
Abstract
A method is proposed for the rapid identification and determination of seven N-nitrosamines in food products by ultra-high-performance liquid chromatography in combination with high-resolution quadrupole-time-of-flight mass spectrometry. The ranges of detectable concentrations of N-nitrosamines are from 0.001–5 to 2–100 ng/mL for liquid (water, beer) and from 2–50 to 4–100 ng/g for solid (meat, fish, mussels, malt, grain, sausage, wieners, and bockwursts) products. The limits of detection are 0.0005–2 ng/mL, 2–5 ng/g for liquid and solid products, respectively. The recovery of analytes is from 62 to 105%. The matrix effect was estimated in determining N-nitrosamines in samples of different nature. The matrix effect is negligible (≤20%) in the determination of N-nitrosamines in water and is significant for beer, meat products, grain, malt, and fish (>20%). A procedure is proposed for the rapid identification and determination of N-nitrosamines by the standard addition method and using matrix calibration. The relative standard deviation of the results of analysis does not exceed 17%. The duration of sample identification is 40 min; the determination of the detected analytes takes 1–1.5 h.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Most N-nitrosamines (NA) have pronounced carcinogenic and mutagenic properties and are included in the lists of priority pollutants of the environment and food products in many countries. In accordance with the Technical Regulations of the Customs Union “On the safety of food products” (ТR CU 021/2011) the maximum permissible level (MPL) of nitrosamines in all types of fish products and marine mammals (including dried products) is 0.003 mg/kg; in canned meat from poultry meat with an addition of sodium nitrite, canned food from offal (including pate) it is 0.002 mg/kg; in brewing malt, 0.015 mg/kg; in raw fat, pork spit, and products from them, 0.002 mg/kg; and in beer, 0.003 mg/kg [1].
Unlike many other toxicants, residual quantities of which may be present in food products and cause significant harm to human health (mycotoxins, pesticides, and polycyclic aromatic hydrocarbons), the task of determining NA by its normative and methodological support has not been sufficiently developed for universal implementation and use in test laboratories conducting routine research on food safety indicators. Therefore, the development of new, more advanced procedures for determining NA is a very urgent task of the analytical chemistry of food raw materials and food products.
Currently, procedures for determining NA can be divided into two groups: the first group includes methods for determining NA derivatives, as a rule, by thin-layer and high-performance liquid chromatography; the second group includes procedures for determining unmodified NA by gas-liquid chromatography (GLC) methods (less commonly by HPLC).
The first group of methods, because of an increase in the number of sample preparation stages and stricter requirements for the purity of the final extract, is more laborious. Guidelines approved in the Russian Federation [2] are based on the separation of volatile NA by steam distillation or under vacuum, extraction of NA with dichloromethane from an aqueous distillate, concentration of the extract, denitrosation of NA with hydrogen bromide in acetic acid, alkylation of amines with 8-methoxy-5-quinolin sulfonyl aziridine, and separation and semi-quantitative determination of fluorescent 8-methoxy-5-[N-(2-N-diethylamino)]-quinolinesulfonamide derivatives in a thin layer of silica gel. HPLC methods for determining NA in food raw materials and food products with fluorimetric [3‒5] and UV detection [6] have been developed. A special feature of the procedures [3–5] is the high concentration sensitivity of detecting residual amounts of NA due to the nature of fluorimetric methods of analysis: the limits of detection reach 0.08–0.075 ng/g [3] and 0.01–0.07 ng/g [5]. To obtain derivatives of the corresponding amines (products of denitrosation of NA), 5-(dimethylamino)naphthalene-1-sulfochloride (dansyl chloride) is usually used [3, 4]. It should be noted that these works described the determination of a small number of NA, despite the fact that residual amounts of different types of NA may be contained in food products [7, 8].
The second group of methods is of priority, as it requires less manipulation with the test sample. Gas-liquid chromatography is increasingly used with thermoenergy, flame ionization (FID) [9, 10], and mass spectrometric (MS) detection for the identification and quantification of NA [7, 11–13]. The low cost of FID makes such devices more available to research laboratories; however the procedures are not sensitive enough for volatile low molecular weight NA.
Because of the complex composition of the matrix of food products and the low concentrations of NA in them, procedures for sample preconcentration and the purification of the obtained extract are necessary [7, 8]. Solid-phase (SPE) or liquid–liquid extraction (LLE), and solid-phase microextraction [7, 13, 14] are used for the extraction of NA from liquid samples (blood, water, etc.). N-nitrosamines are extracted from food samples mainly by steam distillation, and then LLE with slightly polar organic solvents (usually dichloromethane) and SPE are used [14]. Extraction with microwave heating in combination with dispersive liquid-liquid [11, 12] and solid-phase microextraction (carbon molecular sieves are used) [14] is also used to extract NA from food products. Such methods can significantly reduce the time of sample preparation; however, the weight of the sample (usually 1 g) subjected to microwave decomposition imposes additional requirements on the concentration coefficient of the sample, which should be sufficient to detect analytes at the level of MPL.
Of considerable interest is the determination of unmodified NA using ultra-high-performance liquid chromatography with MS detection (UHPLC–MS) [15, 16]. In these works, procedures for determining NA in drinking and waste waters using SPE for the preconcentration and purification of the sample are presented. The prospects for the development of such procedures are due to the peculiarities of the UHPLC–MS method, which include high concentration sensitivity, high selectivity for the determination of analytes of various classes, and low consumption of toxic solvents.
An analysis of the literature shows that at present there are no simple screening procedures that ensure the rapid identification of NA in food products. In addition, the existing procedures for determining NA are very time consuming, multistage, and often require the conversion of the target components to a form suitable for sensitive detection.
In this paper, we propose a simple procedure for the rapid identification and determination of N-nitrosamines in food products by UHPLC with detection by high-resolution quadrupole-time-of-flight MS by exact masses of protonated molecules.
EXPERIMENTAL
Equipment. An UltiMate 3000 ultra-high-performance liquid chromatograph (Thermo Scientific, United States) was used in combination with a maXis 4G quadrupole-time-of-flight mass spectrometry detector and electrospray ionization in an ionBooster device (Bruker Daltonics, Germany). The separation was carried out on columns 30 × 2.1 mm, 50 × 2.1 mm, 100 × 2.1 mm ACQUITY UPLC® BEN C18, 50 × 2.1 mm ACQUITY UPLC® Shield RP18, 50 × 2.1 mm ACQUITY UPLC® Phenyl (1.7 µm) (Waters, United States ), 50 × 2.1 mm Acclaim™ 120 C18 (2.2 and 3.0 μm) (Thermo Scientific, United States) in the gradient elution mode.
Sartorius TE214S I analytical balances of a special accuracy class (Sartorius, Germany), an MPW-260R centrifuge (MPW Med. Instruments, Poland), a Grindomiks GM200 knife mill (Retch, Germany), a Buchi rotary evaporator (Germany), and Corning® membrane filters, 0.20 μm (Germany) were used.
Reagents. A standard solution (2 mg/mL) of a mixture of nitrosamines in dichloromethane EPA 521 Nitrosamine Mix (Supelco Analytical, United States), consisting of N-nitrosomethylethylamine (NMEA), N-nitrosodibutylamine (NDBA), N-nitrosodipropylamine (NDPA), N-nitrosodiethylamine, (NDEA), N‑nitrosodimethylamine (NDMA), N-nitrosopiperidine (NPP), and N-nitrosopyrrolidine (NPR) was used.
CCl4, C2H2Cl4, CHCl3 (Sigma-Aldrich, United States); CH2Cl2 (Scharlab S.L., Spain); and CH3OH (Fisher Chemical, Germany) were used.
Identification and determination. The software product DataAnalysis-4.1, TargetAnalysis (Bruker Daltonics, Germany) was used to identify nitrosamines by the obtained chromatograms, and IsotopePattern (Bruker Daltonics, Germany) was used to compile a picture of the isotopic distribution of analytes. The unknown concentration of the analyte in the sample was determined by the matrix calibration of each of the test products or was calculated by the standard addition method by the equation:
where cadd is the concentration of an additive in the sample, ng/mL (g); Sx, Sx + add are peak areas m/z in the test solution and in the solution with an addition of an analyte, respectively.
Evaluation of matrix effect. To assess the matrix effect (ME), the areas of the chromatographic peaks of analytes with the concentration 5 and 50 ng/g were used; they were obtained under the conditions of analysis of an extract from the analyzed productthat did not contain the compounds under study and deionized water. The ME was calculated by the equation:
where S, S0 are the areas of chromatographic peaks of analytes obtained for extracts from the analyzed product and deionized water, respectively.
The degree of recovery was set at a concentration level for water of 0.001 and 1 ng/mL, for beer 3 and 50 ng/mL, for meat, fish, mussels, malt, grains, sausage, wieners, and bockwursts 5 and 50 ng/g. The degrees of recovery (R) were calculated by the equation:
where X is the found mass concentration of the analyte in the sample, ng/g (mL); X0 is the true value of the mass concentration of the analyte in the sample, ng/g (mL).
Conditions for chromatographic separation and detection. The mobile phase consisted of 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B). Gradient elution was performed: 0 min ‒ 5% B, 0.5 min ‒ 5% B, 2 min ‒ 50% B, 5 min ‒ 100% B, 6 min ‒ 5% B, and 8 min ‒ 5% B. The consumption of the mobile phase was 0.4 mL/min The optimum temperature of the chromatographic column was 50°C, the volume of the injected sample was 50 mL. Temperature of the thermostat of the automatic dispenser was 10°C.
Electrospray ionization was used in the ionBooster device (Bruker Daltonics, Germany). The following optimal values of parameters were found: voltage on the capillary shield 400, 1000 V on the capillary, nitrogen atomizing gas pressure 4.76 atm, nitrogen dehydrator gas flow rate 6 L/min, nitrogen dehydrating gas temperature 200°C, nitrogen gas vaporizer flow rate 250 L/h, the temperature of the nitrogen gas vaporizer 250°C.
The range of recorded ion masses was 50–300 Da. For calibration, a sodium formate solution of 10 mM in a water–isopropanol (1 : 1) mixture was used in the chromatography range 7.5–8 min.
Sample preparation.Beer. A 5-mL portion of beer was placed in a 15-mL centrifuge tube, 5 mL of dichloromethane (DCM) was added, the mixture was shaken manually for 5 min, centrifuged for 5 min at 2700 rpm, and the extract was transferred to an evaporation flask. This operation was repeated twice. The combined extracts were evaporated on a rotary evaporator at 10–15°С to dryness. A 50-mL portion of acetonitrile and 950 mL of water were added to the residue and mixed thoroughly. The obtained solution was filtered through a membrane filter into a microvial and chromatographed.
Water. A 100-mL portion of test water was placed in a 200-mL separatory funnel, 10 mL of DCM was added, and extraction of performed for 5 min; the extract was transferred to an evaporation flask. This operation was repeated twice. The combined extracts were evaporated on a rotary evaporator at 10–15°С to dryness. A 50-μL portion of acetonitrile and 950 μL of water were added to the residue and mixed thoroughly. The obtained solution was filtered through a membrane filter into a microvial and chromatographed.
Sausage, wieners, bockwursts, mussels, meat, fish, grain, and malt. A portion of a crushed test sample weighing 2.0 g was placed in a 50-mL centrifuge tube, 10 mL of DCM was added; the mixture was kept in an ultrasonic bath for 15 min and then centrifuged for 5 min at 2700 rpm. This operation was repeated twice with combining the extracts in a flask for evaporation. The extract was evaporated on a rotary evaporator at 10–15°С to dryness. A 50-μL of acetonitrile and 950 μL of water were added to the residue and mixed thoroughly. The obtained solution was filtered through a membrane filter into a microvial and chromatographed.
RESULTS AND DISCUSSION
Optimization of analysis conditions.Column selection. The properties of the following columns were estimated: 30 × 2.1 mm, 50 × 2.1 mm, 100 × 2.1 mm ACQUITY UPLC® BEN C18, 50 × 2.1 mm ACQUITY UPLC® Shield RP18, 50 × 2.1 mm ACQUITY UPLC® Phenyl (all with a grain diameter of 1.7 μm), and 50 × 2.1 mm Acclaim™ 120 C18, grain diameter of 2.2 μm and 3.0 μm (Thermo Scientific, United States). It was found that the use of any of the above columns does not result in a significant change in the resolution and efficiency of the separation of nitrosamines. The duration of analysis (separation + conditioning) on the columns 5 cm long was 10 min and on a 3-cm column, 8 min. The column 30 × 2.1 mm, ACQUITY UPLC® BEN C18 was selected for the further studied.
Choice of mobile phase. Water, acetonitrile, and methanol without additives and with an addition of formic acid and ammonium formate were used. The following version was chosen the optimal: A ‒ 0.1% aqueous solution of formic acid, B ‒ 0.1% acetonitrile solution of formic acid. The additives of ammonium formate and the use of methanol as the mobile phase did not result in a significant increase in the chromatographic peaks of the analytes.
The flow rate of the mobile phase and the gradient were selected in such a way that the analytes under study were distributed over the retention time from 0.5 to 6 min. The best results were obtained with the flow rate of the mobile phase 0.4 mL/min and the following gradient: 0 min ‒ 5% B, 0.5 min ‒ 5% B, 2 min ‒ 50% B, 5 min ‒ 100% B, 6 min ‒ 5% B, and 8 min ‒ 5% B. As a result, separation time was 6 min, and after conditioning the column for 2 min, we transferred to the next analysis. The total duration of the chromatography of one sample was 8 min.
Choice of temperature and sample volume. The temperature of the thermostat of the chromatography column (30, 40, 50, and 60°C) and the volume of the injected sample (5, 10, 20, 50, 60, and 100 μL) were varied. It was found that, to achieve a high determination sensitivity, the chromatography column temperature should be 50°С (at that the pressure of the mobile phase on the chromatography column was 120–130 bar) and the volume of sample introduced into the dispenser of 50 μL (injection of more than 50 μL resulted in the disruption of the symmetry of the chromatographic peak).
All studied NA under the conditions of electrospray ionization form protonated [M + H]+ molecules (Table 1). To a large extent, the stability of the formation of protonated forms is due to the structure of NA, namely, the presence of nitrogen atoms with lone pairs of electrons, mainly in the amine fragment.
The error in determining the masses of ions did not exceed ± 1 ppm (for NDMA ± 6 ppm) (n = 3). Tables 2–5 present the limits of detection (LOD) and limits of quantification (LOQ) for matrices of different nature, set by the signal-to-noise ratios of 3 and 10, respectively. The limits of detection ranged from 0.0005 to 2 ng/mL for liquid (water, beer) and from 2 to 5 ng/g for solid (meat, fish, mussels, malt, grain, sausage, wieners, and bockwursts) products, which ensures the determination of NA at the MPL level [1].
Sample preparation. Table 1 summarizes data on the instrumental sensitivity of the determination of NA by the proposed method (analysis of standard solutions of NA). One can see that, taking into account the MPLs for various matrices, sample preconcentration is required by selecting the volume and mass of the sample, and the concentration factor. In this regard, for beer (Table 2), the sample selected volume was 5 mL and double extraction with 5 mL of DCM each; for water the sample volume was 100 mL and double extraction with 10 mL of DCM each (Table 3). In these cases, the maximum values of the recovery of analytes (62–105%) were achieved; they are listed in Tables 2, 3. For solid products, double intensification of extraction was necessary by sonication for 15 min (Tables 4, 5).
Evaluation of the matrix effect. The matrix effect is due to the influence of the co-extracted components present in the extract on the ionization of target analytes. Under electrospray ionization conditions, they can both enhance (+) and reduce (–) the intensity of the analyte signal. Tables 2–5 summarize the ME values for various samples. According to the data [17], the ME can be neglected when its values are in the range of ±20%. In this connection, the ME can be neglected for water (Table 3). However, for the other studied products, ME is significant (Tables 4, 5). There are several ways of reducing ME in analyzing samples of biological origin: reducing the volume of the injected sample, dilution of the final extract with water, use of deuterated internal standard, use of the standard additive method, and use of matrix calibration.
We have chosen the last two methods as the simplest and most available. For matrix calibration, the following analyte concentrations were used: 0.001, 0.005, 0.01, 0.05, 0.1, 0.5, 1.0, 2.0, and 5.0 ng/mL for water; 2.0, 3.0, 5.0, 10.0, 20.0, 50.0, 80.0, and 100.0 ng/g for meat and fish products, grain and malt; 0.02, 0.1, 0.5, 1.0, 2.0, 5.0, 10.0, 20.0, 40.0, and 100.0 ng/g for beer. The linearity correlation coefficients of the calibration curves (r) were at least 0.99 (Tables 2–5).
The standard addition method turned out to be a simpler and more effective method for reducing ME. In this case, the effects of matrix components on the analyte and the introduced additive were identical, which helped to prevent an error in estimating the concentration of the compound to be determined in the analyzed extract.
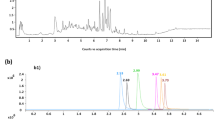
Analysis of real samples. Samples for the study were purchased at local supermarkets. Water for analysis was collected in the reservoirs of the Vladimir region, or from the central water supply of the city of Vladimir. As an example, Fig. 1 presents mass chromatograms of NA obtained in determining them in a sample of beer that does not contain detectable compounds with an addition of 20 ng/mL of each component. Table 6 presents the results of the determination of NA in samples of various origin using the standard addition method. The relative standard deviation of the results of analysis did not exceed 17%. Figure 2 shows mass chromatograms of NDBA, NDEA, and NDPA, which were found in the analysis of the beer sample at the level of 1.7, 1.0, and 0.8 ng/mL, respectively. The duration of sample identification was 40 min; the determination of analytes detected took 1–1.5 h.

Mass chromatograms of N-nitrosamines (addition of 20 ng/mL) for an extract of beer that does not contain N-nitrosamines.

Mass chromatograms of beer extract. Detected N-nitrosamines: (1) N-nitrosodiethylamine, (2) N-nitrosodipropylamine, and (3) N-nitrosodibutylamine.
REFERENCES
TR (Technical Regulations) TS 021/2011: Technical Regulations of the Customs Union “On Food Safety,” Minsk: BelGISS, 2015.
MUK (Methodological Guidelines) 4.4.1.011-93: Determination of Volatile N-Nitrosamines in Food Raw Materials and Foods. Guidelines on Methods of Control, Moscow: Goskomsanepidnadzor RF, 1993.
Cárdenes, L., Ayala, J.H., González, V., and Afonso, A.M., J. Chromatogr. A, 2002, vol. 946, p. 133.
Komarova, N.V. and Velikanov, A.A., J. Anal. Chem., 2001, vol. 56, no. 4, p. 359.)
Lu, S., Wu, D., Li, G., Lv, Z., Gong, P., Xia, L., Sun, Z., Chen, G., Chen, X., You, J., and Wu, Y., Food Chem., 2017, vol. 234, p. 408.
Li, W., Chen, N., Zhao, Y., Guo, W., Muhammd, N., Zhu, Y., and Huang, Z., Anal. Methods, 2018, vol. 10, no. 15, p. 1733.
Crews, C., Qual. Assur. Saf. Crops Foods, 2010, vol. 2, no. 1, p. 2.
Herrmann, S.S., Duedahl-Olesen, L., and Granby, K., J. Chromatogr. A, 2014, vol. 1330, p. 20.
Drabik-Markiewicz, G., Dejaegher, B., De Mey, E., Kowalska, T., Paelinck, H., and Vander Heyden, Y., Food Chem., 2011, vol. 126, p. 1539.
Al-Kaseem, M., Al-Assaf, Z., and Karabeet, F., Pharmacol. Pharm., 2014, vol. 5, p. 195.
Amelin, V.G. and Lavrukhin, D.K., J. Anal. Chem., 2016, vol. 71, no. 4, p. 359.
Kamankesh, M., Ghasemzadeh-Mohammadi, V., and Mohammadi, A., Eur. Food. Res. Technol., 2015, vol. 240, p. 441.
Yuan, Y., Meng, W., Yutian, M., Fang, C., and Xiaosong, H., Int. J. Food Prop., 2015, vol. 18, no. 6, p. 1181. https://doi.org/10.1080/10942912.2014.898652
Huang, M.-C., Chen, H.-C., Fu, S.-C., and Ding, W.-H., Food Chem., 2013, vol. 138, p. 227.
Qian, Y., Wu, M., Wang, W., Chen, B., Zheng, H., Krasner, S.W., Hrudey, S.E., and Li, X.-F., Anal. Chem., 2015, vol. 87, p. 1330.
Ngongang, A.D., Duy, S.V., and Sauve, S., Anal. Methods, 2015, vol. 7, p. 5748.
Ferrer, C., Lozano, A., Agüera, A., Jiménes Girón, A., and Fernández-Alba, A.R., J. Chromatogr. A, 2011, vol. 1218, p. 7634.
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated by V. Kudrinskaya
Rights and permissions
About this article

Cite this article
Amelin, V.G., Bol’shakov, D.S. Rapid Identification and Determination of N-Nitrosamines in Food Products by Ultra-High-Performance Liquid Chromatography–High Resolution Quadrupole-Time-of-Flight Mass Spectrometry by Exact Masses of Protonated Molecules. J Anal Chem 74 (Suppl 1), 39–46 (2019). https://doi.org/10.1134/S1061934819070104
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1061934819070104