
Abstract
The systematics and phylogeny of flatfish is investigated on the basis of an incomplete nucleotide sequence of 16S rRNA and the complete Co-1 and Cyt-b sequences. In total 62 specimens of 14 species of our own collections were submitted to the GenBank/BOLD repositories and studied. Four types of gene trees were reconstructed: Bayesian (BA), maximum likelihood (ML), minimum evolution (ME), and neighbor joining (NJ). These trees showed basically similar topology. Two separate branches on the trees support the previously identified subfamilies Hippoglossoidinae and Pleuronectinae with the monophyletic status of these taxa. The subfamily Pleuronectinae can be considered monophyletic if the tribe Microstomini is excluded from it and genus Lepidopsetta is moved into the tribe Pleuronectini. Three sets of nucleotide sequences were formed and independently studied. One set included all the obtained 16S rRNA gene sequences (291 bp), the second set included a sample of longer 16S rRNA sequences (617 bp), and the third set consisted of three gene sequences: 16S rRNA, Co-1, and Cyt-b (2926 bp). All three data sets gave a similar phylogenetic signal, which is consistent with the traditional concept of the taxonomy of the Pleuronectiformes order; however, the second and third sets provided better resolution of topology.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
The fish family of true flounder Pleuronectidae, to which the main attention is paid in the article, is one of the largest in the order Pleuronectiformes, including 59 nominal fish species of right-sided flounders common in the sea waters of the Northern Hemisphere [1, 2]. In their analysis, J. Cooper and F. Chapleau [1] considered the Pleuronectidae family as a monophyletic taxon, based on ten synapomorphies by morphological characters. An important result obtained by the aforementioned authors is generally consistent with the topology of the branches of the family established in several studies of molecular phylogenetics [3–10]. According to [1], this family includes the subfamilies Hippoglossinae, Eopsettinae, Lyopsettinae, Hippoglossoidinae, and Pleuronectinae, which are represented by genera usually consisting of species with high commercial value (for example, species of the genus of halibut-like flounders, Hippoglossoides). In connection with the fishery importance of these and other flounders and the need to manage such valuable fishery resources, both the precise classification of specimens of individuals and species within the genus and the entire systematics’ relationships among taxa in this family are very important.
Taxonomic studies of the Pleuronectidae have traditionally been based on morphological characters, as follows from the above paragraph. However, the frequent absence of clear evidence of the homology of species’ characters even at low taxonomic levels (within a genus) makes the postulated taxonomic and phylogenetic relationships of many groups of flounders not always convincing if they are justified only by morphology. There are several versions of the classification of flounders, which were proposed by different authors [1, 11–13]. Certain disagreements are also noted regarding the phylogenetic relationships of flounders obtained on the basis of morphological and molecular genetic data [1, 4, 14, 15]. Development of new nuclear and mitochondrial markers based on DNA makes it possible to better identify morphologically similar fish species [16], including many species of flounders. Therefore, it is relevant to search for new or already known, but insufficiently developed, molecular markers for the reconstruction of gene trees, as well as combined or species phylogenetic trees for flounders of the Pleuronectidae family.
In this study, taking into account the above, we present a comparative analysis of incomplete nucleotide sequences (hereinafter referred to as sequences) of the gene 16S rRNA for 14 species belonging to Pleuronectidae, previously not used in such a volume for flounders, in order to assess the success of taxonomic identification of specimens and to establish phylogenetic and taxonomic relationships in this family of flounders. The novelty of this study lies in the fact that the systematics of this group in the authors’ works cited above were not considered on the basis of 16S rRNA. Accordingly, in the presented article, we considered the potential of this marker on a sufficient number of specimens for taxonomic and evolutionary genetic studies of flounders of the Russian Federation.
MATERIALS AND METHODS
A total of 62 sequences of 16S rRNA and additionally 24 sequences of genes 16S rRNA, Co-1, and Cyt-b for 14 species belonging to seven genera of the Pleuronectidae family were analyzed. Latin names are given in accordance with the classification [1]. Sampled fish (2–5 specimens of muscle tissue subjected to ethanol fixation, 95%) were taken from the existing collection of the Laboratory of Molecular Systematics, and voucher specimens of the fish themselves are kept in safe custody at the museum of the A.V. Zhirmunsky National Scientific Center of Marine Biology. DNA isolation was performed using commercial kits (DNA Extran-2, Syntol, Russia).
A fragment of the sequence of gene 16S rRNA was amplified by polymerase chain reaction (PCR) using primers 16Sbr-H and 16Sar-L. The PCR reaction was carried out in a volume of 25 μL of a solution containing the following: distilled deionized water—17.8 μL; dNTP (ZAO Evrogen, Moscow, Russia)—0.5 μL; 5× Buffer (Evrogen)—5 µL; primers at a concentration of 10 μM/μL—0.3 μL for each; Taq polymerase—0.1 μL. The following heat program was used: denaturation at 93°C for 1 min, annealing at 55°C for 1 min, and elongation at 72°C for 1 min for 33 cycles. To determine the localization and arrangement of nucleotides in the sequences, PCR products (DNA specimens) were subjected to cyclic sequencing using the BrightDye Terminator Cycle Sequencing Kit according to the following program: denaturing at 96°C for 10 s, annealing at 45°C for 10 s, elongation at 60°C for 2 min.
Bidirectional DNA strand sequences of genes 16S rRNA were obtained for each DNA specimen. These sequences were then pooled together to generate consensus sequences for each specimen (individual). This procedure was performed using the Geneious, Free Trial software package [17].
Since the length of the obtained sequences varied quite significantly, in the range from 355 base pairs (bp) to 642 bp, two sets of sequences were composed for more accurate further analysis. One set represented all the sequences obtained, while the other included only the longest sequences. According to the data transformation, set 1 contained 62, and set 2 contained 27 sequences. After the alignment procedure and removal of gaps (indels), the length of the sequences of the two sets was 291 and 617 bp, respectively.
Sequence alignment for all taxa was performed using the MEGA-X software package, SP (http://megasoftware.net/) [18] based on the ClustalW [19] module as an integrated MEGA product. The penalties for opening of gaps and for lengthening of gaps were set at 15.0 and 5.0, respectively (for other alignment program settings, the default parameters were used). After the first alignment step, large gaps were manually removed and the final alignment in the second step was done with reduced penalty levels (5.0 and 0.5 for the two options, respectively). All spaces were then manually removed again.
To increase the information capacity, besides the gene 16S rRNA, the analysis included sequences of the genes Co-1 and Cyt-b previously used in the analysis [10]; taken together, these data constituted the third set of sequences, including a total of 24 specimens with a length of 2926 bp.
For further analysis of sequences and construction of gene trees, an optimal model of nucleotide substitutions for the obtained set of sequences was defined. The best evolutionary model that fit the data obtained was evaluated by means of a special module of the MEGA program. For the set of gene 16S rRNA with short sequences (291 bp), the best model was K2P + G (two-parameter model of M. Kimura with a gamma distribution of substitutions) [20]; for the set of this gene with long sequences (617 bp), the best model was JC + G (Jukes–Cantor model with a gamma distribution of substitutions) [21]; for the set of sequences of three genes, the best model was HKY + G (Hasegawa–Kishino–Yano model with a gamma distribution of substitutions) [22].
Gene trees were constructed using four reconstruction methods: Bayesian Analysis (BA), Maximum Likelihood (ML), Neighbor Joining (NJ), and Minimum Evolution (ME). They were implemented in MrBayes 3.2.7 (http://nbisweden.github.io/MrBayes/ download.html) [23, 24] and MEGA-X [18]. The process of tree reconstruction in BA was simulated for one million generations, n (n = 106). The three other reconstructions ML, NJ, and ME were performed with replicates equal to k = 1000 copies of bootstrap (bootstrap support).
The sequence of Platichthys stellatus was chosen as an outgroup when rooting trees, a representative of which, according to the data for the complete mitogenome (mtDNA), was previously assigned to an external branch in the Pleuronectidae family [10]. Phylogenetic trees were visualized and edited, if necessary, using the FigTree software [25] and MEGA-X [18].
All obtained sequences by gene 16S rRNA were registered in GenBank (https://www.ncbi.nlm. nih.gov/); part of previously unpublished sequences of Co-1 and Cyt-b are also included in the article (Table 1).
Statistical analysis of the nucleotide composition was performed using SP MEGA-X. Additionally with the Statistica 6 SP [29], one-way analysis of variance (ANOVA) of the nucleotide composition was conducted separately for each gene.
RESULTS
The analysis was constructed on the basis of the sequences of the three previously presented data sets. The full length of the segment of gene 16S rRNA (full-length sequences “from primer to primer”) is 596–631 bp. The numbers in GenBank of the full-length regions of gene 16S rRNA are as follows: MN888911, MN888895, MN888908, MN888877, MN888901, MN888894, MN888893, MN888892, MN888917, MN888916, MN888915, MN888903, MN888924, MN888918, MN888912, MN888904, MN888905, MN888868, MN888873, MN888898, MN888899, MN888902, MN888927, MN888909, MN888907, MN888884, MN888883, MN888876.
However, not all of the sequences obtained reached their full size. The difference in the length of the sequences is due to poor-quality sequencing of some specimens, which led to a greater “cutoff” of regions near the primers when forming consensus sequences. These sequencing errors are possible owing to the fact that some of the tissue specimens were stored for several years before analysis. However, short fragments are not necessarily bad for assessing variability in closely related taxa and comparing the degree of their similarity-difference for gene tree reconstruction. In this regard, for analysis, the material was divided as explained before into two groups, including long (1) and short (2) gene sequences. In accordance with the outlined approaches (see the Materials and Methods section), four types of trees were constructed: BA, ML, NJ, and ME.
Analysis of all Sequences of 16S rRNA
Figure 1 shows a rooted ML tree derived from a set of sequences of gene 16S rRNA 291 bp in length. The tree nodes support are listed in the following order: BA/ME/NJ/ML.

Rooted gene tree showing phylogenetic relationships based on 62 short nucleotide sequences of 16S rRNA. The topology is presented on the basis of ML reconstruction. The nodes give support values for four tree reconstruction methods in the following order: BA/ME/NJ/ML. For the BA tree, a posteriori probabilities (%, n = 106 generations) are shown, and bootstrap supports are given for the other three reconstructions (k = 1000 replicas).
The branch with specimens of Limanda sakhalinensis became part of the subfamily Hippoglossoidinae, forming a separate, topologically unresolved branch (node) together with Cleisthenes pinetorum of subfamily Hippoglossoidinae. A separate, also unauthorized branch was formed by representatives of three nominal species of halibut-like flounders of the genus Hippoglossoides. Lepidopsetta mochigarei, a representative of the tribe Microstomini, has for all five corresponding branches on the tree unresolved topology. A separate node on the tree is formed by representatives of the Pacific halibut Hippoglossus stenolepis from subfamily Hippoglossinae.
Analysis of Longer Sequences of 16S rRNA
Figure 2 shows a rooted NJ tree derived from a set of gene sequences of 16S rRNA 617 bp in length. The branch Limanda sakhalinensis is included in the subfamily Hippoglossoidinae, located in the same cluster with Cleisthenes pinetorum. The sequence representing the subfamily Hippoglossinae forms a separate node. Lepidopsetta mochigarei, as before for a short set, forms an unresolved node, but topologically it belongs to the tribe Microstomini.

Rooted NJ tree topology showing phylogenetic relationships based on data from 27 sequences of a region of gene 16S rRNA. The nodes give the values of supports for the BA tree (n = 106 generations) and the three other reconstructions in the following general order: BA/ME/ML/NJ (k = 1000 bootstrap replicas for the last three reconstruction methods).
Analysis of the Reconstruction of Gene Trees from the Combined Sequences of Three Genes
For this analysis, the aligned sequences of the gene 16S rRNA region were compared, jointly with genes Co-1 and Cyt-b. The sequences were concatenated using special SP MEGA-X utility and then subjected to further analysis. According to the data obtained, the branch Limanda sakhalinensis is included in the subfamily Hippoglossoidinae (Fig. 3). The branch Cleisthenes pinetorum is also included in the subfamily Hippoglossoidinae (Fig. 3).

Rooted ML tree showing phylogenetic relationships based on data from 24 concatenated sequences of gene 16S rRNA, Co-1, and Cyt-b. The nodes give the values of supports for the BA tree (n = 106 generations) and for the three other reconstructions in the following general order: BA/NJ/ME/ML (k = 1000 bootstrap replicas for the last three reconstruction methods).
Nucleotide Composition
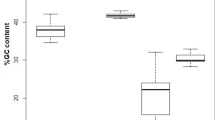
The ratio of pyrimidines (T, C) and purines (A, G) in genes 16S rRNA, Co-1, and Cyt-b deviated from the ratio of 50 : 50 (Appendix, Fig. 4). In sequences of 16S rRNA, there are no large differences in the ratio of pyrimidines (T, C) and purines (A, G), but general heterogeneity of the nucleotide composition with a predominance of C and A nucleotides can be observed (Fig. 4a). For Co-1 and Cyt-b, there is a statistically significant deviation in the ratio of pyrimidines to purines with a predominance of pyrimidines (Figs. 4b, 4c).

Average values of the composition (%) of four nucleotides in 24 studied sequences of genes 16S rRNA (a), Co-1 (b), and Cyt-b (c). According to the results of one-way analysis of variance (ANOVA). The vertical lines represent the 95% confidence interval.
ANOVA for each gene found that the differences for four nucleotides were statistically significant: for 16S rRNA, F = 2147.9, d.f. = 3; 92, P < 0.0001; for Co-1, F = 3673.3, d.f. = 3; 92, P < 0.0001; for Cyt-b, F = 4320.7, d.f. = 3; 92, P < 0.0001. The ratio of (T + C) : (A + G) for 16S rRNA, Co-1, and Cyt-b was 45.4 : 54.6, 56.5 : 43.5, and 61.1 : 38.9%, respectively.
DISCUSSION
As noted in the Introduction, the largest subfamily in the family is Pleuronectinae. This subfamily is represented by two tribes, Microstomini and Pleuronectini. According to the data obtained by 16S rRNA, they do not form monophyletic branches (see Figs. 1, 2). Thus, the taxonomy at the subfamily level needs further clarification. For example, for a larger information signal, it is necessary to increase the number of both nuclear and mitochondrial markers in the study. This will help reduce the number of topologically unresolved nodes in the resulting trees.
As noted above, J. Cooper and F. Chapleau [1], in their revision of this family based on traditional morphological characters, substantiated that Pleuronectidae is a monophyletic group. The monophyly of flounders established on the basis of the classical approach corresponds in many cases to molecular phylogenetic reconstructions in studies of this family for such markers as 12S rRNA and 16S rRNA, as well as for genes Co-1 and Cyt-b [4–6, 8, 9] and the complete mitogenome [10].
The subfamily Hippoglossoidinae, in the most representative material in the work, included two of the three genera Cleisthenes (C. pinetorum) and Hippoglossoides (H. dubius Schmidt, 1904, H. elassodon Jordan & Gilbert, 1180, H. robustus Gill & Townsend, 1897) (Fig. 3). Species of the genus Hippoglossoides form a mixed cluster on the BA tree (Fig. 3). On the basis of this, it can be assumed that two taxa H. elassodon and H. robustus are synonyms of a single species. Synonymy of H. elassodon and H. robustus was already proposed earlier on the basis of morphological and molecular-phylogenetic data [4, 27–31]. By the principle of seniority, a taxon of the species rank, H. elassodon Jordan & Gilbert, 1880 can be accepted as valid, and H. robustus Gill & Townsend, 1897 can be considered a junior synonym for this species. The proposal of synonymy of H. elassodon and H. robustus has was already made previously, as noted above. However, K.A. Vinnikov et al. [31] in their two-sided analysis (morphology + genetics) propose to reintroduce synonymy. But synonymy was never introduced. In the databases, these two taxa still appear as separate species. One of the tasks of the article is to sharpen this issue in order to finally resolve this case in a special publication.
The data for the genus Limanda deserve a separate discussion. Sequences of the species Limanda sakhalinensis occur, as noted in the results and shown in Figs. 1–3, and are included in the branch of the subfamily Hippoglossoidinae. In a comparative anatomical study by J. Cooper and F. Chapleau [1], monophyly of this genus was not confirmed. In our study, as in previous molecular phylogenetic studies [4, 5, 10], Limanda sakhalinensis Hubbs, 1915 was included in the subfamily Hippoglossoidinae. Considering all these data, it is quite appropriate to recommend revising the position of Limanda sakhalinensis Hubbs, 1915, moving it to the genus Hippoglossoides with the name Hippoglossoides (Limanda) sakhalinensis in the composition of subfamily Hippoglossoidinae. Accordingly, it is necessary to revise the morphology and species characteristics, as well as diagnostic keys, which is supposed to be done in independent work.
Genus Lepidopsetta (L. mochigarei) was included in the tribe Pleuronectini, subfamily Pleuronectinae by our data, while in [1] this genus was considered exclusively within the tribe Microstomini of the subfamily Pleuronectinae. In molecular phylogenetic studies based on Co-1 and Cyt-b [4, 5, 10], genus Lepidopsetta was considered within the tribe Pleuronectini. Thus, it is preferable to consider the genus Lepidopsetta as a part of the tribe Pleuronectini. However, this question, given the weak topological signal for this branch by the marker 16S rRNA in the work, requires further refinement using a larger number of genes.
Deviation in the equality in ratio (T + C): (A + T) is well described in the literature for many protein-coding genes [4, 32]. The presented analysis (Fig. 4, Appendix) shows that the bias for genes Co-1 and Cyt-b in the ratio of purines to pyrimidines is significantly different from the bias for the gene 16S rRNA. Obviously, the detected displacement of the nucleotide composition for the structural genes studied in this work reflects the hydrophobic properties of the proteins encoded by them [33]. Discovery of the cause of the heterogeneity of the nucleotide composition in the sequences of 16S rRNA requires further research.
REFERENCES
Cooper, J.A. and Chapleau, F., Monophyly and intrarelationships of the family Pleuronectidae (Pleuronectiformes), with a revised classification, Fish. Bull., 1998, vol. 96, no. 4, pp. 686—726.
Keast, A. and Chapleau, F., A phylogenetic reassessment of the monophyletic status of the family Soleidae, with comments on the suborder Soleoidei (Pisces; Pleuronectiformes), Canad. J. Zool., 1988, vol. 66, no. 12, pp. 2797—2810. https://doi.org/10.1139/z88-408
Berendzen, P.B. and Dimmick, W.W., Phylogenetic relationships of Pleuronectiformes based on molecular evidence, Copeia, 2002, vol. 3, pp. 642—652. https://doi.org/10.1643/0045-8511(2002)002[0642:PRO-PBO]2.0.CO;2
Kartavtsev, Y.P., Park, T-J., Vinnikov, K.A., et al., Cytochrome b (Cyt-b) gene sequence analysis in six flatfish species (Teleostei, Pleuronectidae), with phylogenetic and taxonomic insights, Mar. Biol., 2007, vol. 152, no. 4, pp. 757—773. https://doi.org/10.1007/s00227-007-0726-9
Kartavtsev, Y.P., Sharina, S.N., Goto, T., et al., Cytochrome oxidase 1 gene sequence analysis in six flatfish species (Teleostei, Pleuronectidae) of Far East Russia with inferences in phylogeny and taxonomy, Mitochondrial DNA, 2008, vol. 19, pp. 479—489. https://doi.org/10.1080/19401730802570934
Pardo, B.G., Machordom, A., Foresti, F., et al., Phylogenetic analysis of flatfish (order Pleuronectiformes) based on mitochondrial 16S rDNA sequences, Sci. Mar., 2005, vol. 69, no. 4, pp. 531—543.
Sharina, S.N. and Kartavtsev, Y.P., Phylogenetic and taxonomic analysis of flatfish species (Teleostei, Pleuronectiformes) inferred from the primary nucleotide sequence of cytochrome oxidase 1 gene (Co-1), Russ. J. Genet., 2010, vol. 46, no. 3, pp. 356—361. https://doi.org/10.1134/S1022795410030130
Betancur, R.R., Munroe, T.A., Orti, G., et al., Addressing gene tree discordance and non-stationarity to resolve a multi-locus phylogeny of the flatfishes (Teleostei: Pleuronectiformes), Syst. Biol., 2013, vol. 62, pp. 763—785. https://doi.org/10.1093/sysbio/syt039
Betancur, R.R. and Ortí, G., Molecular evidence for the monophyly of flatfishes (Carangimorpharia, Pleuronectiformes), Mol. Phylogenet. Evol., 2014, vol. 73, pp. 18—22. https://doi.org/10.1016/j.ympev.2014.01.006
Kartavtsev, Y.Ph., Sharina, S.N., Saitoh, K., et al., Phylogenetic relationships of Russian Far Eastern flatfish (Pleuronectiformes, Pleuronectidae) based on two mitochondrial gene sequences, Co-1 and Cyt-b, with inferences in order phylogeny using complete mitogenome data, Mitochondrial DNA, 2014, vol. 27, no. 1, pp. 667—678. https://doi.org/10.3109/19401736.2014.913139
Norman, I.R., A Systematic Monograph of the Flatfishes (Heterosomata), vol. I: Psettodidae, Bothidae, Pleuronectidae, London: British Museum, 1934.
Sakamoto, K., Interrelationships of the family Pleuronectidae (Pisces: Pleuronectiformes), Mem. Fac. Fish., Hokkaido Univ., 1984, vol. 31, pp. 95—215.
Lindberg, G.U. and Fedorov, V.V., Fishes of Japan Sea and Nearby Parts of Okhotsk and Yellow Seas, part 6: Teleostomi. Osteichthyes. Actinopterigii, XXXI: Pleuronectiformes, St. Petersburg: St. Petersburg Univ. Press, 1993.
Chapleau, F., Pleuronectiform relationships: a cladistic reassessment, Bull. Mar. Sci., 1993, vol. 52, no. 1, pp. 516—540.
Vernau, O., Moreau, C., Catzeflis, F.M., and Renaud, F., Phylogeny of flatfishes (Pleuronectiformes): comparisons and contradictions of molecular and morpho-anatomical data, J. Fish Biol., 1994, vol. 45, no. 4, pp. 685—696. https://doi.org/10.1111/j.1095-8649.1994.tb00934.x
Keith, P., Lord, C., Lorion, J., et al., Phylogeny and biogeography of Sicydiinae (Teleostei: Gobiidae) inferred from mitochondrial and nuclear genes, Mar. Biol., 2011, vol. 158, pp. 311—326. https://doi.org/10.1007/s00227-010-1560-z
Kearse, M., Moir, R., Wilson, A., et al., Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data, Bioinformatics, 2012, vol. 28, no. 12, pp. 1647—1649. https://doi.org/10.1093/bioinformatics/bts199
Tamura, K., Peterson, D., Peterson, N., et al., MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods, Mol. Biol. Evol., 2011, vol. 28, pp. 2731—2739. https://doi.org/10.1093/molbev/msr121
Thompson, J.D., Higgins, D.G., and Gibson, T.J., CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice, Nucleic Acids Res., 1994, vol. 22, no. 22, pp. 4673—4680. https://doi.org/10.1093/nar/22.22.4673
Kimura, M., A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences, J. Mol. Evol., 1980, vol. 16, pp. 111—120. https://doi.org/10.1007/bf01731581
Jukes, T.H. and Cantor, C.R., Evolution of protein molecules, in Mammalian Protein Metabolism, New York: Academic, 1969, vol. III, pp. 21—132. https://doi.org/10.1016/B978-1-4832-3211-9.50009-7.
Hasegawa, M., Kishino, H., and Yano, T., Dating of the human—ape splitting by a molecular clock of mitochondrial DNA, J. Mol. Evol., 1985, vol. 22, pp. 160—174. https://doi.org/10.1007/bf02101694
Hulsenbeck, J.P. and Rondquist, F., MrBAYES: Bayesian inference of phylogenetic trees, Bioinformatics, 2001, vol. 17, no. 8, pp. 754—755. https://doi.org/10.1093/bioinformatics/17.8.754
Ronquist, F. and Huelsenbeck, J.P., MrBayes 3: Bayesian phylogenetic inference under mixed models, Bioinformatics, 2003, vol. 19, no. 12, pp. 1572—1574. https://doi.org/10.1093/bioinformatics/btg180
http://tree.bio.ed.ac.uk/software/figtree/.
STATISTICA for Windows (Computer Program Manual), Tulsa: StatSoft, 1999. http://statsoft.ru/products/STATISTICA_Base/.
Ivankov, V.N., Vinnikov, K.A., Borisovets, E.E., and Kartavtsev, Y.P., Taxonomic relations between Pseudopleuronectes yokohamae and P. schrenki, Conference for Educated Students—Abstracts, Vladivostok: Far Eastern University Press, 2002, pp. 69—70.
Kartavtsev, Yu.F., Sviridov, V.V., Hanzawa, N., and Sasaki, T., Genetic divergence of Far Eastern dace species of the genus Tribolodon (Pisces: Cyprinidae) and closely related taxa, Russ. J. Genet., 2002, vol. 38, no. 11, pp. 1285—1297.
Vinnikov, K.A., Taxonomic status of flatfishes, Hippoglossoides elassodon and H. robustus (morphometric analysis), VI Regional’naya konferentsiya po aktual’nym problemam ekologii, morskoy biologii i biotekhnologii studentov, aspirantov, molodykh prepodavateley i sotrudnikov vuzov i nauchnykh organizatsiy Dal’nego Vostoka Rossii: tezisy dokladov (VI Regional Conference on Current Problems of Ecology, Marine Biology and Biotechnology for Ordinary Students, Graduate Students, Young Teachers and Employees of Universities and Scientific Organizations of the Russian Far East: Abstracts), Vladivostok: Dal’nevost. Univ., 2003, pp. 25—26.
Vinnikov, K.A., Ivankov, V.N., and Pitruk, D.L., Taxonomic status of Pseudopleuronectes yokohamae and P. schrenki (Pleuronectidae sensu Cooper and Chapleau, 1998), J. Ichthyol., 2006, vol. 46, no. 3, pp. 301—310. https://doi.org/10.1134/S0032945206040035
Vinnikov, K.A., Thomson, R.C., and Munroe, T.A., Revised classification of the righteye flounders (Teleostei: Pleuronectidae) based on multilocus phylogeny with complete taxon sampling, Mol. Phylogenet. Evol., 2018, vol. 125, pp. 147—162. https://doi.org/10.1016/j.ympev.2018.03.014
Kim, I.C., Kweon, H.S., Kim, Y.J., et al., The complete mitochondrial genome of the javeline goby Acanthogobius hasta (Perciformes, Gobiidae) and phylogenetic considerations, Gene, 2004, vol. 336, no. 2, pp. 147—153. https://doi.org/10.1016/j.gene.2004.04.009
Nailor, G.J., Collins, T.M., and Brown, W.M., Hydrophobicity and phylogeny, Nature, 1995, vol. 373, no. 6515, pp. 565—566. https://doi.org/10.1038/373565b0
Funding
This study was financially supported by the Russian Foundation for Basic Research under grant 15-29-02456-ofi in the direction of research of the genetic foundations of biodiversity, as well as by the Far Eastern Branch of the Russian Academy of Sciences under grant DV no. 18-4-040 “Comprehensive Study of the Biodiversity of Fish and Invertebrates Based on Dna Bar Coding and Development and Support of Databases and Biobanking”.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest. The authors declare that they have no conflict of interest.
Statement on the welfare of animals. All applicable international, national, and/or institutional guidelines for animal care and use have been followed.
Appendix
Appendix
Nucleotide composition for genes 16S rRNA, Co-1, and Cyt-b in 24 flounder fish sequences
Species/Mean | Nucleotides, all positions (% of total) | ||||
|---|---|---|---|---|---|
T | C | A | G | сumulative (bp) | |
16S rRNA | |||||
Cleisthenes pinetorum 78-07 | 16.8 | 28.9 | 29.9 | 24.4 | 291 |
Cleisthenes pinetorum 79-07 | 16.8 | 28.9 | 29.9 | 24.4 | 291 |
Hippoglossoides elassodon 33 | 16.8 | 28.9 | 30.2 | 24.1 | 291 |
Hippoglossoides elassodon 34 | 16.8 | 28.9 | 29.9 | 24.4 | 291 |
Hippoglossoides robustus 286 | 16.8 | 28.9 | 30.2 | 24.1 | 291 |
Hippoglossoides robustus 288 | 17.2 | 28.5 | 30.2 | 24.1 | 291 |
Hippoglossoides robustus 289 | 16.8 | 28.9 | 30.2 | 24.1 | 291 |
Hippoglossoides robustus 31 | 16.8 | 28.9 | 30.2 | 24.1 | 291 |
Limanda sakhalinensis 71 | 16.8 | 28.9 | 29.9 | 24.4 | 291 |
Limanda sakhalinensis 72 | 16.8 | 28.9 | 29.9 | 24.4 | 291 |
Liopsetta pinnifasciata 13 | 17.5 | 27.8 | 31.6 | 23.0 | 291 |
Liopsetta pinnifasciata 19-07 | 17.5 | 27.8 | 31.6 | 23.0 | 291 |
Liopsetta pinnifasciata 20-07 | 17.5 | 27.8 | 31.6 | 23.0 | 291 |
Liopsetta pinnifasciata 2k | 17.5 | 27.8 | 31.6 | 23.0 | 291 |
Myzopsetta punctatissima 37 | 17.2 | 27.5 | 32.3 | 23.0 | 291 |
Myzopsetta punctatissima 86-07 | 17.2 | 27.5 | 32.3 | 23.0 | 291 |
Platichthys stellatus 18-07 | 17.2 | 27.8 | 31.3 | 23.7 | 291 |
Platichthys stellatus 4k | 17.2 | 27.8 | 31.6 | 23.4 | 291 |
Pseudopleuronectes herzensteini 05-07 | 17.5 | 27.8 | 32.0 | 22.7 | 291 |
Pseudopleuronectes herzensteini 6k | 17.5 | 27.8 | 32.0 | 22.7 | 291 |
Pseudopleuronectes herzensteini 7k | 17.5 | 27.8 | 32.0 | 22.7 | 291 |
Pseudopleuronectes obscurus 09-07 | 17.2 | 28.2 | 32.0 | 22.7 | 291 |
Pseudopleuronectes obscurus 45 | 16.8 | 28.2 | 32.0 | 23.0 | 291 |
Pseudopleuronectes obscurus 46 | 17.2 | 28.2 | 32.0 | 22.7 | 291 |
Mean | 17.14 ± 0.13 | 28.26 ± 0.13 | 31.1 ± 0.13 | 23.5 ± 0.13 | – |
Co-1 | |||||
Cleisthenes pinetorum 78-07 | 29.4 | 27.5 | 23.6 | 19.5 | 1540 |
Cleisthenes pinetorum 79-07 | 29.4 | 27.3 | 23.7 | 19.5 | 1540 |
Hippoglossoides elassodon 33 | 29.3 | 27.6 | 24.0 | 19.2 | 1540 |
Hippoglossoides elassodon 34 | 29.1 | 27.6 | 24.0 | 19.4 | 1540 |
Hippoglossoides robustus 286 | 29.0 | 27.8 | 24.0 | 19.2 | 1540 |
Hippoglossoides robustus 288 | 29.2 | 27.5 | 24.0 | 19.3 | 1540 |
Hippoglossoides robustus 289 | 29.2 | 27.4 | 24.1 | 19.3 | 1540 |
Hippoglossoides robustus 31 | 29.0 | 27.8 | 24.0 | 19.2 | 1540 |
Limanda sakhalinensis 71 | 28.8 | 27.1 | 24.4 | 19.7 | 1540 |
Limanda sakhalinensis 72 | 28.6 | 27.3 | 24.4 | 19.7 | 1540 |
Liopsetta pinnifasciata 13 | 29.4 | 27.0 | 24.8 | 18.8 | 1540 |
Liopsetta pinnifasciata 19-07 | 29.4 | 27.0 | 24.8 | 18.8 | 1540 |
Liopsetta pinnifasciata 20-07 | 29.4 | 27.0 | 24.8 | 18.8 | 1540 |
Liopsetta pinnifasciata 2K | 29.4 | 27.0 | 24.8 | 18.8 | 1540 |
Myzopsetta punctatissima 37 | 28.8 | 27.2 | 24.7 | 19.2 | 1540 |
Myzopsetta punctatissima 86-07 | 28.8 | 27.2 | 24.7 | 19.2 | 1540 |
Platichthys stellatus 18-07 | 29.0 | 27.2 | 24.8 | 19.0 | 1540 |
Platichthys stellatus 4K | 28.8 | 27.3 | 24.8 | 19.0 | 1540 |
Pseudopleuronectes herzensteini 05-07 | 28.8 | 28.0 | 23.7 | 19.5 | 1540 |
Pseudopleuronectes herzensteini 6K | 28.7 | 28.0 | 23.7 | 19.6 | 1540 |
Pseudopleuronectes herzensteini 7K | 28.7 | 28.0 | 23.6 | 19.7 | 1540 |
Pseudopleuronectes obscurus 09-07 | 29.3 | 26.9 | 24.5 | 19.2 | 1540 |
Pseudopleuronectes obscurus 45 | 29.3 | 27.0 | 24.5 | 19.2 | 1540 |
Pseudopleuronectes obscurus 46 | 29.4 | 27.1 | 24.5 | 19.1 | 1540 |
Mean | 29.09 ± 0.07 | 27.38 ± 0.07 | 24.29 ± 0.07 | 19.25 ± 0.07 | – |
Cyt-b | |||||
Cleisthenes pinetorum 78-07 | 30.8 | 30.9 | 22.5 | 15.9 | 1095 |
Cleisthenes pinetorum 79-07 | 30.8 | 30.8 | 22.6 | 15.8 | 1095 |
Hippoglossoides elassodon 33 | 30.3 | 31.2 | 22.2 | 16.3 | 1095 |
Hippoglossoides elassodon 34 | 30.3 | 31.2 | 22.2 | 16.3 | 1095 |
Hippoglossoides robustus 286 | 30.3 | 31.3 | 22.2 | 16.2 | 1095 |
Hippoglossoides robustus 288 | 30.6 | 31.1 | 22.1 | 16.3 | 1095 |
Hippoglossoides robustus 289 | 30.6 | 31.1 | 22.2 | 16.2 | 1095 |
Hippoglossoides robustus 31 | 30.4 | 31.3 | 22.1 | 16.2 | 1095 |
Limanda sakhalinensis 71 | 30.8 | 30.1 | 23.4 | 15.7 | 1095 |
Limanda sakhalinensis 72 | 30.8 | 30.2 | 23.3 | 15.7 | 1095 |
Liopsetta pinnifasciata 13 | 29.9 | 30.9 | 22.9 | 16.3 | 1095 |
Liopsetta pinnifasciata 19-07 | 29.7 | 31.0 | 22.9 | 16.4 | 1095 |
Liopsetta pinnifasciata 20-07 | 29.5 | 31.3 | 23.1 | 16.1 | 1095 |
Liopsetta pinnifasciata 2K | 29.8 | 30.9 | 23.0 | 16.3 | 1095 |
Myzopsetta punctatissima 37 | 30.8 | 29.8 | 22.0 | 17.4 | 1095 |
Myzopsetta punctatissima 86-07 | 30.8 | 29.9 | 22.1 | 17.3 | 1095 |
Platichthys stellatus 18-07 | 30.0 | 30.6 | 22.6 | 16.7 | 1095 |
Platichthys stellatus 4K | 29.8 | 30.8 | 22.9 | 16.5 | 1095 |
Pseudopleuronectes herzensteini 05-07 | 29.9 | 31.4 | 21.4 | 17.4 | 1095 |
Pseudopleuronectes herzensteini 6K | 30.0 | 31.0 | 21.6 | 17.4 | 1095 |
Pseudopleuronectes herzensteini 7K | 30.0 | 31.2 | 21.4 | 17.4 | 1095 |
Pseudopleuronectes obscurus 09-07 | 30.5 | 30.1 | 22.6 | 16.7 | 1095 |
Pseudopleuronectes obscurus 45 | 30.4 | 30.3 | 22.2 | 17.1 | 1095 |
Pseudopleuronectes obscurus 46 | 30.4 | 30.1 | 22.6 | 16.9 | 1095 |
Mean | 30.29 ± 0.1 | 30.77 ± 0.1 | 22.42 ± 0.1 | 16.52 ± 0.1 | – |
Rights and permissions
About this article

Cite this article
Redin, A.D., Kartavtsev, Y.P. Phylogenetic Relationships of Flounders from the Family Pleuronectidae (Ostichties: Pleuronectiformes) Based on 16S rRNA Gene. Russ J Genet 57, 348–360 (2021). https://doi.org/10.1134/S102279542103011X
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S102279542103011X