Adam et al. (using a more complex catalytic system also containing cobalt ions). One should emphasize that electrochemical detection (and characterization) has many advantages as far as cysteine-rich proteins such as the MT proteins are concerned, since many of them do not show a signal in traditional UV detectors (as they do not contain any aromatic residues) and are also difficult to analyze otherwise [5]
Unfortunately, the use of such electrodes for analytical purposes has several drawbacks. First of all, it is obvious that the adsorption process used to “capture” the sulfur species on the electrode surface severely affects its “chemistry”, including its redox properties. Rather than measuring, for instance, the “true” thiol/disulfide redox potential or monitoring the reduction of a polysulfane (RS x R, R ≠ H, x ≥ 3) to perthiol (RSxH, R ≠ H, x ≥ 2), one is de facto investigating surface-bound sulfur-gold or sulfur-mercury species [1, 27]. The latter obviously exhibit their very own redox behavior, which is likely to differ considerably from one of the “free” sulfur species in solution.
Not surprisingly, there have been some attempts in the past to minimize the influence of adsorption phenomena and to optimize the electrochemistry of sulfur- (and selenium-, tellurium-) containing substances. On the one hand, alternative, coupled, or indirect methods have been employed which measure redox behavior by different means. Willem Koppenol and colleagues, for instance, have applied CV to equilibria of chalcogen species with a redox dye and combined this setup with pulse radiolysis to inch closer to the various oxidation and reduction potentials of selenocysteine [18]. Other groups have considered alternative electrodes, including amalgamated copper electrodes and carbon electrodes [4, 28]. While amalgamated electrodes still suffer from adsorption phenomena, as solid rather than liquid electrodes, they nonetheless provide more flexibility when it comes to practical applications. Carbon electrodes may also be used, as they are also easy to handle and are less prone to adsorption phenomena, yet these electrodes do not monitor the classical, biologically relevant thiol/disulfide redox behavior, but rather a kind of radical chemistry which is not directly relevant in biological systems [26].
While the thiol/disulfide redox pair - despite some drawbacks - is in principle accessible electrochemically, other sulfur modifications and transformations are more difficult to monitor. Here, one must bear in mind that sulfur is a true redox chameleon in Biology, where it occurs in more than ten different oxidation states (including fractional ones) and in various sulfur “chemotypes”. Each of these chemotypes exhibits its own chemical properties. While most of them are also redox-active, the mechanisms at the center of their respective redox chemistries may vary widely.
Figure 7.3 summarizes the most common sulfur chemotypes (Fig. 7.3a) and some of the underlying reaction mechanisms (Fig. 7.3b) [29]. It is fairly obvious from this figure that despite the fact that there are plenty of redox processes involved in sulfur (bio-)chemistry, few of them involve electron transfer and hence are directly accessible by electrochemical methods. Indeed, nucleophilic exchange and atom transfer mechanisms devoid of direct electron transfer dominate the redox behavior of most sulfur species. It is therefore doubtful that an electrochemical method would easily be able to reveal the redox behavior of sulfur species such as sulfenic and sulfinic acids, thiosulfinates, thiosulfonates, sulfoxides, and sulfones. Nonetheless, these sulfur species should not be ignored outrightly, as they occur inside most living cells and play a pivotal role in intracellular redox signal, response, and control.

Selection of biochemically relevant Reactive Sulfur Species formally derived from the thiol (RSH) chemotype (center). Clockwise from the top: thiyl radical, sulfenic acid, sulfinic acid, sulfonic acid, disulfide, trisulfide, thiosulfinate, thiosulfonate (a); sulfur-based redox reactions may proceed via different redox mechanisms, some of which do not involve direct electron transfer and hence are difficult to analyze using electrochemical techniques (b)
Electrochemistry becomes even more difficult when moving from simple sulfur-containing compounds to sulfur proteins and enzymes. Here, the last decade has revealed a central role of cysteine (and, to a lesser extent, methionine) redox chemistry in cell signaling and control. We are now aware of numerous proteins and enzymes which contain redox-active cysteine residues, whose thiol groups can be modified posttranslationally to disulfides, sulfenic, sulfinic, and sulfonic acids as well as to S-nitrosothiols [30]. Such modifications can have a significant impact on protein function and enzyme activity and may ultimately result in a decisive cellular response, such as an antioxidant response or cell death via apoptosis. Unfortunately, electrochemical methods are still not specific enough to determine the redox properties of individual cysteine residues in larger proteins and also fall short of measuring redox properties of proteins which experience sulfenic or sulfinic acid modification, or S-nitrosation (see Sects. 5 and 6).
Electrochemistry of chalcogen-containing compounds and proteins becomes even more complicated when considering selenium- and tellurium-based redox systems. This is rather unfortunate since the last couple of decades has revealed the existence and paramount importance of quite a few (mammalian) selenoproteins and enzymes, including glutathione peroxidase (GPx), the human form of thioredoxin reductase (TrxR), thyroxine deiodinase (T(4)-5′-deiodinase), selenoprotein P (SelP), and selenoprotein W (SelW). While the selenocysteine (SeCys) residues in these proteins and enzymes exhibit a distinct redox chemistry, very little is known so far regarding their oxidation and reduction potentials. To the best of our knowledge, electrochemical techniques such as CV or DPP have not yet been applied in the context of these selenoproteins, despite the fact that the selenol/diselenide redox couple provides a fairly specific and easily distinguishable electrochemical response [17].
While research so far seems to have shied away from employing electrochemical methods to study the redox properties of selenoproteins, various methods have been developed to investigate selenium- and tellurium-containing compounds. During the last two decades, there has been considerable interest in the development of small-molecule synthetic mimics of GPx and of related enzymes [26, 31–36]. These agents, which often exhibit pronounced antioxidant activity in vitro, have also been studied by electrochemistry (see Sect. 7). Methods such as CV have been at the forefront of these investigations, and electrodes employed include various types of platinum, carbon, and mercury electrodes [25, 26, 32]. Platinum working electrodes have been particularly useful (and easy to use) in organic solvents [25]. In order to investigate such compounds in a more realistic, physiologically relevant scenario, aqueous, buffered systems whose exact composition may in part depend on the solubility properties of the substance under investigation have also been used. In this case, carbon and mercury working electrodes have been most effective [26, 37–39].
After this brief introduction to the “electrochemical tool kit” to analyze chalcogen-centered redox behavior, we will now turn our attention to individual applications which currently are of particular interest to biological redox chemists. Here, we will take a “top-down” approach, starting with whole cell cultures, then moving to the electrochemistry at or near single cells and then considering specific intracellular target molecules, such as sulfur- and selenoproteins and enzymes. The latter also form a prime target for chalcogen-based redox agents, which will be discussed briefly, with a focus on employing electrochemical methods as part of rational drug design.
7.3 Watching Cells Grow
Cell culture studies form a central part of modern Life Sciences, from microbiology to the development of drugs and pesticides. Here, the growth rate and metabolism of the cell/organism is often one of the most significant determinants. Using such studies, one may, for instance, establish the survival and “fitness” of a particular mutant or screen for the activity of a particular drug prototype. Unfortunately, traditional methods to determine the rate of cell proliferation, such as absorbance readings (OD600) and various staining techniques (e.g., sulforhodamine B, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) staining, Neutral Red uptake), are rather time consuming and are often marred by artifacts and poor reproducibility. Not surprisingly, alternative methods based on the measurement of cell growth, such as the colony forming unit (CFU) or colony forming assay, are also commonly used in cell culture studies [40]. Unfortunately, all of these methods are based on discontinuous monitoring, i.e., the analysis after certain fixed periods of time, and generally provide only a handful of data points. As time-resolution is low, such studies may miss subtle yet important changes in growth rate, such as initial growth phases and short rest phases. Indeed, cell growth and the effects of agents on cell growth are not continuous and linear. The biological activity of a given compound may, for instance, differ at different time points due to complicated metabolic processes. Arsenite, for example, does not show the same toxicity over a prolonged period of time [41]. This circumstance is not necessarily a drawback but may result in additional, extremely valuable data: Amazingly, if recorded and analyzed properly, the time-dependent profile of cell growth in response to a drug can even be used to compare different drugs with each other and ultimately to derive some information regarding drug action. While kinetic information on the dynamics of cell growth clearly is of paramount importance, it cannot be obtained easily by using classical absorbance methods [42].
Within this context, electrochemical methods offer several interesting alternatives. Electrical impedance measurements, for instance, are non-invasive and enable continuous monitoring of cell growth and subsequent (bioinformatic) profiling. This method has been pioneered by the studies of Giaever and Keese [43]. Using impedance measurements to study the spreading, motion, and cell density of mammalian fibroblasts on evaporated gold electrodes, the authors could show that the impedance signals measured were a direct consequence of cell growth, spreading, and motility, as signals disappeared when cytochalasin (a drug that prevents motion) was added to the medium [43].
This method has been developed into several commercial systems which employ multi-well plates, which at first sight resemble classical plastic 96-well plates yet feature integrated (micro-) electrodes at the bottom of each well (see Fig. 7.4). As cells growing on the bottom of these wells increase the electrical resistance of the system, these multi-well plates can be used to monitor the growth of cultured cells in real time and under normal growth conditions (i.e., in the incubator). As long as the cells remain adsorbed to the bottom of the well, it is possible to measure how the cell layer grows and if cells die and detach from the electrode surface, for instance in response to a cytotoxic compound. In this case, the resulting decrease in impedance [44] can be monitored quite precisely and in real time [45]. Although the original method - which is still widely used - is limited to adhesive cell lines, it has provided a major step forward and has already revealed several “secrets” in cell growth behavior which otherwise may have escaped attention (see Fig. 7.4).

Monitoring cell growth in real time by non-invasive impedance measurements. Several cell samples can be monitored in parallel and the effects of drugs can be measured continuously. This technique has many advantages compared to traditional staining methods. As the “growth” curves for the control (in black) and the drug-injected sample (in grey) show, there is a clear effect due to the injection of the drug. This effect, however, would be entirely missed by a traditional method which would perform an aliquot-based reading after 1 and 4 h. Furthermore, the growth curves can be profiled and compared to the curves of other drugs in order to pin down similarities and hence help to identify possible (related/different) cellular modes of action and targets. Please note that it is also possible to work with non-adhesive cells as long as there is a specific “anchor” present at the electrode surface
Since then, impedance has been applied by several groups with different aims and experimental designs. With impedance sensors it is possible to determine whether a drug causes short-term or long-term cellular responses [46, 47]. In the same context, such methods have proved to be very fast and powerful in establishing the cytotoxicity of test compounds in cancer cells [40, 48–51]. Here, their ability to monitor continuously how a treatment with a known or supposed cancer agent affects to cell population and cell growth has turned out to be of considerable advantage, especially in the context of profiling and comparing effects on cell growth induced by different drugs (for specific examples of applications see below). Presently, attempts are underway to expand the method and its capabilities even further, for instance to answer questions related to cell metabolism or modes of cell death (such as apoptosis) [51]. Some of these additional features will be discussed below.
The application of impedance measurements is not restricted to mammalian cells. Several studies have used impedance to detect fairly selectively pathogenic bacterial cells, using biorecognition elements such as antibodies attached on the electrode surface [52–54]. In this particular case, the cells in question do not grow on the electrode area (as in the original method), but are “fished” and subsequently anchored to the surface by specific recognition antibodies. Once captured at the electrode surface, these bacteria also increase electric resistance which can be measured. Related electric methods, such as the determination of dielectric properties inside a bioreactor through capacitance and conductance measurements, have been used in microbial, fungal, and yeast fermentations in reactors to monitor growth with satisfactory results [55].
Today it is possible to reduce the size of the impedance sensors and to combine them with other electrochemical, microelectrode-based sensors, such as an ion-sensitive field-effect transistor (ISFET) for pH measurement and conductometric sensors for carbon dioxide in well plates of different sizes and geometries [56]. For instance, pH measurements are often crucial in tissue culture, and online, continuous monitoring is desired. Here, novel types of electrodes have recently been tested, including metal (antimony) oxide microelectrodes [57]. It is even possible to interdigital a set of sensors, such as an amperometric sensor (for oxygen consumption) with ISFET and interdigital electrode structure (IDES) sensors in a silicon chip. The values subsequently obtained by such a device are reproducible and reasonable when compared with established methods [58].
While initial studies had to be performed with “home made” electrodes freshly prepared before each experiment [43], several commercial systems are nowadays available which can be employed routinely in daily research. Here, the Bionas 2500 analysis system with IDES and other sensors (ISFET and Clark electrodes) is used widely. Abarzua et al., for instance, have used this system to study the anticancer activity of plant extracts [49]. Similarly, Ceriotti et al. have determined the cytotoxicity of known anticancer molecules and observed that values were comparable with the results obtained by classical methods [50].
A second commercial system, the Real-Time Cell Electronic Sensing System (RT-CES), has been developed by ACEA Biosciences. This device measures the impedance in all wells of a 16- or 96-well microplate in parallel, therefore accelerating data collection enormously. Applications of this advanced, automated technology are very broad and include proliferation studies in different cell lines, drug cytotoxicity determination in cancer cells, induction of apoptosis, receptor tyrosine kinase activity, and protective effects of antagonists of drugs or biological molecules [51, 59].
Besides the two systems mentioned here, there are numerous others which provide the user with a wealth of applications (e.g., the systems developed by MDS Analytical Technologies and Roche Applied Science). Currently, 96-well and even 384-well plates are available for high-throughput assays which ultimately combine these electrochemical sensing techniques with bioinformatics tools. Abassi and collaborators, for instance, have been able to perform a time-resolved screening of 2,000 compounds with known anticancer potential in two cancer cell lines to ascertain which of the compounds present short-term or long-term responses (or both). After determining the impedance-based time-dependent cell response profiles (TCRPs), they clustered the structures into families according to the TCRPs associated with each response. Ultimately, the analysis of similar activities enables the prediction of applications of compounds which may not have been realized previously [60].
7.4 Monitoring Redox Processes at the Cellular Level
As mentioned in the previous section, most traditional cell culture methods used in drug discovery involve the selection and subsequent modification of a target and hence may not be sufficiently predictive because of the interferences/modifications required. As for cell proliferation, label-free technologies able to use native systems instead of the modified ones to monitor (bio-) chemical processes in, at, or near cells are particularly interesting. Such techniques are non-destructive and allow the continuous monitoring of certain processes. They also do not require expensive markers, enzymes, or antibodies, which is a major issue in biochemical research.
While there are various “sensors” available to monitor cellular processes, such as metabolism, at microscopic scale (e.g., pH, dioxygen, hydrogen sulfide), we will focus on electrochemical approaches which may be used to monitor redox changes close to or even inside the intact cell. From the perspective of biological redox processes, the detection and quantification of physiologically relevant redox-active species is of particular interest. Some of these chemical species have already been summarized in Table 7.1, which is clearly selective and far from complete. As already mentioned, ROS, RNS, and sulfur-containing species play a major role in normal metabolism, the maintenance of the cellular redox homeostasis, intracellular redox signaling, and host defense [61–64].
In the past, the analysis of intracellular levels of ROS and RNS, but also of GSH and cellular thiol content, in essence has relied on two methods: (a) a rather crude method which involves breaking down the cell and analyzing ROS, RNS, GSH, and thiol levels in the homogenate; (b) various staining methods which involve the application of specific stains to “visualize” and quantify the intracellular level of oxidative stress (OS), individual ROS, and GSH [65]. Both methods have their merits. The first method can be employed rather successfully, for instance, to indirectly determine the formation of •NO by quantifying its follow-on product nitrite via the Griess reaction [66] or to measure the total intracellular thiol content with 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB, Ellman’s reagent) [67, 68]. This method can also be combined with HPLC, which enables the quantitative determination of several reactive species in the same sample [69, 70]. Staining techniques are somewhat more sophisticated and often rely on expensive equipment, such as fluorescent microplate readers and confocal fluorescence microscopy. These methods can be combined to provide an “intracellular diagnostic platform” which in essence allows the monitoring of processes inside intact cells or cellular organelles and over time [71, 72].
Nonetheless, these methods also suffer from certain drawbacks. Both methods still cannot reliably distinguish between different ROS or different thiols. For instance, 2′,7′-dichlorodihydrofluorescein acetate, which is widely used to establish intracellular levels of OS, is fairly unspecific and detects various oxidizing species, including H2O2 and •OH radicals [69]. While there are numerous attempts to develop more specific stains [70], the stains available so far are still rather unspecific. At the same time, various ROS are chemically too reactive and/or unstable and hence escape analysis in cell extracts after lysis.
Here, electrochemistry may provide a reliable and in many ways straightforward alternative in the detection of certain ROS and RNS. Indeed, there have been numerous attempts to measure different ROS in biological samples, often employing modified electrodes which provide some specificity for individual ROS or RNS. We will consider these reactive species in the increasing order of their respective cytotoxicity.
The superoxide radical anion (O2 •−) is a byproduct of aerobic mitochondrial respiration which is turned into hydrogen peroxide and dioxygen by one of the various superoxide dismutase enzymes [73–76]. Allen Hill and his colleagues at Oxford were the first to detect O2 •− electrochemically in a cell-based system employing opsonized microelectrodes of graphite or gold [77, 78]. This pioneering work has subsequently been followed up by studies employing more “sophisticated” electrodes, such as modern carbon fiber microelectrodes and gold electrodes modified with cytochrome c [79].
Hydrogen peroxide (H2O2) is formed as part of mitochondrial respiration (see above) and also by immune cells as part of host defense. Its particular toxicity mostly results from its participation in the Fenton reaction, whereby H2O2 is reduced to the highly aggressive hydroxyl (•OH) radical [75, 76]. Its stability and appropriate half-life allows electrochemical detection, for instance on platinized surfaces of carbon electrodes and platinized carbon fiber microelectrodes [80–83].
The most reactive ROS is OH•, which is highly damaging for the cells - it indiscriminately reacts with proteins, DNA, and membrane lipids. As the lifetime of •OH in a biological system is in the order of a few nanoseconds, diffusion across membranes and/or toward more remote electrodes (e.g., placed outside the cell) is not feasible [81]. Nonetheless, Zhu et al. have recently described a rather innovative method to indirectly detect and quantify •OH radicals. This method exploits the highly aggressive behavior of •OH. It measures the impedance at a coated electrode, which decreases once •OH radicals gradually damage the coating and expose the electrode surface [84].
The RNS nitric oxide (•NO) [76, 85] and peroxynitrite (ONOO−) can be detected directly by electrochemical methods due to their specific redox characteristics. Nitric oxide is determined with the help of polymer-coated carbon microelectrodes [86, 87]. In contrast, peroxynitrite can be detected rather selectively electrochemically using a carbon fiber microelectrode modified by a film of Mn(III)-[2, 2]paracyclophenylporphyrin [88, 89]. Other reactive RNS are very difficult to detect or to distinguish electrochemically due to their high reactivity and short half-life time.
Some of the detection methods for the more common reactive species, however, have already been turned into rather sophisticated sensing devices, such as a biocompatible sensor for •NO, which employs carbon-based screen-printed electrodes [90]. Frequently, these systems are not only specific and sensitive but also suitable to analyze rather low sample volumes (“lab on a chip”), such as a microfluidic device designed to measure OS generated by macrophages [91] or a device to measure •NO release from cells grown directly on the surface of a sensor [92].
Indeed, Christian Amatore and colleagues at the University Paris Descurtes, France have recently developed an electrochemical method which allows researchers to measure various reactive species at the level of a single cell [93]. As most (but not all) ROS and RNS diffuse out of the cell, it is possible to quantify these species outside the cell using specially designed (ultra-) microelectrodes and a specific set of potentials as indicative measuring points - and to subsequently calculate back to intracellular concentrations (see Fig. 7.5a). Most ROS possess a characteristic electrochemical potential, and such methods are able to capture otherwise elusive ROS and to distinguish between them (e.g., H2O2, ONOO−, •NO, and NO2 − can be detected and quantified by measuring at 300, 450, 650, and 850 mV vs. the Ag/AgCl electrode (SSCE), respectively) [20]. These methods are particularly suitable for investigating ROS generating cells, such as macrophages, and may even be employed to monitor events associated with a single cell. The recent study by Hu et al. on a single MG63 osteosarcoma cell submitted to mechanical stress, for instance, reflects the enormous potential of this method [20]. Additional publications that describe simultaneous detections of different ROS and RNS have been reviewed by Borgmann [88].

(a) Single cell electrochemical measurements to monitor ROS/RNS production at/near a single MG53 macrophage. As measurements are taken in the growth medium close to the macrophage, ROS/RNS need to diffuse out of the macrophage in order to be detected. (b) Prospective future measurements of ROS/RNS inside cells. In contrast to the current state-of-the-art setup depicted in part a, this proposed method measures ROS/RNS inside the cell using a tiny electrode able to penetrate the cell membrane. This method does no longer require the diffusion of reactive species out of the cells, yet relies on very small electrodes and hence minute currents. Please note that this kind of intracellular electrochemical monitoring is still in its early developmental phase and not yet available for routine measurements. It may also encounter additional complications due to the presence of numerous (redox-active) biomolecules inside the cell
Nonetheless, one should emphasize that such an approach also has its limitations. It is not possible to measure all ROS or RNS outside the cell because of the short lifetimes of the most reactive ones, such as the •OH radical. The latter is unable to diffuse out of the cell, where the microelectrode is placed, as it is too reactive and will be “lost” before it can reach the electrode. One alternative would involve the application of nanoscopic electrodes which may be inserted into intact cells (proposed in Fig. 7.5b). Unfortunately, electrodes which would be small enough to operate within cells without interfering with normal cell metabolism and, at the same time, would also be large enough to provide a measurable current, are not yet available.
7.5 Characterization of Cysteine-Containing Proteins and Enzymes
While the determination and in some cases also the quantification of ROS and RNS at or near living cells has witnessed considerable progress during the last decade, electrochemical methods to characterize the prime cellular targets of these reactive species, i.e., redox-sensitive cysteine- and selenocysteine-containing proteins and enzymes, are still in their infancy. This lack of appropriate methods to describe the redox behavior of cysteine- and selenocysteine proteins is out of step with the recent, dramatic progress in the field of sulfur-based intracellular redox signaling and regulatory systems. During the last decade, numerous studies have provided a rather detailed insight into the cellular “thiolstat”, a pivotal, overarching regulatory system involved in cellular life and death decisions, including proliferation, differentiation, and apoptosis [30, 94].
Today, there are just a few methods available to appropriately characterize such regulatory processes at the level of proteins. Frequently, sophisticated staining, antibody, and proteomic techniques are employed to identify posttranslational cysteine modifications in the cell, which include disulfide bond formation (intra- and intermolecular disulfides, S-thiolation, S-glutathiolation), S-nitrosation (leading to RSNO species), and sulfenic acid (RSOH) formation (for a review see [94]). Nonetheless, these detection methods still provide a rather static picture. They may answer the question which (cysteine) residues are modified in proteins and enzymes, yet do not refer to the why. To answer the question why specific residues are prone to modification, one may need to consider the reactivity or oxidation potential of the (cysteine, selenocysteine) residues in question and to compare them to other residues. Such Epa values of thiols may explain and in the future even predict which particular cysteine residues in which particular proteins become oxidized/modified first and under which conditions, assuming the residues in question are accessible to oxidation and also come into contact with oxidants.
It is rather disappointing, and in stark contrast to the thriving area of electrochemistry of metalloproteins, that few electrochemical investigations have been performed so far on cysteine- and selenocysteine-containing proteins and enzymes. Indeed, the zinc/sulfur protein metallothionein (MT-1 and MT-2), which contains seven zinc ions bound to 20 cysteine residues in two zinc/sulfur clusters (Zn4Cys11 and Zn3Cys9), seems to be the only protein which has been detected and whose redox properties have been analyzed so far using electrochemical techniques [12, 13, 95]. Here, the cysteine residues (rather than the metal ions) are investigated. The same also applies to related cysteine-based peptides [4, 5, 10, 96]. As these techniques rely on mercury-based electrodes, they are obviously also complicated by adsorption phenomena as discussed in Sect. 2. Nonetheless, these (and related) studies provide the first examples of electrochemical investigations of cysteine-redox behavior in proteins. Apart from these examples, and to the best of our knowledge, electrochemistry so far has not been employed to study the redox behavior of cysteine (or selenocysteine) residues in proteins. As a consequence, a reliable redox scale of sulfur and selenium proteins is not available yet, despite the fact that the thiol/disulfide and the selenol/diselenide redox pairs can both be detected at metal (mercury) electrodes (see Sect. 2).
Indeed, only a handful of redox potentials are available for cysteine residues in peptides, proteins, and enzymes so far, for instance for glutathione and the active site cysteine residues in human thioredoxin (Trx), human glutaredoxin (Grx), human protein disulfide isomerase (PDI), and TrxR (see Table 7.2) [97]. These values have been obtained by indirect methods, such as equilibrium redox titrations using the GSH/GSSG couple in conjunction with the Nernst equation.
Apart from oxidation and reduction potentials estimated for these usual suspects (i.e., proteins and enzymes which are well-known to be involved in the maintenance of the intracellular redox balance), our knowledge regarding Epa values of cysteine residues is extremely limited. Since such E values are currently unavailable, most biochemists exploring the cellular thiolstat use pKa values instead. While there is some merit in using acidity of a given cysteine thiol as a measure for its reactivity (the thiolate form is considerably more reactive than the thiol), the pKa value is a poor and too simplistic substitute for true redox parameters.
7.6 Electrochemistry of Sulfur-Containing Natural Products
While the electrochemistry of cysteine proteins and enzymes is far from trivial, sulfur-containing natural products, such as thiols, disulfides, and polysulfanes, can be characterized electrochemically with comparable ease. Such studies have been used rather extensively in the context of redox-active antioxidants, chemopreventive agents, and pro-oxidant redox modulators, some of which interfere with cellular signaling [4, 5, 96].
Table 7.3 provides a brief and necessarily incomplete overview of such - electrochemically accessible - sulfur species which play a role in Biology and on occasion also form part of pharmaceutical research. In most instances, such products can be studied using CV and DPP in conjunction with a metal electrode (often mercury, see Sect. 2).
Examples of particular interest include the various natural sulfur compounds found in garlic and related Allium species, such as diallyl disulfide (DADS), diallyl trisulfide (DATS), and diallyl tetrasulfide (DATTS) [27, 29, 98]. While the quality of data obtained by such methods is limited because of adsorption phenomena, CV still provides some crucial information which is lacking otherwise, including aspects of the reversibility of reduction/oxidation and electrochemical potentials. Indeed, CV of disulfides and polysulfanes is superior to alternative, non-electrochemical methods, such as redox titrations with redox dyes, as CV is a direct method and provides fairly precise, extensive, and reproducible information. In the case of the diallyl polysulfanes, for instance, the electrochemical studies reported by our group have shed some light on the redox properties of these unusual sulfur compounds [27, 29, 30, 98, 99]. They have also questioned the notion that polysulfanes are strong oxidants and ultimately have supported the idea that such compounds - once activated - act via a reducing mechanism. Here, electrochemical data supports the concept of Munday and colleagues, which explains the exceptional biological activity of diallyl polysulfanes (and related polysulfanes) with the ability of their reduced forms to reduce dioxygen to O2 •− and the subsequent occurrence of an oxidative insult [100]. Similarly, the electrochemical studies of naturally occurring 3-ethenyl-3,4-dihydro-1,2-dithiine (1,2-DT) have underlined the rather distinct redox properties of this and related α,β-unsaturated disulfide compounds, which sets them apart from “normal” disulfides (see Fig. 7.6) [101].

Characterization of sulfur-containing natural products using CV in conjunction with a dropping mercury electrode, as exemplified by the natural compound 3-ethenyl-3,4-dihydro-1,2-dithiine (1,2-DT). This kind of analysis provides valuable information regarding the redox behavior of such compounds, including oxidation and reduction potentials and reversibility (a). Such measurements are also capable of distinguishing between different sulfur compounds in the same sample and can be run in the presence of a reference compound, such as GSH (b). Voltammograms were recorded for 1,2-DT in the abscence and presence of GSH in phosphate buffer of pH 7.4 at a scan rate of 250 mV/s using a Hg working, Pt wire counter, and Ag/AgCl reference electrode (Khairan et al., unpublished results)
Similar electrochemical studies are certainly possible for related disulfide-based compounds, including, for instance, thiuram disulfides (such as the drug disulfiram) and 3-dithiolethiones (such as the drug Oltipraz). At the same time, the release of hydrogen sulfide from certain natural products or drug molecules, the biological effects of inorganic polysulfides (S x 2−), and sulfur-metal interactions can also be investigated electrochemically.
In contrast, some of the higher sulfur oxidation states, such as sulfenic, sulfinic, and sulfonic acids, as well as thiosulfinates and thiosulfonates, may not be accessible directly using electrochemical techniques. As already mentioned, such sulfur species undergo redox transformations via substitution mechanisms. Since these reactions do not involve electron transfer, it may be difficult to access such redox systems electrochemically, even if a particular “indicator” reaction is employed (e.g., the electrochemical potential of one-electron abstraction as possible indicator of nucleophilicity or electrophilicity, see Sect. 2). Ultimately, biologically interesting sulfur species, such as sulfenic and sulfinic acids as well as thiosulfinates, may escape electrochemical analysis. Their redox behavior may have to be measured and described by other means, for instance, by equilibrium reactions with GSH or via the kinetics of the underlying exchange reactions.
7.7 Electrochemistry as Part of QSAR
Despite the fact that many sulfur-containing natural products escape a thorough electrochemical analysis, the research performed with disulfides (and related structures) has shown that electrochemistry may provide a fast and reliable tool to describe the redox properties of such compounds and subsequently also explain, rationalize, and even predict biological activity (or the lack thereof). Not surprisingly, methods such as CV and DPP have also been used to characterize the redox properties of redox-active selenium- and tellurium agents, especially those used in the context of intracellular redox modulation [25, 26, 32, 102–104]. If redox processes are able to control cell proliferation, differentiation, and apoptosis, so the idea, then agents able to modulate such processes should qualify as highly effective and maybe even selective drugs for a wide range of human diseases [105, 106]. Indeed, chemoprevention by antioxidants, aging and oxidative stress, inflammation and antioxidants, host defense against invading microorganisms via an oxidative burst, and cancer cells going into apoptosis due to crossing the internal redox threshold are all important and current topics related—in one way or another—to redox control [23].
It is therefore hardly surprising that research into redox modulating agents is currently booming and methods such as CV and DPP provide a wide range of opportunities to analyze and describe such compounds (Fig. 7.7). During the 1990s, Ian Cotgreave, Lars Engman, and colleagues at the Karolinska Institute, Stockholm, Sweden have employed CV for the characterization of various GPx mimics, i.e., structurally related selenium and tellurium compounds [25, 102]. These studies, conducted at platinum-button working electrodes in dichloromethane (Bu4NClO4 as electrolyte), have, for instance, revealed a certain correlation between the oxidation potentials of structurally related chalcogen compounds and their antioxidant capacity in biological test systems [25]. Indeed, “oxidisability” can be related to the structure of such compounds and also has a profound effect on the compound’s antioxidant capacity [102]. A similar link between the molecular structure of redox-active chalcogen compounds, their oxidation potential, and subsequent biological activity has also been observed by Giles et al. using a set of structurally related selenium and tellurium compounds and an in vitro (catalytic) antioxidant assay [26, 32, 107]. In these studies, the first oxidation potential (Epa1) again seems to be indicative of the “oxidisability”, and hence chemical redox (re-)activity, which in turn seems to be responsible for biological activity.

Prospective role of electrochemical analysis in modern QSAR. Methods such as CV and polarography can be used to obtain valuable information regarding the redox properties of prospective drug molecules which exert their biological effects via a redox-based reaction (e.g., oxidative modification of target proteins). The chemical structure determines electrochemical properties (e.g., Epa values), which in turn may influence chemical reactivity, control the biochemical mode of action, and subsequently may explain aspects of biological activity
Many aspects of these early, pioneering electrochemical investigations into the emerging “chemical structure-electrochemical potential-biological activity relationship” could since be confirmed in organic solvents as well as in aqueous solutions. The electrochemical parameters clearly occupy a prominent position in this three-way relationship: On one hand, they reflect structural features of the compounds, such as a particular redox mechanism or the presence of electron donating or withdrawing substituents. On the other hand, they are indicative of a particular redox behavior which plays a central role in the compound’s biological activity (see Fig. 7.7). While it is hardly surprising that agents acting as redox modulators also exhibit a redox chemistry which in some ways is the cause and hence also predictive of the biological activity, it is indeed surprising to notice that many of these compounds have not yet been studied properly by electrochemical methods. As the therapeutic interest in redox modulation continues to grow, one may anticipate additional, maybe even pioneering electrochemical work in this area of pharmaceutical research.
7.8 Outlook and Conclusions
The previous sections have shown that electrochemical methods already play a significant role in the analysis of chalcogen-centered biological redox processes, from monitoring the formation of ROS and RNS at the single cell level and the characterization of the redox properties of cysteine proteins and enzymes to the elucidation of structure-potential-activity relationships for diverse natural sulfur compounds and synthetic selenium- and tellurium-based redox modulators. Nonetheless, our discussion of these examples has also demonstrated that there are many questions that still need to be addressed and subsequently answered. Here, we would like to point toward a few areas of biological redox chemistry which are still in desperate need of proper analysis, and where electrochemical methods may play a significant role in the future.
First of all, the appropriate description of the complex redox behavior of cysteine proteins and enzymes comes to mind. As an increasing number of redox-active, regulatory, and signaling cysteine proteins of the cellular thiolstat is emerging, there is an urgent need to determine the precise redox properties of these proteins, including their susceptibility toward oxidation and S-thiolation as well as reversibility of such processes. These posttranslational modifications often control protein function and enzyme activity. They do not occur randomly, but rather seem to target particular cysteine (and selenocysteine) residues in specific proteins, probably on the basis of low oxidation potential of the given residue and/or its accessibility for the oxidizing agents. Not surprisingly, electrochemical studies would assist enormously in determining which of these proteins are especially prone toward oxidation/modification, which residues are primarily affected and if such modifications are reversible or not.
Another area of interest is the characterization of sulfur-, selenium-, and tellurium-containing agents which may be beneficial as redox modulating drugs (e.g., as antioxidants, cytostatic/cytotoxic drugs). The last two decades have witnessed considerable progress in this area, yet considerably more studies with more substances need to be performed to establish appropriate structure-potential-activity relationships. Ultimately, electrochemistry may play a central role in this field of research, as the electrochemical potentials reflect the structural properties and also form the basis for biological activity. In the end, Epa and Epc values may not only be considerably more precise, reliable, and reproducible when compared to “activities” in the various antioxidant and redox assays (such as the ferric reducing antioxidant power (FRAP) assay, the total antioxidant capacity (TAC) assay, or the oxygen radical absorption capacity (ORAC) assay) they may also be much easier to obtain and faster to measure.
In any case, the biological chemistry of sulfur, selenium, and tellurium provides ample opportunities for innovative electrochemical investigations in the near and medium future.
References
Armstrong FA, Hill HAO, Walton NJ. Direct electrochemistry of redox proteins. Acc Chem Res. 1988;21:407–13.
Kolb DM, Amatore C, Compton RG. Modern electrochemistry: interdisciplinary research at the forefront of science. Chemphyschem. 2010;11:2655–6.
Park S, Boo H, Chung TD. Electrochemical non-enzymatic glucose sensors. Anal Chim Acta. 2006;556:46–57.
Stenken JA, Puckett DL, Lunte SM, et al. Detection of N-acetylcysteine, cysteine and their disulfides in urine by liquid chromatography with a dual-electrode amperometric detector. J Pharm Biomed Anal. 1990;8:85–9.
Adam V, Fabrik I, Kohoutkova V, et al. Automated electrochemical analyzer as a new tool for detection of thiols. Int J Electrochem Sci. 2010;5:429–47.
Stetter JR, Penrose WR, Yao S. Sensors, chemical sensors, electrochemical sensors, and ECS. J Electrochem Soc. 2003;150:11–6.
Pohanka M, Skladai P. Electrochemical biosensors—principles and applications. J Appl Biomed. 2008;6:57–64.
Feinberg BA, Lau YK. The electrochemistry of high-potential iron-sulfur proteins and their novel brdicka waves. Bioelectrochem Bioenerg. 1980;7:187–94.
Hu NF. Direct electrochemistry of redox proteins or enzymes at various film electrodes and their possible applications in monitoring some pollutants. Pure Appl Chem. 2001;73:1979–91.
Mendieta J, Rodriguez AR. Electrochemical study of the binding properties of a metallothionein I related peptide with cadmium or/and zinc. Electroanalysis. 1996;8:473–9.
Seiwert B, Karst U. Analysis of cysteine-containing proteins using precolumn derivatization with N-(2-ferroceneethyl)maleimide and liquid chromatography/electrochemistry/mass spectrometry. Anal Bioanal Chem. 2007;388:1633–42.
Krizkova S, Zitka O, Adam V, et al. Possibilities of electrochemical techniques in metallothionein and lead detection in fish tissues. Czech J Anim Sci. 2007;52:143–8.
Adam V, Baloun J, Fabrik I, et al. An electrochemical detection of metallothioneins at the zeptomole level in nanolitre volumes. Sensors. 2008; 8:2293–305.
Gerritsen M, Jansen JA, Kros A, et al. Influence of inflammatory cells and serum on the performance of implantable glucose sensors. J Biomed Mater Res. 2001;54:69–75.
Mang A, Pill J, Gretz N, et al. Biocompatibility of an electrochemical sensor for continuous glucose monitoring in subcutaneous tissue. J Diabetes Sci Technol. 2005;7:163–73.
Keenan DB, Mastrototaro JJ, Voskanyan G, et al. Delays in minimally invasive continuous glucose monitoring devices: a review of current technology. J Diabetes Sci Technol. 2009;3:1207–14.
Jacob C, Giles GL, Giles NM, et al. Sulfur and selenium: the role of oxidation state in protein structure and function. Angew Chem Int Ed Engl. 2003;42:4742–58.
Nauser T, Dockheer S, Kissner R, et al. Catalysis of electron transfer by selenocysteine. Biochemistry. 2006;45:6038–43.
Koppenol WH, Stanbury DM, Bounds PL. Electrode potentials of partially reduced oxygen species, from dioxygen to water. Free Radic Biol Med. 2010;49:317–22.
Hu R, Guille M, Arbault S, et al. In situ electrochemical monitoring of reactive oxygen and nitrogen species released by single MG63 osteosarcoma cell submitted to a mechanical stress. Phys Chem Chem Phys. 2010;12:10048–54.
Jacob C, Doering M, Burkholz T. The chemical basis of biological redox-control. In: Jacob C, Winyard PG, editors. Redox signaling and regulation in biology and medicine. Weinheim: Wiley-VCH; 2009. p. 63–122.
Doering M, Ba LA, Lilienthal N, et al. Synthesis and selective anticancer activity of organochalcogen based redox catalysts. J Med Chem. 2010; 53:6954–63.
Jacob C, Winyard P. Redox signalling and regulation in biology and medicine. Weinheim: Wiley-VCH; 2009.
Amatore C, Arbault S, Bruce D, et al. Characterization of the electrochemical oxidation of peroxynitrite: relevance to oxidative stress bursts measured at the single cell level. Chemistry. 2001;7:4171–9.
Cotgreave IA, Moldeus P, Engman L, et al. The correlation of the oxidation potentials of structurally related dibenzo[1,4]dichalcogenines to their antioxidance capacity in biological systems undergoing free radical-induced lipid peroxidation. Biochem Pharmacol. 1991;42:1481–5.
Giles GI, Tasker KM, Johnson RJK et al. (2001) Electrochemistry of chalcogen compounds: prediction of antioxidant activity. Chem Commun. 2490–2491
Anwar A, Burkholz T, Scherer C, et al. Naturally occurring reactive sulfur species, their activity against Caco-2 cells, and possible modes of biochemical action. J Sulfur Chem. 2008;29:251–68.
Hason S, Simonaho SP, Silvennoinen R, et al. Detection of phase transients in two-dimensional adlayers of adenosine at the solid amalgam electrode surfaces. J Electroanal Chem. 2004;568:65–77.
Jacob C, Anwar A. The chemistry behind redox regulation with a focus on sulphur redox systems. Physiol Plant. 2008;133:469–80.
Jacob C. Redox signalling via the cellular thiolstat. Biochem Soc Trans. 2011;39:1247–53.
Mugesh G, Panda A, Singh HB, et al. Glutathione peroxidase-like antioxidant activity of diaryl diselenides: a mechanistic study. J Am Chem Soc. 2001;123:839–50.
Giles GI, Giles NM, Collins CA et al. (2003) Electrochemical, in vitro and cell culture analysis of integrated redox catalysts: implications for cancer therapy. Chem Commun. 2030–2031
Jacob C, Knight I, Winyard PG. Aspects of the biological redox chemistry of cysteine: from simple redox responses to sophisticated signalling pathways. Biol Chem. 2006;387:1385–97.
Engman L, McNaughton M, Gajewska M, et al. Thioredoxin reductase and cancer cell growth inhibition by organogold(III) compounds. Anticancer Drugs. 2006;17:539–44.
Kumar S, Engman L. Microwave-assisted copper-catalyzed preparation of diaryl chalcogenides. J Org Chem. 2006;71:5400–3.
Engman L, Al-Maharik N, McNaughton M, et al. Thioredoxin reductase and cancer cell growth inhibition by organotellurium compounds that could be selectively incorporated into tumor cells. Bioorg Med Chem. 2003;11:5091–100.
Collins CA, Fry FH, Holme AL, et al. Towards multifunctional antioxidants: synthesis, electrochemistry, in vitro and cell culture evaluation of compounds with ligand/catalytic properties. Org Biomol Chem. 2005; 3: 1541–6.
Mecklenburg S, Collins CA, Doring M, et al. The design of multifunctional antioxidants against the damaging ingredients of oxidative stress. Phosphorus Sulfur Silicon Relat Elem. 2008;183:863–88.
Schneider T, Baldauf A, Ba LA, et al. Selective antimicrobial activity associated with sulfur nanoparticles. J Biomed Nanotechnol. 2011;7:395–405.
Ceriotti L, Ponti J, Colpo P, et al. Assessment of cytotoxicity by impedance spectroscopy. Biosens Bioelectron. 2007;22:3057–63.
Komissarova EV, Saha SK, Rossman TG. Dead or dying: the importance of time in cytotoxicity assays using arsenite as an example. Toxicol Appl Pharmacol. 2005;202:99–107.
Repetto G, del Peso A, Zurita JL. Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nat Protoc. 2008;3:1125–31.
Giaever I, Keese CR. Monitoring fibroblast behavior in tissue culture with an applied electric field. Proc Natl Acad Sci USA. 1984;81:3761–4.
Du D, Cai J, Ju H, et al. Construction of a biomimetic zwitterionic interface for monitoring cell proliferation and apoptosis. Langmuir. 2005;21:8394–9.
Asphahani F, Zhang M. Cellular impedance biosensors for drug screening and toxin detection. Analyst. 2007;132:835–41.
Zhu J, Wang X, Xu X, et al. Dynamic and label-free monitoring of natural killer cell cytotoxic activity using electronic cell sensor arrays. J Immunol Methods. 2006;309:25–33.
Atienza JM, Zhu J, Wang X, et al. Dynamic monitoring of cell adhesion and spreading on microelectronic sensor arrays. J Biomol Screen. 2005; 10:795–805.
Asphahani F, Thein M, Veiseh O, et al. Influence of cell adhesion and spreading on impedance characteristics of cell-based sensors. Biosens Bioelectron. 2008;23:1307–13.
Abarzua S, Drechsler S, Fischer K, et al. Online monitoring of cellular metabolism in the MCF-7 carcinoma cell line treated with phytoestrogen extracts. Anticancer Res. 2010;30:1587–92.
Ceriotti L, Kob A, Drechsler S, et al. Online monitoring of BALB/3T3 metabolism and adhesion with multiparametric chip-based system. Anal Biochem. 2007;371:92–104.
Solly K, Wang X, Xu X, et al. Application of real-time cell electronic sensing (RT-CES) technology to cell-based assays. Assay Drug Dev Technol. 2004;2:363–72.
Radke SM, Alocilja EC. A high density microelectrode array biosensor for detection of E. coli O157:H7. Biosens Bioelectron. 2005;20:1662–7.
Varshney M, Li Y. Interdigitated array microelectrode based impedance biosensor coupled with magnetic nanoparticle-antibody conjugates for detection of Escherichia coli O157:H7 in food samples. Biosens Bioelectron. 2007;22:2408–14.
Yang L, Li Y, Griffis CL, et al. Interdigitated microelectrode (IME) impedance sensor for the detection of viable Salmonella typhimurium. Biosens Bioelectron. 2004;19:1139–47.
Olsson L, Nielsen J. On-line and in situ monitoring of biomass in submerged cultivations. Trends Biotechnol. 1997;15:517–22.
van Leeuwen M, Li X, Krommenhoek EE, et al. Quantitative determination of glucose transfer between concurrent laminar water streams in a H-shaped microchannel. Biotechnol Prog. 2009;25:1826–32.
Wang M, Ha Y. An electrochemical approach to monitor pH change in agar media during plant tissue culture. Biosens Bioelectron. 2007; 22: 2718–23.
Brischwein M, Motrescu ER, Cabala E, et al. Functional cellular assays with multiparametric silicon sensor chips. Lab Chip. 2003;3:234–40.
Atienza JM, Yu NC, Kirstein SL, et al. Dynamic and label-free cell-based assays using the real-time cell electronic sensing system. Assay Drug Dev Technol. 2006;4:597–607.
Abassi YA, Xi B, Zhang WF, et al. Kinetic cell-based morphological screening: prediction of mechanism of compound action and off-target effects. Chem Biol. 2009;16:712–23.
Halliwell B. Oxygen and nitrogen are pro-carcinogens. Damage to DNA by reactive oxygen, chlorine and nitrogen species: measurement, mechanism and the effects of nutrition. Mutat Res. 1999;443:37–52.
Szabo KE, Gutowski N, Holley JE, et al. Redox control in human disease with a special emphasis on the peroxiredoxin-based antioxidant system. In: Jacob C, Winyard PG, editors. Redox signaling and regulation in biology and medicine. Weinheim: Wiley-VCH; 2009. p. 409.
den Hertog J. Protein tyrosine phosphatases as mediators of redox signaling. In: Jacob C, Winyard PG, editors. Redox signalling and regulation in biology and medicine. Weinheim: Wiley-VCH; 2009. p. 197.
Charlier E, Piette J, Gloire G. Redox regulation of apoptosis in immune cells. In: Jacob C, Winyard PG, editors. Redox signalling and regulation in biology and medicine. Weinheim: Wiley-VCH; 2009. p. 385.
Hansen RE, Winther JR. An introduction to methods for analyzing thiols and disulfides: reactions, reagents, and practical considerations. Anal Biochem. 2009;394:147–58.
Fox JB. Kinetics and mechanisms of the griess reaction. Anal Chem. 1979;51:1493–502.
Hamilton CJ, Saravanamuthu A, Eggleston IM, et al. Ellman’s-reagent-mediated regeneration of trypanothione in situ: substrate economical microplate and time-dependent inhibition assays for trypanothione reductase. Biochem J. 2003;369:529–37.
Woodward JJ. The effects of thiol reduction and oxidation on the inhibition of NMDA-stimulated neurotransmitter release by ethanol. Neuropharmacology. 1994;33:635–40.
Murrant CL, Reid MB. Detection of reactive oxygen and reactive nitrogen species in skeletal muscle. Microsc Res Tech. 2001;55:236–48.
Tarpey MM, Wink DA, Grisham MB. Methods for detection of reactive metabolites of oxygen and nitrogen: in vitro and in vivo considerations. Am J Physiol Regul Integr Comp Physiol. 2004;286:R431–44.
Li L, Chen CY, Chun HK, et al. A fluorometric assay to determine antioxidant activity of both hydrophilic and lipophilic components in plant foods. J Nutr Biochem. 2009;20:219–26.
Kalivendi SV, Kotamraju S, Zhao H, et al. Doxorubicin-induced apoptosis is associated with increased transcription of endothelial nitric-oxide synthase. Effect of antiapoptotic antioxidants and calcium. J Biol Chem. 2001;276:47266–76.
Boveris A, Chance B, Oshino N. The cellular production of hydrogen-peroxide. Biochem J. 1972;128(3):617–30.
Gough DR, Cotter TG. Hydrogen peroxide: a Jekyll and Hyde signalling molecule. Cell Death Dis. 2011;2:e213.
Thomas C, Mackey MM, Diaz AA, et al. Hydroxyl radical is produced via the Fenton reaction in submitochondrial particles under oxidative stress: implications for diseases associated with iron accumulation. Redox Rep. 2009;14:102–8.
Gutteridge JMC, Halliwell B. Free radicals and antioxidants in the year 2000—A historical look to the future. Ann N Y Acad Sci. 2000;899:136–47.
Green MJ, Hill HAO, Tew DG, et al. An opsonized electrode—The direct electrochemical detection of superoxide generated by human-neutrophils. FEBS Lett. 1984;170:69–72.
Hill HAO, Tew DG, Walton NJ. An opsonized microelectrode—Observation of the respiratory burst of a single human neutrophil. FEBS Lett. 1985;191:257–63.
Tammeveski K, Tenno TT, Mashirin AA, et al. Superoxide electrode based on covalently immobilized cytochrome c: modelling studies. Free Radic Biol Med. 1998;25:973–8.
Roberts JG, Hamilton KL, Sombers LA. Comparison of electrode materials for the detection of rapid hydrogen peroxide fluctuations using background-subtracted fast scan cyclic voltammetry. Analyst. 2011;136:3550–6.
Amatore C, Arbault S, Guille M, et al. Electrochemical monitoring of single cell secretion: vesicular exocytosis and oxidative stress. Chem Rev. 2008;108:2585–621.
Cho SH, Jang A, Bishop PL, et al. Kinetics determination of electrogenerated hydrogen peroxide (H2O2) using carbon fiber microelectrode in electroenzymatic degradation of phenolic compounds. J Hazard Mater. 2010;175:253–7.
Arbault S, Pantano P, Jankowski JA, et al. Monitoring an oxidative stress mechanism at a single human fibroblast. Anal Chem. 1995;67:3382–90.
Zhu AW, Liu Y, Rui Q, et al. Selective and sensitive determination of hydroxyl radicals generated from living cells through an electrochemical impedance method. Chem Commun. 2011;47:4279–81.
Ferrer-Sueta G, Radi R. Chemical biology of peroxynitrite: kinetics, diffusion, and radicals. ACS Chem Biol. 2009;4:161–77.
Katrlik J, Zalesakova P. Nitric oxide determination by amperometric carbon fiber microelectrode. Bioelectrochemistry. 2002;56:73–6.
Santos RM, Lourenco CF, Piedade AP, et al. A comparative study of carbon fiber-based microelectrodes for the measurement of nitric oxide in brain tissue. Biosens Bioelectron. 2008;24:704–9.
Borgmann S. Electrochemical quantification of reactive oxygen and nitrogen: challenges and opportunities. Anal Bioanal Chem. 2009;394:95–105.
Kubant R, Malinski C, Burewicz A, et al. Peroxynitrite/nitric oxide balance in ischemia/reperfusion injury-nanomedical approach. Electroanalysis. 2006;18:410–6.
Miserere S, Ledru S, Ruille N, et al. Biocompatible carbon-based screen-printed electrodes for the electrochemical detection of nitric oxide. Electrochem Commun. 2006;8:238–44.
Amatore C, Arbault S, Chen Y, et al. Electrochemical detection in a microfluidic device of oxidative stress generated by macrophage cells. Lab Chip. 2007;7:233–8.
Trouillon R, O’Hare D, Chang SI. An electrochemical functional assay for the sensing of nitric oxide release induced by angiogenic factors. BMB Rep. 2011;44:699–704.
Amatore C, Arbault S, Koh AC. Simultaneous detection of reactive oxygen and nitrogen species released by a single macrophage by triple potential-step chronoamperometry. Anal Chem. 2010;82:1411–9.
Jacob C, Ba LA. Open season for hunting and trapping post-translational cysteine modifications in proteins and enzymes. Chembiochem. 2011;12:841–4.
Hidalgo J, Aschner M, Zatta P, et al. Roles of the metallothionein family of proteins in the central nervous system. Brain Res Bull. 2001;55:133–45.
Ralph TR, Hitchman ML, Millington JP, et al. The electrochemistry of L-cystine and L-cysteine.1. Thermodynamic and kinetic-studies. J Electroanal Chem. 1994;375:1–15.
Ren XL, Bjornstedt M, Shen B, et al. Mutagenesis of structural half-cystine residues in human thioredoxin and effects on the regulation of activity by selenodiglutathione. Biochemistry. 1993;32:9701–8.
Jacob C, Anwar A, Burkholz T. Perspective on recent developments on sulfur-containing agents and hydrogen sulfide signaling. Planta Med. 2008;74:1580–92.
Schneider T, Ba LA, Khairan K, et al. Interactions of polysulfanes with components of red blood cells. MedChemComm. 2011;2:196–200.
Munday R, Munday JS. Comparative haemolytic activity of bis(phenylmethyl) disulphide, bis(phenylethyl) disulphide and bis(phenylpropyl) disulphide in rats. Food Chem Toxicol. 2003;41:1609–15.
Sarakbi M-B. Natural products and related compounds as promising antioxidants and antimicrobial agents. Saarbruecken: University of Saarland; 2009.
Andersson CM, Hallberg A, Linden M, et al. Antioxidant activity of some diarylselenides in biological-systems. Free Radic Biol Med. 1994;16:17–28.
Jacob C, Lancaster JR, Giles GI. Reactive sulphur species in oxidative signal transduction. Biochem Soc Trans. 2004;32:1015–7.
Giles NM, Gutowski NJ, Giles GI, et al. Redox catalysts as sensitisers towards oxidative stress. FEBS Lett. 2003;535:179–82.
Fry FH, Jacob C. Sensor/effector drug design with potential relevance to cancer. Curr Pharm Des. 2006;12:4479–99.
Jamier V, Ba LA, Jacob C. Selenium- and tellurium-containing multifunctional redox agents as biochemical redox modulators with selective cytotoxicity. Chemistry. 2010;16:10920–8.
Giles GI, Collins CA, Stone TW, et al. Electrochemical and in vitro evaluation of the redox-properties of kynurenine species. Biochem Biophys Res Commun. 2003;300:719–24.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Álvarez, E.D., Viswanathan, U.M., Burkholz, T., Khairan, K., Jacob, C. (2013). Bio-Electrochemistry and Chalcogens. In: Schlesinger, M. (eds) Applications of Electrochemistry in Medicine. Modern Aspects of Electrochemistry, vol 56. Springer, Boston, MA. https://doi.org/10.1007/978-1-4614-6148-7_7
Download citation
DOI: https://doi.org/10.1007/978-1-4614-6148-7_7
Published:
Publisher Name: Springer, Boston, MA
Print ISBN: 978-1-4614-6147-0
Online ISBN: 978-1-4614-6148-7
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)