
Abstract
Theoretical aspects of low-carbon steel corrosion in H3PO4 solutions containing FePO4 are considered. In the system under study, reactions of iron with the acid solution and Fe(III) salt are thermodynamically allowed. The oxidizing power of this medium, characterized by the Fe(III)/Fe(II) couple redox potential, is mainly determined by its anionic composition. Phosphate anions of a corrosive medium bind Fe(III) cations into complex compounds, reducing their oxidizing ability. In H3PO4 solutions containing FePO4 and Fe3(PO4)2, the dependence of the system’s redox potential on the Fe(III) and Fe(II) cation relative content is poorly described by the Nernst equation, which is due to the nonequivalent complex formation of these cations with phosphate anions. Analysis of the effect of the studied media convection on the low-carbon steel electrode reactions allowed revealing some of their features. In a FePO4-containing H3PO4 solution, kinetically controlled partial reactions of iron anodic ionization and H+ cathodic reduction, as well as diffusion-controlled Fe(III) cation cathodic reduction, occur on the steel. The FePO4 accelerating effect on the steel corrosion in H3PO4 solution is due only to the Fe(III) reduction but does not affect the H+ reduction and the iron ionization. The value of the Fe(III)-cation diffusion coefficient in the studied corrosive medium was experimentally determined from the data of cyclic voltammetry of the Pt electrode therein and the results of the studying of the cathodic reaction of a steel disk electrode at different rotation velocities. The data on the low-carbon steel corrosion in the flow of the studied media, obtained from the metal samples mass loss, are in full agreement with the results of the study of the electrode partial reactions. An accelerating effect of FePO4 on the steel corrosion in H3PO4 solutions is observed. In this environment, steel corrosion is determined by the convective factor, which is typical of processes with diffusion control. The empirical dependence of the steel corrosion rate on the medium flow intensity is described by the linear dependence k = kst + λw1/2, where kst is the steel corrosion rate in a static medium, w is the rotation velocity of the propeller stirrer that creates the medium flow, λ is the empirical coefficient.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
INTRODUCTION
The phosphoric acid solutions are a perspective alternative to the hydrochloric and sulfuric acid solutions that are used in the iron and steel work and housing and utilities sector for the steel work and production equipment cleaning from heating furnace cinder, the corrosion products, and mineral deposits. An important technological advantage of the H3PO4 solutions, as compared with the hydrochloric and sulfuric media, is the high rate of dissolution of the iron oxides (FeO, Fe3O4, Fe2O3) forming the heating furnace cinder [1]. During operation, the H3PO4 solutions accumulate Fe(III) phosphates, mainly due to their interaction with the heating furnace cinder and corrosion products. The latter change significantly the media properties and their aggressiveness toward their contacting steels. The Fe(III) phosphate is water-insoluble. The Fe(III) phosphate solubility in H3PO4 solutions is the result of its chemical interaction with the acid, which leads to the formation of a mixture of acid phosphates of complex composition [2]. In this work, we shall formally regard such systems as a FePO4-containing H3PO4 solution.
We see it fit revealing special features of the low-carbon steel corrosion mechanism in the H3PO4 solutions containing the Fe(III) phosphate. To better understand the processes occurring in the steel/Fe(III)-phosphate-containing H3PO4-solution corrosion systems, one must analyze some thermodynamic and kinetic characteristics of both the corrosive medium per se (the Fe(III)-salt-containing phosphoric acid solution) and the corrosion system. Indispensable is to evaluate the aggressive-medium convection effect on both the steel corrosion integrally and its separate stages. The studying of the effect of the aggressive medium hydrodynamic parameters on the steel corrosion is of importance both in theoretical and in practical respects. Under production-line conditions, the exploiting of acidic solutions often occurs in the liquid streams or is accompanied with a significant natural convection as a result of the hydrogen gas evolution.
To prognose thermodynamic possibility of the metal corrosion in aqueous media and determining the reaction products, the Pourbaix diagrams (E–pH) used to be applied [3–5]. A diagram [6] form has been chosen that described equilibria momentarily occurring in the Fe–H2O system when iron hydroxides unstable with respect to the oxide phases are formed therein (Fig. 1). This approach reflects more accurately the equilibria that can establish in corrosion media. When the acidity of an aqueous corrosion medium is lowered, the Fe cations in the first instance will form thermodynamically unstable hydroxide phases, rather than the iron oxides. The iron oxide phase formation from hydroxides is a lengthy process.

A fragment of E vs. pH diagram of the Fe-metal and Fe(III)-cation stability field in water at 25°С and 101.3 kPa of full pressure [6]: (1) the Fe-metal stability field boundary, (2, 3) the Fe(III)-cation stability field boundaries, (4, 5) the water stability limit lines. The solid phases are only Fe, Fe(OH)2, and Fe(OH)3. The stability fields are given only for the cases log aFe(III) = log aFe(II); they correspond to the values of –6, –4, –2, and 0.
The iron metal and Fe(III) cation stability fields in this system are spaced-apart. This points to the impossibility of coexistence of these components under equilibrium conditions in the corrosion system. In acidic medium, the iron-metal limits of stability in the E–pH diagram are determined by the curve 1 that corresponds to the following equilibrium:
whereas the Fe(III) salt solubility, by the curve 2:
where \(E_{{{{{\text{Fe(III)}}} \mathord{\left/ {\vphantom {{{\text{Fe(III)}}} {{\text{Fe(II)}}}}} \right. \kern-0em} {{\text{Fe(II)}}}}}}^{^\circ }\) is the Fe(III)/Fe(II) redox-couple standard electrode potential, whose value is 0.771 V at 25°С, aFe(III) and aFe(II) is the Fe(III) and Fe(II) active concentrations in the solution. At the equality of aFe(III) and aFe(II), the value of the Fe(III)/Fe(II) redox-couple potential corresponds to its \(E_{{{{{\text{Fe(III)}}} \mathord{\left/ {\vphantom {{{\text{Fe(III)}}} {{\text{Fe(II)}}}}} \right. \kern-0em} {{\text{Fe(II)}}}}}}^{^\circ },\) which is shown in Fig. 1. Under conditions of real corrosion process in discussed aggressive media, the aFe(III) and aFe(II) ratio can be different, which affects the system’s redox-potential significantly. For example, when the aFe(III)-to-aFe(II) ratio is 9, the EFe(III)/Fe(II) value comes to 0.827 V. At aFe(III)/aFe(II) = 99 we have EFe(III)/Fe(II) = 0.889 V. By contrast, at the aFe(III)/aFe(II) = 99–1 the value EFe(III)/Fe(II) = 0.653 V. Thus, the oxidizing ability of the discussed system depends significantly on the Fe(III) and Fe(II) soluble salt concentrations ratio therein. The increase in the Fe(III) relative content can lead to a significant increase of the system’s redox-potential.
Also, the stability limits in the discussed system of the Fe(III) salts are determined by the vertical part of line 3, which characterizes the process of the Fe(III) soluble compounds’ transition to insoluble form, thanks to lowering of the medium acidity:
Thus, in the Fe(III)-salt-containing solutions of acids the iron and steel corrosion occurs in accordance with the following equation:
Concurrently, the process
is allowed thermodynamically because the water stability lower limit (curve 4)
lies at higher potentials than the equilibrium (1).
The analysis of the Pourbaix diagrams allows predicting the mediated pathway of the Fe(III) salts accumulation in corrosion medium as a result of Fe(II) salts oxidation in the solution with ambient oxygen. The water stability upper limit (curve 5)
lies at the potentials much more positive than those of the transition (2) even in the case when the oxygen partial pressure р(О2) = 0.2 atm (which is characteristic of air). This path for the Fe(III) salt accumulate in the acidic solution is discussed in works [7, 8].
The analysis of the Pourbaix diagram of the Fe–H2O system allowed obtaining important information concerning the studied case of steel corrosion. However, this information is to a great extent formalized because in the discussed system the effect of the anions present in aqueous medium on thermodynamic characteristics is not taken into consideration [9]; the latter fact should be investigated further.
In addition to thermodynamic aspects of the low-carbon steel corrosion in the Fe(III)-salt-containing acidic solutions, it is important to consider specific kinetic features of the process. The low-carbon steel corrosion in inorganic acid can be described in simplified form by the summary reaction (5), which is the result of a predominant passing of the following partial reactions [10]:
the iron anodic dissolution:
and the hydrogen cathodic evolution:
The reaction (9) [10] includes both the H+ delivering from the acid bulk to the metal surface
and the charge transfer stage (the Volmer reaction)
It is followed by the chemical reaction stage (the Tafel process)
or the electrochemical recombination (the Heyrovsky process)
The stages (11) and (12) in aggregate is the Volmer–Tafel mechanism; the stages (11) and (13), the Volmer–Heyrovsky mechanism. It is recognized that both mechanisms are realized at the steel surfaces during the hydrogen evolution [11].
The iron anodic ionization mechanism suggested by Heusler [10] believed to have the FeOHad compound formation in the course of the Fe crystal lattice atom reaction with the adsorbed OH− anions:
This compound catalyzed the further reaction of the Fe(II) ion transfer through the electrical double layer:
In its turn, the FeOH+ compound decomposed slowly:
By contrast, Bockris [12] regarded FeOH as an intermediate product in the stagewise passing of the reaction:
The iron dissolution in phosphate solutions was shown [13] to involve OH– and \({{{\text{H}}}_{{\text{2}}}}{\text{PO}}_{4}^{ - },\) at that, the \({{{\text{H}}}_{{\text{2}}}}{\text{PO}}_{4}^{ - }\) ions participated in the anodic process only at рН > 4. Later, in solutions with larger phosphate-anion summary content, their participation in the anodic reaction at the Armco iron was observed at lower pH values [14]:
At low-carbon steels, sometimes, another mechanism can be realized:
In solutions of acids containing such a strong oxidant as the Fe(III) salt, the process (4) can be realized in parallel; it includes an anodic [equation (8)] and a cathodic
reactions [15, 16]. The cathodic reaction at the low-carbon steel in these media is comprised of the parallel and independent processes of hydrogen evolution under kinetic control and the Fe(III)-cations reduction to Fe(II), passing under diffusion control.
The above-discussed mechanisms of hydrogen cathodic evolution and iron anodic ionization are the common notion, therefore, their refining is out of scope of this work. By contrast, the involvements of the Fe(III) salts dissolved in acids in the low-carbon steel corrosion is disregarded in literature. Because the steel corrosion in these media is controlled by diffusion at one of its stages, of primary importance is the taking into consideration f the convective factor that can contribute markedly to the metal disruption.
EXPERIMENTAL
In the solutions’ preparation, we used H3PO4 (reagent grade) and distilled water. The H3PO4 solutions containing the Fe(III) phosphate were obtained in the reaction of excess H3PO4 with Fe(OH)3 deposited from FeCl3 solution under the action of NaOH (reagent grade). To prepare the Fe(III) chloride solution, FeCl3⋅6H2O (chemically pure) was used. Because of technical difficulties in the preparation of H3PO4 solutions containing the Fe(II) phosphates, which can be used for potentiometric measurements, they were replaced by solutions containing equivalent FeSO4 concentration. With this approach, sometimes, the \({\text{SO}}_{4}^{{2 - }}\) and \({\text{PO}}_{4}^{{3 - }}\) content unaccounted for was maximally 4.8% each from the summary anion concentration in the solution, which allowed neglecting this factor in further discussion.
The potentiometric measurements in 2 M H3PO4 solution containingй 0.1 M Fe(III) + Fe(II) were carried out in thermostatted electrochemical glass cell with reserved zone for saturated silver/silver chloride reference electrode. The working electrode was made of smooth platinum (1.5 сm2); the auxiliary electrode was a saturated silver/silver chloride electrode. The potential difference between the working electrode and reference electrode was controlled using a PI-50 potentiostat.
The platinum electrode cyclic voltammetry in the studied media was applied using an EL-02.061 potentiostat in a thermostatted three-electrode glass cell with separated electrode compartments. The working electrode was platinum wire (S = 15.9 mm2); the reference electrode, silver/silver chloride electrode; the auxiliary electrode, platinum plate (S = 1.5 сm2). The working Pt-electrode potential was scanned from 1.4 to 0.0 V, then, from 0.0 to 1.4 V; the potential scanning rate was 0.05, 0.10, and 0.20 V/s.
The presence of an oxidant, a dissolved ambient oxygen, in the studied aqueous media can affect the results of potentiometric and cyclic-voltammetry studies carried out therein. To remove the dissolved oxygen from the studied aqueous media, they were pre-deaerated for 30 min with argon gas (reagent grade). The mean gas delivery rate was 1 mL/s. The electrochemical measurements were carried out in a static medium upon the stopping of the argon gas bubbling through the studied solution. Prior to the performing of the experiment, the working Pt-electrode was degreased with acetone, then kept in the concentrated HNO3 for 3 min and washed with distilled water.
Electrochemical measurements at the St3 low-carbon steel (the composition, wt %: С, 0.14–0.22; P, 0.04; Si, 0.15–0.33; Mn, 0.40–0.65; S, 0.05; Cr, 0.3; Ni, 0.3; N, 0.008; Cu, 0.3; As, 0.08; the rest, Fe) were carried out using a rotating disc electrode (n = 460 rpm) in the 2 М H3PO4 solution at t = 25°С. The steel potential was measured against silver/silver chloride electrode. The steel electrode was ground with emery paper (М20) and degreased with acetone. Polarization curves were taken using an EL-02.061 potentiostat at a potential scanning rate of 0.0005 V/s. Prior to the applying the polarization, the electrode was kept in the studied solution for 30 min, to set the free corrosion potential Ecor, then the anodic and cathodic polarization curves were taken. Upon their taking, the dependence of cathodic current at Е = ‒0.30 V on the electrode rotation velocity (n = 0, 460, 780, 1090, and 1400 rpm) was studied. In the case of the steel corrosion in the Fe(III)-salt-containing H3PO4 solutions, the cathodic process includes the reaction (9). Its character can depend on the hydrogen gas pressure in the system. To obtain stable results of electrochemical measurements, the dissolved oxygen gas was removed from the studied media by their deaeration with gaseous hydrogen, rather than with argon. This allowed performing electrochemical measurements at a constant hydrogen gas pressure in the system. The solutions were deaerated for 30 min prior to the beginning of the studies. The hydrogen was obtained in electrolyzer from NaOH solution. The mean gas flow rate was 1 mL/s. During the performing of electrochemical measurements, no hydrogen bubbling through solution was carried out.
The Fe(III) phosphate effect on the electrode reactions was evaluated by the value of the acceleration coefficient
where i0 and iFe(III) is the current densities in the supporting solution and in the solution added with the Fe(III) phosphate.
The lowest of the above-given electrode rotation velocity values (460 rpm) was taken as a basic rotation velocity of the steel disc electrode in the electrochemical studies. At this rotation velocity we obtained the closest values of the kinetic and diffusion current densities characterizing the cathodic reaction realized at the electrode under the experimental conditions. This holds out the promise of the most correct evaluating of the effect of the Fe(III) phosphate additives on the steel partial cathodic reactions.
The electrode potentials are given in the standard hydrogen scale.
The corrosion rate of the 08PS steel (the composition, wt %: С, 0.08; Mn, 0.5; Si, 0.11; P, 0.035; S, 0.04; Cr, 0.1; Ni, 0.25; Cu, 0.25; As, 0.08; the rest, Fe) in 2 М H3PO4 solution at a temperature of 20 ± 2°С was determined by the mass loss (≥5 per point) of samples sized 50 mm × 20 mm × 0.5 mm, calculated on the basis of 50 mL of the acid solution per sample:
here Δm is the change of the sample mass, g; S is the sample surface area, m2; τ is the corrosion test time, h; the test durability is 2 h. The studies were carried out both in static and dynamic corrosion medium at the magnetic stirrer rotation velocity w = 250, 420, 750, and 1080 rpm. Prior to the experiment, the samples were cleaned out at a grinding wheel (ISO 9001, the abrasive grit 60) and degreased by acetone.
The effect of the presence of the Fe(III) phosphate in the acid and the corrosion medium flow mode on the steel corrosion rate were estimated by the corrosion loss increments
And the corrosion acceleration coefficient increments
where kFe(III) and k0 is the rates of the steel corrosion in the acid solution in the presence and in the absence of the Fe(III) salt; kdyn and kst is the corrosion rates in the dynamic and static media.
RESULTS AND DISCUSSION
The first step in the understanding of the processes occurring in the aggressive medium/metal system is the study of the aggressive medium thermodynamic and kinetic characteristics. Important information on the effect of the aqueous corrosion medium anionic composition in the presence of oxidative metal cations therein on some its thermodynamic characteristics can be obtained from potentiometric studies [9, 17]. The redox-potential of the discussed system is determined by the equilibrium reaction (2); it can be described by the Nernst equation:
where R is the universal gas constant; T is the absolute temperature; z is the number of electrons involved in the redox-process [z = 1 for the Fe(III)/Fe(II) couple]; F is the Faraday constant. It is challenging to use the Nernst equation in its present form, the more so, at high electrolyte concentrations, because the activity of any type of potential-determining ions in solutions is by no means connected with their concentration by a simple interrelation. The non-complexed iron cation active concentrations in the solution can be found by using activity coefficients calculated by the Debye–Hueckel equation and the equilibrium constants characterizing the Fe complex compounds. It is highly unlikely that the discussed problem can be solved because it is very complicated and its chemical composition is rather indeterminate.
From a practical standpoint, the real potential is more suitable to describe processes occurring in the Fe-cation-containing acid solutions. It can be interpreted as a potential of redox-system settled in a particular solution under the equality of the initial concentrations of the oxidized and reduced forms of the potential-determining ions uncorrected for the complex-formation, hydrolysis, and like processes [9]. For the studied system, its real potential is most suitable for the qualitative interpreting of experimental data connected with the potential-determining ion complex-formation.
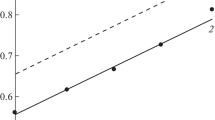
To better understand the processes involving the potential-determining ion in the studied system, we determined its real potential for the 2 M H3PO4 + 0.05 M Fe(III) + 0.05 M Fe(II) solution. The similar parameter was determined for 2 M H3PO4 solution containing 0.1 М Fe(III) + Fe(II), at СFe(III)/СFe(II) = 99, 9, 9–1, and 99–1 (Fig. 2). When no deviation from the ideal state of the 2 M H3PO4 + 0.05 M Fe(III) + 0.05 M Fe(II) solution occurs, the real potential must be equal to \(E_{{{{{\text{Fe(III)}}} \mathord{\left/ {\vphantom {{{\text{Fe(III)}}} {{\text{Fe(II)}}}}} \right. \kern-0em} {{\text{Fe(II)}}}}}}^{^\circ }.\) Also, the dependence of the system’s potential on the СFe(III)/СFe(II) ratio is described by the Nernst equation (Fig. 2, curve 1). The experimentally determined values of the redox-potentials of the Pt-electrode in the Fe(III)- and Fe(II)-salt containing 2 M H3PO4 solution (Fig. 2, points) lie much lower than those expected for a system non-deviated from its ideal state. The observed effect is a result of the potential-determining ion interaction with the corrosion medium components. The Fe(III)-cations are bound with the phosphate-anions to complex compounds whose oxidative ability is lower than that of Fe(III) hydrated ions. The formation of the Fe(III) phosphate complexes in the phosphate media is evidenced by their stability constants (Table 1). The Fe(III)-phosphate-complexes’ composition and structure in the H3PO4 solutions is discussed in works [19, 20].

The Pt-electrode potentials in argon-deaerated 2 М H3PO4 solution containing 0.10 М Fe(III) + Fe(II) as a function of the Fe(III)- and Fe(II)-ion concentration ratio. Here (1) the dependence plotted based on the hypothetic suggestion on the absence of the concerned system deviation from ideal state, (2) the experimental dependence. The theoretical dependence is obtained by using the Nernst equation.
There is still an open question, whether it is to be expected that the Nernst equation is formally fulfilled in the studied systems when the linear dependence of the system redox-potential on the logarithm of the ratio of the potential-determining particles’ active concentrations has been retained. To solve the problem with due allowances made for the Nernst equation we plotted a model dependence crossing the experimentally determined value of the Pt-electrode potential obtained at СFe(III) = СFe(II) = 0.05 М (Fig. 2, curve 2). The comparison of experimental data with the model (curve 2) showed the experimental points approaching the theoretical line at СFe(III) > СFe(II). By contrast, at СFe(III) < СFe(II) the experimental points lied at larger Es as compared with the theoretical line, which is most pronounced at СFe(III)/СFe(II) = 99–1. The character of this phenomenon, to our view, is mainly based on the non-equivalent complex formation of the Fe(III-) and Fe(II)-cations with phosphate-anions. In spite of the lesser stability of the Fe(II) complexes with phosphate-anions (Table 1), at larger СFe(II) in H3PO4 solutions the Fe(II)-cations are mainly bound into the complexes, which is beneficial in the increase of the potential.
The cyclic voltammetry is a more informative method for the studying of the properties of the discussed corrosion medium. Voltammograms of a Pt-electrode in FePO4-containing 2 М H3PO4 solution have two peaks (Fig. 3, Table 2). The first (cathodic) peak observed at more negative potentials corresponds to the Fe(III)-cation reduction at the Pt-electrodeе:
whereas the second (anodic) one, to the oxidation of the formed Fe(II)-cations:

Cyclic voltammogram of Pt-electrode taken at 25°C in argon-deaerated 2 M H3PO4 solution containing 0.1 М FePO4 at the potential scanning rate, V/s: (1) 0.05; (2) 0.10; (3) 0.20.
The oxidative ability of the Fe(III)-cations in phosphoric-acid medium is determined by the half-wave potential:
which is widely judged as the system’s redox-potential [21]. Here Epc, Epa is the cathodic and anodic peak potential, respectively. At that, in the studied system the E1/2 values approach the EFe(III)/Fe(II) value determined by the potentiometric method at СFe(III) = СFe(II), which also points to the presence of the Fe(III)-cations in the H3PO4 solution as their complex compounds with the phosphate-anions.
The maximal cathodic current in the current–voltage curves is given by the Randles–Ševčik equation [21]:
where S is the electrode surface area (m2); С is the electroactive substance concentration (mol/m3); D is the electroactive particle diffusion coefficient (m2/s); v is the potential scanning rate (V/s); P is the parameter, a function of zFvτ/RT, where τ is the time. Based on equation (37) and using the experimentally determined values of the maximal cathodic currents, we found the Fe(III)-cation diffusion coefficient in 2 М H3PO4 (Table 2). The obtained DFe(III) mean value in 2 M H3PO4 is 110 ± 10 μm2 s–1, which is close to DFe(III) = 120 μm2 s–1 in 1 M H3PO4 at 20°С [22].
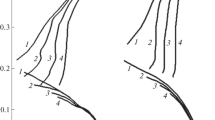
The visions of the corrosion medium properties are necessary for the understanding of processes occurring in the corrosion medium at the aggressive medium/metal interface. The most relevant information on the steel corrosion mechanism in acidic solutions is obtained in the course of the revelation of specific features of electrode reactions occurring thereat. In 2 M H3PO4 the form of the low-carbon steel polarization curves is characteristic of the corrosion occurring in the area of the steel active dissolution (Fig. 4, Table 3). In this medium, the slope of the steel cathodic polarization (bс) approached that theoretically predicted for iron (0.120 V). However, the metal anodic polarization (ba) appeared being higher than the theoretical value (0.035 V) [14]. The increase in the steel slope ba resulted from the formation of a visually observed mud layer at its surface. The presence of the FePO4 acid in the solution shifted the steel free corrosion potential (Ecor) toward more positive values, which is a result of the cathodic reaction counterinhibition by this additive. The Fe(III)-cations practically do not affect the anodic process; however, the cathodic reaction order with respect to their concentration is positive. As the FePO4 content in the corrosion medium increased, the cathodic reaction acceleration coefficient also grows. The initial section of the cathodic polarization curves is characterized by a limiting current (ilim). By contrast, the slope of the anodic polarization curves corresponds to the background dependence.

Polarization curves of the St3 steel in FePO4-containing 2 M H3PO4 solution, М: (1) 0; (2) 0.01; (3) 0.02; (4) 0.05; (5) 0.10. n = 460 rpm; t = 25°C.
The presence of FePO4 additives in the H3PO4 solution practically does not affect the character of the anodic reaction which passed in accordance with equation (8) both in the absence and in the presence of the Fe(III) phosphate. The form of cathodic polarization curves points to the Fe(III) involvement in the cathodic reaction. In concentrated solutions of acids (рН < 2) the cathodic reaction corresponding to equation (9) passed under kinetic control [23], which is confirmed by the character of cathodic polarization curves. In the presence of Fe(III) phosphate they are complicated by a limiting current, which points to the change of the cathodic reaction mechanism. The observed limiting current can be caused by diffusion limitations in the delivery of oxidants present in the acid solution—H+ and Fe3+—to the steel surface. Because the H+ concentration is larger by more than one order of magnitude than СFe(III), the limiting current is rather the result of the diffusion limitations in the delivery of Fe(III)-cations to the steel surface. To confirm this suggestion, it is necessary to study the effect of the electrolyte solution flow on the steel cathodic reaction rate. Such studies used to be carried out with the rotating disc electrode. By varying its rotation velocity, the regime of the liquid flow near the metal surface can be controlled [24, 25].
The cathodic process at steel including the reaction (9) passing under the kinetic control and the reaction (26) controlled by diffusion can be characterized by the following expression:
where ik and id is the kinetic and diffusion current densities. In the case of laminar liquid flow near the rotating metal disc electrode surface, the id value is proportional to the square root from the disc electrode rotation velocity (n). Therefore, expression (38) can be rewritten as:
In the 2 M H3PO4 + FePO4 solution, the ic vs. n1/2 experimental dependence is linear (Fig. 5). However, in 2 M H3PO4 there is no cathodic current response to the change in the steel disc rotation velocity, which points to the kinetic nature of the reaction (9). In the presence of FePO4 the cathodic current kinetics component is the same as in the absence thereof, which points to the mutual independence of the reactions (9) and (26). Moreover, it is clear that the reaction (9) is under kinetic control; the reaction (26), under diffusion control.

Dependence of the cathodic current density on the steel disc rotation velocity in FePO4-containing 2 M H3PO4 solution, М: (1) 0; (2) 0.01; (3) 0.02; (4) 0.05; (5) 0.10. Е = –0.30 V, t = 25°C.
The diffusion current of the Fe(III) reduction at a steel cathode at the laminar liquid flow is described by the following formula [23]:
Here C* is the Fe(III) concentration in the solution bulk, η is the kinematic viscosity of the liquid (0.011 сm2/s [26]), n is the in the steel disc angular rotation velocity. By using equation (40), it is possible to calculate DFe(III) in 2 M H3PO4 (Table 4). The obtained DFe(III) values are in good agreement with the data, found by the cyclic voltammetry method (Table 2). The difference between the DFe(III) value obtained by cyclic voltammetry at Pt-electrode and the mean value of the same quantity calculated from the measurements at the rotating disc electrode does not exceed 18%. To our view, the cyclic voltammetry method produced more correct values of DFe(III). The DFe(III) value determined from the measurements in real corrosion medium is less accurate because several parallel processes occur therein. The larger DFe(III) values obtained from the measurements at the rotating steel disc electrode are mainly due to the hydrogen bubbles formed at the metal surface, as the hydrogen is evolved at cathodic potentials. These bubbles adhered to the electrode are likely to facilitate turbulent flows near the electrode surface during its rotation, which violates the laminar regime of the corrosion-medium motion.
The revelation of some thermodynamic and kinetic parameters of the studied corrosion medium allowed predicting the low-carbon steel corrosion character. The results of potentiometry showed that the presence of the Fe(III) phosphate in the corrosion medium increased its redox-potential and, as a consequence, its aggressivity toward the steel. In spite of the decrease in the Fe(III) cation oxidative ability because of its binding to the complexes with the phosphate-anions, it remained well perceptible. The EFe(III)/Fe(II) value is 0.323 V even at low Fe(III)-cation concentration (0.001 M). The presence of the Fe(III) salt in H3PO4 solution increased the systems oxidative ability. The higher the Fe(III) content, the more noticeable is the effect. It is logical to assume that the increase of the Fe(III) phosphate content in the studied aggressive medium will have a detrimental effect on low-carbon steel corrosion stability. In addition, the steel corrosion in the FePO4-containing 2 M H3PO4 passed through the stages characterized by both kinetic control and diffusion limitations. Correspondingly, the steel corrosion in such systems must depend on the aggressive medium convection type significantly.
The postulated suggestions were confirmed in the studies of the low-carbon steel corrosion in the FePO4-containing 2 M H3PO4 flow, with regard to the metal samples’ mass loss (Fig. 6).

Dependence of the 08PS steel corrosion rate on the propeller mixer rotation velocity in corrosion medium at 20 ± 2°C in FePO4-containing 2 M H3PO4 solution, M: (1) 0; (2) 0.005; (3) 0.01; (4) 0.02; (5) 0.05; (6) 0.10. The results are given both noncorrected (а) and corrected for the natural convection (b). The experiment duration is 2 h.
In particular, the low-carbon steel corrosion in the FePO4-containing 2 M H3PO4 is intensified upon the increasing of the salt content. In all studied media, we observed a clear response of the corrosion process to the corrosion medium agitating intensity. The experimental dependence of the low-carbon steel corrosion rate on the propeller mixer rotation velocity used in the medium forced convection can be described by the following formula:
where kst is the low-carbon steel corrosion rate in the static medium, w is the mixer rotation velocity, λ is the empirical coefficient (Table 5). Equation (41) formally corresponds to expression (39) that characterized electrode reactions passed under diffusion control.
When analyzing experimental dependences, it should be borne in mind that according to the diffusion kinetics laws, only kinetic component of the corrosion process must occur in the static medium, whereas the diffusion component cannot be realized because of the absence of the liquid motion. Correspondingly, the kst values obtained in the FePO4-containing media must equal the kst value in 2 M H3PO4 where the process occurs strictly in the kinetic regime. However, the kst actual values observed in the FePO4-containing media are significantly larger than the value in 2 M H3PO4. This effect results from the natural convection occurring in the static medium. The effect is still more intensified as a result of the hydrogen gas bubbles plentiful evolution in the course of the corrosion process at a rather large steel surface area because the pop up hydrogen bubbles agitate the aggressive medium. Nonetheless, when passing from static medium to dynamic one, this effect is leveled out as the propeller mixer rotation velocity increased. With reference to the above mentioned, the obtained experimental results are presented in two extreme dependences, both without taking into account and with consideration to the natural convection processes. It is to be noted that in the FePO4-free 2 M H3PO4 we observed but quite insignificant response from the corrosion process to the increase of the medium flow rate, which resulted from the presence of air oxygen therein. The effect is negligibly small, however.
The analysis of experimental data showed that the increase of FePO4 content in aggressive medium accelerated the low-carbon steel corrosion (Table 6). Also, in the presence of FePO4 in the phosphoric acid solution the intensity of the steel corrosion increased when passing from the static medium to dynamic one. For example, at СFe(III) = 0.1 М in 2 M H3PO4 solution the passing from the static medium to dynamic one (w = 1080 rpm) was accompanied by the increment of the corrosion loss Δk = 36.6 g/(m2 h). At that, the steel corrosion rate in the static medium is kst = 9.2 g/(m2 h), which is much lower than the increment of the corrosion loss observed in dynamic medium.
CONCLUSIONS
(1) The low-carbon steel corrosion in FePO4-containing H3PO4 solutions can be realized through the iron reactions with the acid and the Fe(III) salt. The oxidative ability of the FePO4-containing H3PO4 solutions is lower than that predicted theoretically because of the Fe(III) binding into complexes with phosphate-anions.
(2) In the FePO4- and Fe3(PO4)2-containing H3PO4 solutions the dependence of the system’s redox-potential on the Fe(III)- and Fe(II)-cation relative content is unsatisfactorily described by the Nernst equation which is due to non-equilibrium complex-formation of these cations with phosphate-anions.
(3) In the FePO4-containing 2 M H3PO4 solution, three partial reactions are realized at the steel: the iron anodic ionization, H+ and Fe(III) cathodic reduction. The first two reactions occurred under kinetic control; the last one, under diffusion control. The increase in the FePO4 concentration in the corrosion medium accelerated the cathodic reduction reaction.
(4) In the FePO4-containing H3PO4 solutions, the Fe(III) diffusion coefficient (DFe(III)) can be determined experimentally by the Pt-electrode cyclic voltammetry or by the measurements of the dependence of Fe(III)-cation reduction current on the steel disc electrode rotating velocity; both methods provided close results.
(5) The data on the low-carbon steel corrosion in the H3PO4 solution flow obtained from the metal sample mass loss is in good agreement with the results of electrochemical studies. A FePO4 accelerating effect on the steel corrosion in 2 M H3PO4 solutions is found. The empirical dependence of the steel corrosion rate on the medium flow intensity (under agitation with a propeller mixer agitation) can be presented as a linear dependence:
where kst is the steel corrosion rate in static medium; w is the propeller mixer rotation velocity; λ is the empirical coefficient.
REFERENCES
Kuzin, A.V., Gorichev, I.G., Shelontsev, V.A., Kuzmenko, A.N., Plakhotnaia, O.N., and Ovsyannikova, L.V., The Role of a Complex Formation in the Dissolution of Iron Oxides in Orthophosphoric Acid, Moscow Univ. Chem. Bull., 2021, vol. 76, no. 6, p. 398. https://doi.org/10.3103/S0027131421060055
Prodan, I.E., Yeshchenko, L.S., and Pechkovsky, V.V., Study of the crystallization of iron phosphates in the system iron(III)—phosphoric acid—water, Russ. J. Inorg. Chem. (in Russian), 1989, vol. 34, no. 7, p. 1860.
Barthel, J. and Deiss, R., The limits of the Pourbaix diagram in the interpretation of the kinetics of corrosion and cathodic protection of underground pipelines, Mater. and Corros., 2021, vol. 72, no. 3, p. 434. https://doi.org/10.1002/maco.202011977
Huang, H.-H., The E h–pH Diagram and Its Advances, Metals, 2016, vol. 6, no. 1, p. 23. https://doi.org/10.3390/met6010023
Perry, S.C., Gateman, S.M., Stephens, L.I., Lacasse, R., Schulz, R., and Mauzeroll, J., Pourbaix Diagrams as a Simple Route to First Principles Corrosion Simulation, J. Electrochem. Soc., 2019, vol. 166, no. 11, p. C3186. https://doi.org/10.1149/2.0111911jes
Pourbaix, M., Atlas of Electrochemical Equilibria in Aqueous Solutions, 2nd English Edition, Houston: National Association of Corrosion Engineers, 1974, p. 307.
Wermink, W.N. and Versteeg, G.F., The Oxidation of Fe(II) in Acidic Sulfate Solutions with Air at Elevated Pressures. Part 1. Kinetics above 1 M H2SO4, Ind. Eng. Chem. Res., 2017, vol. 56, no. 14, p. 3775. https://doi.org/10.1021/acs.iecr.6b04606
Wermink, W.N. and Versteeg, G.F., The Oxidation of Fe(II) in Acidic Sulfate Solutions with Air at Elevated Pressures. Part 2. Influence of H2SO4 and Fe(III), Ind. Eng. Chem. Res., 2017, vol. 56, no. 14, p. 3789. https://doi.org/10.1021/acs.iecr.6b04641
Zakharov, V.A., Songina, O.A., and Bekturova, G.B., Real potentials of oxidation–reduction systems (Overview), Zh. Anal. Khim. (in Russian), 1976, vol. 31, no. 11, p. 2212.
Kaesche, H., Die Korrosion der Metalle. Physikalischchemische Prinzipien und Aktuelle Probleme, Berlin: Springer, 1979.
Antropov, L.I. Theoretical Electrochemistry (in Russian), Moscow, Vysshaya Shkola, 1965, p. 348–380.
Bockris, J.O'M., Drazic, D., and Despic, A.R., The electrode kinetics of the deposition and dissolution of iron, Electrochim. Acta, 1961, vol. 4, no. 2–4, p. 325. https://doi.org/10.1016/0013-4686(61)80026-1
Katrevich, A.N., Florianovich, G.M., and Kolotyrkin, Ya.M., Elucidation of the kinetic parameters of the reaction of active dissolution of iron in phosphate solutions, Prot. Met. (in Russian), 1974, vol. 10, no. 4, p. 369.
Reshetnikov, S.M. and Makarova, L.L., Kinetics and mechanism of cathodic and anodic processes that determine acid corrosion of metals in the area of active dissolution, In: Redox and Adsorption Processes on the Solid Metal Surfaces (in Russian), Udmurt State Univ., Izhevsk, 1979, p. 25–49.
Avdeev, Ya.G. and Andreeva, T.E. Characteristics of the Mechanism of Corrosion of Low-Carbon Steels in Acid Solutions Containing Fe(III) Salts, Russ. J. Phys. Chem. A., 2021, vol. 95, no. 6, p. 1128. https://doi.org/10.1134/S0036024421060029
Avdeev, Ya.G. and Andreeva, T.E., Mechanism of Steel Corrosion in Inhibited Acid Solutions Containing Iron(III) Salts, Russ. J. Phys. Chem. A., 2022, vol. 96, no. 2, p. 423. https://doi.org/10.1134/S0036024422020030
Avdeev, Ya.G., Andreeva, T.E., Panova, A.V., and Kuznetsov, Yu.I., Effect of anionic composition of solutions of mineral acids containing Fe(III) on their oxidizing properties, Int. J. Corros. Scale Inhib., 2019, vol. 8, no. 1, p. 139. https://doi.org/10.17675/2305-6894-2019-8-1-12
Lurie, Yu.Yu., Spravochnik po analiticheskoy khimii (Handbook of Analytical Chemistry) (in Russian), Moscow, Khimiya, 1971, p. 255–265.
Kim, M.S., Kim, C.H., and Sohn, Y.S., Complex Formation Between Ferric Ion and Phosphoric Acid, J. Korean Chem. Soc., 1975, vol. 19, no. 5, p. 325.
Filatova, L.N., Vendilo, A.G., and Sandu, R.A., Chemical Forms of Iron(III) in Solutions of Orthophosphoric Acid as Probed by Electronic Absorption Spectroscopy, Russ. J. Inorg. Chem., 2012, vol. 57, no. 9, p. 1272. https://doi.org/10.1134/S0036023612090057
Plambeck, J.A. Electroanalytical Chemistry: Basic Principles and Applications, New York : Wiley, 1982.
Belqat, B., Laghzizil, A., Elkacimi, K., Bouhaouss, A., and Belcadi, S., Fluoride effect on the electrochemical behaviour of the Fe(III)/Fe(II) system in H3PO4 + H2O + HF, J. Fluorine Chem., 2000, vol. 105, p. 1. https://doi.org/10.1016/S0022-1139(00)00256-6
Pleskov, Yu.V. and Filinovskii, V.Yu., The Rotating Disk Electrode, New York: Consultants Bureau, 1976.
Du, C., Tan, Q., Yin, G., and Zhang, J., Rotating Disk Electrode Method, In Rotating Electrode Methods and Oxygen Reduction Electrocatalysts, Eds. Xing, W., Yin, G., and Zhang, J., Elsevier B.V., 2014, p. 171–198. https://doi.org/10.1016/B978-0-444-63278-4.00005-7
Jia, Z., Yin, G., and Zhang, J., Rotating Ring-Disk Electrode Method, In Rotating Electrode Methods and Oxygen Reduction Electrocatalysts, Eds. Xing, W., Yin, G., and Zhang, J., Elsevier B.V., 2014, p. 199–229. https://doi.org/10.1016/B978-0-444-63278-4.00006-9
Xing, W., Yin, M., Lv, Q., Hu, Y., Liu, C., and Zhang, J., Oxygen solubility, diffusion coefficient, and solution viscosity, In Rotating Electrode Methods and Oxygen Reduction Electrocatalysts, Eds. Xing, W., Yin, G., and Zhang, J., Elsevier B.V., 2014, p. 1–31. https://doi.org/10.1016/B978-0-444-63278-4.00001-X
Funding
This work is carried out as R&D “Chemical material resistance, protection of metals and other materials against corrosion and oxidation” (2022–2024), the Integrated National Information System reg. no. 122011300078-1, the inventory no. FFZS-2022-0013.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflict of interest.
Additional information
Translated by Yu. Pleskov
Rights and permissions
About this article

Cite this article
Avdeev, Y.G., Panova, A.V. & Andreeva, T.E. Corrosion of Low-Carbon Steel in a Flow of Phosphoric Acid Solution Containing Iron(III) Phosphate. Russ J Electrochem 59, 512–523 (2023). https://doi.org/10.1134/S1023193523070030
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1023193523070030