
Abstract
This study aims to know the efficiency of the partial sequence of COI and 16S rRNA genes for the identification of crab species and to determine their phylogenetic relationships. A total of 36 sequences of 14 different species of crab were generated, belonging to 9 genera and 7 families. The average %GC content was higher in the sequences of the COI gene compared to the 16S rRNA. A large variation of the GC content was found at the 3rd codon position of the COI sequences. All the species were discriminated by COI and 16S rRNA when an intraspecific threshold of 2% K2P was used. The mean congeneric divergence was 72 and 94-fold higher than mean conspecific divergence for the COI and 16S rRNA markers, respectively. However, K2P% between closely related species was higher in COI compared to 16S rRNA. The Maximum Likelihood (ML) phylogenetic tree constructed based on COI and 16S rRNA gene sequences showed that the same species were clustered together under a single clade, supporting that both the markers were efficient in discriminating crab species. The ML tree of the COI sequences showed long-branch attraction and clustering of the species from different genus together. Such incongruence was not found in the tree topology of the 16S rRNA, providing phylogenetic relationships among species with a true divergence rate. The study revealed that the COI gene would be much efficient in discriminating closely related species, and 16S rRNA would be superior to COI in phylogenetic analysis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Crabs are the members of decapod crustaceans belonging to the Brachyura suborder with short stalked eyes; short, broad and more or less flattened bodies (carapace) with small abdomens that are folded under the thorax; inhabiting marine, brackish, or freshwater. Crab fishery is now emerging as an important sector in Bangladesh. The country exports mainly three crab species which includes mud crabs, Scylla serrata or other Scylla species; three spot swimming crabs, Portunus sanguinolentus and blue swimming crabs, Portunus pelagicus (Roy et al. 2012). In the fiscal year 2018–2019, Bangladesh has exported 470.23 metric tons of crabs after fulfilling the local demand (DoF 2019). A total of 38 crab species under 11 families are recorded from Bangladesh among them 18 are categorized as Data Deficient, 15 Least Concern and five Vulnerable (IUCN Bangladesh 2015). Most of them are either heavily exploited or under intense pressure from habitat destruction as well as anthropogenic and natural activities.
The authentic identification of organisms is crucial for biodiversity assessment and conservation. The identification of the brachyuran crabs is usually based on the morphometric and meristic characteristics. Traditional morphological identification sometimes becomes unable to discriminate look alike or damaged specimens. Molecular characterization enables to discriminate closely related species or cryptic species (Bezeng and van der Bank 2019), damaged specimens, eggs, larva (Brandão et al. 2016), or any stages of life where morpho taxonomy is incompetent. One of the key factors for the successful application of DNA barcoding is the availability of reliable sequences in reference libraries. Newly generated DNA barcodes can be checked for taxonomic conflicts, species identification and products analysis by comparing their sequences against this barcode reference library. The ambiguity in taxonomic identification of some crab’s species, description of new species and even mislabeling detection of crab species in the markets have successfully been resolved from different geographic locations (Abbas et al. 2016; Balasubramanian et al. 2014; Lai et al. 2010; Knowlton and Leray 2015; Raupach et al. 2015; Van der Meij et al. 2015) using the molecular approach.
DNA barcoding, using COI as a universal gene region and a standard analytical technique, greatly facilitated species discovery and identification in a wide variety of lineage (Hebert et al. 2003a, 2003b, 2004a, 2004b; Hajibabaei et al. 2006; Lopez-Vaamonde et al. 2021; Montes et al. 2017; Ševčík et al. 2016; Ward et al. 2005). In most organisms, the COI gene has been suggested as the standard barcoding marker, and the genetic distance and phylogenetic tree-based analysis are suggested as the ideal barcoding approaches (Hebert et al. 2003a; Ratnasingham and Hebert 2007; Ward et al. 2005). High species-level identification rates are well defined for many species based on COI barcoding, including 98% for marine fishes and 93.6% for birds (Ward 2009), 95.27% for northwestern Pacific mollusca (Sun et al. 2016), with increases in species diversity observed in many regions (Puckridge et al. 2013; Ward et al. 2008; Zemlak et al. 2009). However, some complexity, such as the hybridization and introgression of species and the discrimination of recently segregated species, concerns the use of COI barcoding (Moritz and Cicero 2004; Ward 2009; Ward et al. 2005).
As an alternative candidate barcode, mitochondrial markers like 16S ribosomal RNA could be considered. The 16S rRNA sequence as a conserved gene can measure the true divergences between distantly related organisms and can be easily amplified and sequenced across various animals (Lakra et al. 2009; Ma et al. 2015, 2013). In many organisms it has been used successfully to distinguish specific species, including Zoantharia (Sinniger et al. 2008), hydrozoans (Zheng et al. 2013), fishes (Chakraborty and Iwatsuki 2006; Lee et. al. 2014), and amphibians (Vences et al. 2005). The combination of conserved and variable regions makes the gene popular for reconstructing animal phylogenies (Vences et al. 2005), allowing the study of the old evolutionary relationship and also recent separation events.
The present study aims to explore the use of the COI and 16S rRNA gene in DNA barcoding of crabs of Bangladesh. We focus on interpreting pros and cons of two candidate barcodes in studying genetic divergence and understanding phylogenetic relationships among species. Understanding the effectiveness will allow us to make the definite use of the marker genes in diversity, evolution and conservation study.
Materials and methods
Specimen collection and species identification
The study was conducted in the southern region of Bangladesh. Sample specimens include adult crabs collected mainly from the Cox’s Bazar (21.43 N 91.82 E), Moheshkhali (21.29 N 91.53 E), Banshkhali (21.99 N 91.95 E), Hatiya (22.30 N 91.06 E) and Patuakhali (22.36 N 90.33 E) coastal areas, between July 2017 to December 2019. Immediately after collection, the crab specimens were kept in the cool icebox and carried to the Advanced Fisheries and DNA Barcoding Laboratory, Department of Zoology, University of Dhaka. Morphological identification of the collected species was preliminarily done during the field sampling and then validated based on the published taxonomic literature (Carpenter 2002; IUCN Bangladesh 2015; Ahmed et al. 2008). Tissue from the claws of each fresh specimen was dissected out with a sterile blade and preserved in 90% ethanol for further molecular analysis. The voucher specimens were deposited at the Dhaka University Zoology Museum (DUZM) and tagged with DUZM voucher ID.
DNA extraction, PCR amplification and DNA sequencing
DNA was isolated from a 5 mg tissue sample of each specimen using Invitrogen™ PureLink™ Genomic DNA Mini Kit. DNA was extracted following the manufacturer’s protocol. The quality and quantity of the extracted DNA was measured using NanoDrop™ spectrophotometer. COI and 16S rRNA gene sequences were amplified by polymerase chain reaction with the primer LCO-1490 (forward) 5’ TCAACAAATCATAAGGACATTGG 3’ and HCO-2198 (reverse) 5’ TAAACTTCAGGGTGTCCAAAGAATCA 3’ (Folmer et al. 1994) for COI and primer 16Sar (forward) 5’ CGCCTGTTTATCAAAAACAT 3’ and 16Sbr (reverse) 5’ CCGGTCTGAACTCAGATCATGT 3’ (Palumbi et al. 1991) for 16S rRNA sequences. The PCR was conducted in 25 µl volumes containing 23 µl of PCR Master Mix and 2 µl of DNA sample, mixed and spun for 30 s for homogenization of the mixture. PCR Master Mix consists of 12.5 µl Taq Polymerase, 8.5 µl Nano Pure water, 1 µl forward primer and 1 µl reverse primer. For both COI and 16S rRNA, the annealing temperature used was 54 ℃ for 30 s. The PCR amplifications were performed on Applied Biosystems Thermal Cycler (Thermo Fisher Scientific) under the following conditions: an initial denaturation at 95 ℃ for 5 min followed by 41 cycles of 95 ℃ for 30 s, 54 ℃ for 30 s, 72 ℃ for 1 min, and a final extension at 72 ℃ for 5 min. To protect the amplified gene from the damage the PCR products were kept at room temperature for 15 min, and then stored at -26 ℃ until further downstream application. PCR products were separated in 1% agarose gel, and purified using PureLink™ PCR purification kit. The good quality purified PCR products of DNA concentration > 10 ng/µl were sent to First BASE laboratories, Malaysia for sequencing. Sequencing was done by Sanger dideoxy sequencing technology using ABI PRISM 3730xl Genetic Analyzer exploiting the BigDye R Terminator v3.1 cycle sequencing kit chemistry.
Bioinformatics analysis
The assembled contigs were prepared by the CAP3 DNA assembly program using bioinformatics software Unipro Ugene (Okonechnikov et al. 2012). Analysis includes 36 DUZM COI and 16S rRNA sequences, along with 49 sequences of similar species retrieved from NCBI GenBank database including an outgroup Thenus indicus. All the sequences of COI and 16S rRNA were aligned automatically using MUSCLE and then adjusted manually (Edgar 2004). The boxplot distribution of the %GC content was constructed with the help of Rstudio platform (Team 2015). For distance-based method, genetic pairwise divergence for each marker was determined by calculating Kimura two-parameter (K2P) (Kimura 1980) distance using MEGA X (Kumar et al. 2018). Nucleotide saturation was tested by calculating the substitution saturation index using DAMBE version 7.0.35 (Xia 2018; Xia et al. 2003). Phylogenetic trees were constructed for COI and 16S rRNA sequences using Mega X (Kumar et al. 2018) based on Maximum Likelihood statistical method, where robustness of clustering was determined by bootstrap analysis with 1000 replicates.
Results
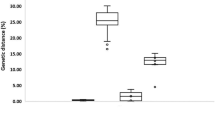
A total of 36 sequences were generated (20 COI and 16 16S rRNA) from 14 species of crabs belonging to 7 families. Among them Galene bispinosa (Family Galenidae), Charybdis japonica and Portunus reticulatus (Family Portunidae) were the newly recorded species from Bangladesh (Ahmed et al. 2021). Morphological identification of 14 species were further validated by molecular characterization based on both COI and 16S rRNA sequences, where the nucleotide Blast showed ≥ 96% identity with the available sequences and then deposited in the NCBI GenBank (Table 1). The average length of the aligned sequences was 596 bp and 483 bp for COI and 16S rRNA, respectively. 16S rRNA sequence of Zosimus aeneus was however shorter than others, with low identity coverage in Blast, which might be due to poor sequencing or DNA extraction. The alignment of the partial COI sequences showed a Maximum Likelihood estimate of the transition/transversion (R) bias 1.85. The nucleotide frequencies of the COI sequences were 25.84% (A), 36.37% (T/U), 20.69% (C), and 17.1% (G). The Maximum Likelihood estimate of the transition/transversion (R) bias was 3.135 for the 16S rRNA sequences, and the nucleotide frequencies were 34.8% (A), 34.72% (T/U), 11.38% (C), and 19.1% (G). The GC content calculated was summarized as boxplot distribution in Fig. 1, representing the %GC at the species level of the COI and 16S rRNA sequences. Among all the three codon positions of the COI sequences, large variation with the highest SEM value 1.185 was observed at the 3rd codon position. The overall %GC content was higher for the COI sequences with the mean value of 37.79 ± 2.02 in comparison with the 16S rRNA sequences of mean value 30.48 ± 1.26.

Boxplot distribution of the %GC content of the COI and 16S rRNA sequences at the species level
Genetic divergence pattern analyses
The K2P% genetic distances within each taxonomic level were summarized in Table 2. The average genetic distance for COI gene within species, genus and family were 0.234 ± 0.353, 16.89 ± 4.108 and 21.83 ± 2.360, respectively. In contrast, for 16S rRNA the average divergence within species, genus and family were 0.052 ± 0.197, 4.886 ± 1.311 and 9.799 ± 1.824, respectively. The pattern of K2P% divergence at different taxonomic ranks within species, genus, and family was plotted in Fig. 2. In both the markers, genetic divergence increased progressively with higher taxonomic level, which supports a marked change in genetic divergence at the species boundary.

Histogram of the K2P% divergence within species, genus and family of the a COI and the b 16S rRNA sequences
Saturation test
To identify saturation, the substitution saturation index Iss value was compared with the critical Iss.c value. For the COI sequences, Iss < Iss.c at 1st and 2nd codon position and Iss > Iss.c at 3rd codon position for both symmetrical and asymmetrical tree construction, indicating saturation at 3rd codon position. In the 16S rRNA sequences, Iss < Iss.c for the symmetrical and asymmetrical tree topology, suggesting little or no saturation.
Phylogenetic tree analysis
The intraspecific monophyletic clustering with high bootstrap percentage of 99–100% BP was observed for both markers, reflecting accurate taxonomic assignment of the species. However, in phylogeny within genus 16S rRNA has comparatively higher clade support than COI. Moreover, the phylogenetic tree of the COI sequences showed long-branch attraction (LBA) artifact, as interfamilial species were found to be in monophyly (Fig. 3). On the other hand, congeneric and confamilial sequences were clustered together with no phylogenetic discordant in the ML tree of the 16S rRNA (Fig. 4).

Maximum Likelihood (ML) phylogenetic tree constructed based on COI sequences. The sequences of the present study were represented as DUZM

Maximum Likelihood (ML) phylogenetic tree constructed based on 16S rRNA sequences. The sequences of the present study were represented as DUZM
Discussion
In this study, 36 partial sequences (20 COI and 16 16S rRNA) of 14 different crab species were successfully generated using two widely recognized identifying markers, COI and 16S rRNA. A series of comparative analysis were conducted for both the marker genes to clarify their strengths and drawbacks in species identification.
The mean of overall %GC content calculated where COI has 37.79 ± 2.02% GC (range: 34.6%-42.1%, SEM: 0.452) which was significantly higher than the 30.48 ± 1.26% GC of 16S rRNA (range: 28.3%-32.9%, SEM: 0.315) (p value < 0.0001). GC rich region has been proved to create incongruences in phylogenetic tree topology (Romiguier et al. 2010; Spencer 2006), as these regions have higher rate of evolution (Roux et al., 2016), which likely to cause long-branch attraction artifacts and issues related to heterotachy-driven biases (Philippe et al., 2005). Among COI sequences, the average %GC content was 50.67 ± 2.05, 41.70 ± 0.526 and 20.93 ± 5.30 for the 1st, 2nd and 3rd codon position, respectively (1st > 2nd > 3rd) (Fig. 1). However, the variation range was highest at 3rd codon among three codon positions of the COI sequences, ranging from 12.7–32.7% with SEM value 1.185, a similar pattern was observed in decapods (Matzen et al. 2011). The range of 1st and 2nd codon was however 46.6–53.6% and 40.9–42.9% with SEM value 0.459 and 0.118, respectively.
The pattern of mean K2P% within species < within genus < within family represents increased divergence with higher taxonomic levels for both the COI and 16S rRNA sequences. All the species could be discriminated efficiently for both the marker with a threshold of 2% divergence within the species (Hebert et al. 2003b). For the COI sequences, the mean K2P% divergence of individuals within species was 0.234 compared to 16.89 for species within the genus. Thus, congeneric species were approximately 72 times more divergent than conspecific individuals. Within genus the highest divergence of 22.68% was found between P. reticulatus and P. sanguinolentus and the lowest was 7.99% between P. reticulatus and P. pelagicus. Within family, the highest divergence was 27.65% between P. pelagicus and C. natator and the lowest was 18.24% between P. pelagicus and C. feriata (Table 2). In case of 16S rRNA marker, the mean divergence within genus was 4.886% which was 94-fold higher than the mean divergence 0.052% within species. The highest congeneric divergence was 5.93% between C. natator and C. feriata and the lowest was 2.459% between P. pelagicus and P. reticulatus. The highest confamilial divergence was 12.18% between P. reticulatus and C. natator and lowest was 5.979% between P. pelagicus and S. olivacea (Table 2). Higher mean divergence in congeneric COI sequences and large genetic divergence between closely related species indicated that COI could be better in discriminating against intragenic species than 16S rRNA. The utility of the species discrimination relies on the principle of the barcoding gap, estimated from the difference between the maximum K2P% within species and minimum K2P% within the genus. The value of the barcoding gap was 6.55% and 1.67% in COI and 16S rRNA, respectively.
Genetic saturation of each marker gene was studied for better understanding their efficiency in providing the phylogenetic signal. Similar to high GC content, genetic saturation is also responsible for creating long-branch attraction (Lartillot et al. 2007). The substitution saturation index was measured in COI and 16S rRNA sequences to test the saturation. At the 1st and 2nd codon position of the COI sequences, Iss 0.4307 was significantly lower than Iss.c 0.7056 (p < 0.0001). However, Iss 0.9929 was significantly higher than Iss.c 0.6265 (p < 0.0001) at the 3rd codon, making COI incongruous for phylogenetic tree construction (Fig. 3). In contrast, 16S rRNA had Iss value 0.4197 less than Iss.c 0.7207 (p < 0.0001), indicating no saturation, making the gene sequences more efficient for constructing species phylogeny.
Phylogenetic analysis among crab species have been reported in a number of studies (Haye et al. 2002; Hernández et al. 2019; Ocampo et al. 2013; Schubart et al. 2001; Scott Harrison 2004). However, it was difficult to contrast our results with those from other authors where different species were studied. Here we attempt to understand the evolutionary relationship among crabs commonly found in Bangladesh and also identify which marker is effective enough in providing true phylogenetic signal. Maximum Likelihood (ML) was chosen as statistical method for the phylogenetic analysis, due to their robustness. The lowest BIC and AICc value reveal the best fit substitution model GTR + G + I, and HKY + G for COI and 16S rRNA, respectively. Thus, the respective evolutionary model was chosen for the phylogenetic tree construction of each gene. In the ML analyses of the COI and 16S rRNA sequences, all the morphologically assigned species formed monophyletic clusters with strong bootstrap support (Figs. 3 and 4). No taxonomic deviation at the species level confirmed the reliability of the sequences and the efficiency of both the marker genes in species discrimination. Our study includes species mostly from family Portunidae, where genus Scylla was in paraphyly with Charybdis. Comparing the clade support within genus, 16S rRNA showed moderate to high percentage, with 87% BP within Charybdis and 98% BP within Portunus. In COI sequences, although P. pelagicus and P. reticulatus were grouped with maximum support, the clade within genus Charybdis was poorly supported by 43% BP. Also, within Charybdis, C. natator and C. japonica were closely clustered compared to C. feriata. Furthermore, in the COI phylogenetic tree, M. planipes, (family Matutidae) was clustered with P. sanguinolentus of a different family Portunidae, sharing their recent common ancestor with 52% BP (Fig. 3). This inefficiency in providing true relationship might results from the saturation at the 3rd codon position of the COI sequences. In contrast, the species of the same genus and family were grouped together with no branch length inconsistency observed in the ML tree topology of the 16S rRNA (Fig. 4). This evident that 16S rRNA would be much efficient in delineating species at the species, genus and family level and determining true divergence and evolutionary relationship among crab species compared to the COI. Whereas COI deficiently resolves the relationship between highly associated congeners due to the high GC content and substitution saturation, it was, however, better in differentiating closely related species when other markers show inadequate variability.
Conclusion
The present study demonstrated that both the COI and 16S rRNA genes could efficiently discriminate at species level. COI was better at distinguishing closely related crab species, showing a wide range of divergence within the genus and family. However, saturation and high %GC content at the 3rd codon position of the COI sequences, make the marker inefficient in providing true phylogenetic signal. Contrarily, 16S rRNA showed no substitution saturation and low %GC content, thus, proved to establish fewer incongruities in the phylogenetic tree construction. Further study with other crustaceans such as shrimps, lobsters, crayfish, prawns, krill, etc. might be performed to develop a strong conclusion for COI and 16S rRNA gene efficiency in the identification and phylogenetic delineation of crustaceans.
Data availability
The sequence data have been submitted to the NCBI GenBank database (https://www.ncbi.nlm.nih.gov/genbank/) under the accession numbers MT219299-MT219313, MN200404, MN200409-MN200411, MN200414, MT192548-MT192558, MT192560, MT192561, MT193621-MT193623, which have open public access.
References
Abbas EM, Abdelsalam KM, Mohammed-Geba K, Ahmed HO, Kato M (2016) Genetic and morphological identification of some crabs from the Gulf of Suez, Northern Red Sea. Egypt the Egyptian Journal of Aquatic Research 42(3):319–329. https://doi.org/10.1016/j.ejar.2016.08.003
Ahmed ATA, Kabir SMH, Ahmad M, Rahman AKA, Haque EU, Ahmed ZU, Begum ZNT, Hassan MA, Khondker M (2008) Encyclopedia of Flora and Fauna of Bangladesh. Volume 18. Part II. Arthropoda: Crustacea. Asiatic society of Bangladesh, Dhaka. pp 226
Ahmed MS, Afrin T, Barua A (2021) New distributional record of Charybdis japonica, Coenobita violascens, Galene bispinosa, and Portunus reticulatus (Crustacea: Decapoda) from Bangladesh waters of the Bay of Bengal. Regi Stud Marine Sci. https://doi.org/10.1016/j.rsma.2021.101785
Balasubramanian CP, Cubelio SS, Mohanlal DL, Ponniah AG, Kumar R, Bineesh KK, Ravichandran P, Gopalakrishnan A, Mandal A, Jena JK (2014) DNA sequence information resolves taxonomic ambiguity of the common mud crab species (Genus Scylla) in Indian waters. Mitochondrial DNA 27(1):270–275. https://doi.org/10.3109/19401736.2014.892076
IUCN Bangladesh (2015) Red list of Bangladesh volume 6: Crustaceans. IUCN, International Union for Conservation of Nature, Bangladesh Country Office, Dhaka, Bangladesh, pp xvi+256
Bezeng BS, van der Bank HF (2019) DNA barcoding of southern African crustaceans reveals a mix of invasive species and potential cryptic diversity. PLoS ONE 14(9):0222047. https://doi.org/10.1371/journal.pone.0222047
Brandão MC, Freire AS, Burton RS (2016) Estimating diversity of crabs (Decapoda: Brachyura) in a no-take marine protected area of the SW Atlantic coast through DNA barcoding of larvae. Syst Biodivers 14(3):288–302. https://doi.org/10.1080/14772000.2016.1140245
Carpenter KE (Ed) (2002) The living marine resources of the Western Central Atlantic. Volume 1: Introduction, molluscs, crustaceans, hagfishes, sharks, batoid fishes, and chimaeras. FAO Species Identification Guide for Fishery Purposes and American Society of Ichthyologists and Herpetologists. Special Publication No. 5. Rome, FAO, pp 1–600
Chakraborty A, Iwatsuki Y (2006) Genetic variation at the mitochondrial 16s rRNA gene among Trichiurus lepturus (Teleostei:Trichiuridae) from various localities: Preliminary evidence of a new species from west coast of Africa. Hydrpbiologia 563(1):501–513. https://doi.org/10.1007/s10750-006-0105-4
DoF (2019) Yearbook of Fisheries Statistics of Bangladesh, 2018–19 Fisheries Resources Survey System (FRSS), Department of Fisheries. Bangladesh. Ministry of Fisheries and Livestock 36:135
Edgar RC (2004) MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32(5):1792–1797. https://doi.org/10.1093/nar/gkh340
Folmer OM, Black M, Hoeh R, Lutz R, Vrijehoek R (1994) DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotech 5:304–313
Hajibabaei M, Janzen D, Burns J, Hallwachs W, Hebert PDN (2006) DNA barcodes distinguish species of tropical lepidoptera. PNAS 103(4):968–971. https://doi.org/10.1073/pnas.0510466103
Haye PA, Tam YK, Kornfield I (2002) Molecular phylogenetics of mole crabs (Hippidae: Emerita). J Crustac Biol 22(4):903–915
Hebert PDN, Cywinska A, Ball SL, deWaard JR (2003a) Biological identifications through DNA barcodes. Proceedings Biological Sciences 270(1512):313–321. https://doi.org/10.1098/rspb.2002.2218
Hebert PDN, Ratnasingham S, deWaard JR (2003b) Barcoding animal life: Cytochrome C oxidase subunit I divergences among closely related species. Proceedings Biological Sciences 270(Suppl 1):S96-99. https://doi.org/10.1098/rsbl.2003.0025
Hebert PDN, Stoeckle MY, Zemlak TS, Francis CM (2004a) Identification of birds through DNA barcodes. PLoS Biol 2(10):e312. https://doi.org/10.1371/journal.pbio.0020312
Hebert PDN, Penton EH, Burns JM, Janzen DH, Hallwachs W (2004b) Ten species in one: DNA barcoding reveals cryptic species in the neotropical skipper butterfly Astraptes fulgerator. Proc Natl Acad Sci USA 101(41):14812–14817. https://doi.org/10.1073/pnas.0406166101
Hernández M, Martín MV, Herrador-Gómez PM, Jiménez S, Hernández-González C, Barreiro S, López-Abellán LJ (2019) Mitocondrial COI and 16S rDNA sequences support morphological identification and biogeography of deep-sea red crabs of the genus Chaceon (Crustacea, Decapoda, Geryonidae) in the Eastern Central and South Atlantic Ocean. PLoS ONE 14(2):e0211717. https://doi.org/10.1371/journal.pone.0211717
Kimura M (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16(2):111–120. https://doi.org/10.1007/bf01731581
Knowlton N, Leray M (2015) Exploring Coral Reefs Using the Tools of Molecular Genetics. Coral Reefs in the Anthropocene. https://doi.org/10.1007/978-94-017-7249-5_6
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549
Lai JCY, Ng PKL, Davie PKF (2010) A revision of the Portunus pelagicus (Linnaeus, 1758) species complex (Crustacea: Brachyura: Portunidae), with the recognition of four species. The Raffles Bulletin of Zoology 58(2):199–237
Lakra WS, Goswami M, Gopalakrishnan A (2009) Molecular identification and phylogenetic relationships of seven Indian sciaenids (Pisces: Perciformes, Sciaenidae) based on 16s rRNA and Cytochrome C oxidase subunit I mitochondrial genes. Mol Biol Rep 36(5):831–839. https://doi.org/10.1007/s11033-008-9252-1
Lartillot N, Brinkmann H, Philippe H (2007) Suppression of long-branch attraction artefacts in the animal phylogeny using a site-heterogeneous model. BMC Evol Biol 7:S4. https://doi.org/10.1186/1471-2148-7-S1-S4
Lee M, Munroe TA, Shao K (2014) Description of a new cryptic, shallow water tonguefish (Pleuronectiformes: Cynoglossidae: Symphurus) from the Western North Pacific Ocean. J Fish Biol 85(3):563–585. https://doi.org/10.1111/jfb.12440
Lopez-Vaamonde C, Kirichenko N, Cama A, Doorenweerd C, Godfray C, Guiguet A, Gomboc S, Huemer P, Landry JF, Lastuvka A, Lastuvka Z, Kyungmin L, Lees D, Mutanen M, van Nieukerken E, Segerer A, Triberti P, Wieser C, Rougerie R (2021) Evaluating DNA barcoding for species identification and discovery in European gracillariid moths. Front Ecol Evol. https://doi.org/10.3389/fevo.2021.626752
Ma H, Ma C, Li X, Xu Z, Feng N, Ma L (2013) The complete mitochondrial genome sequence and gene organization of the mud crab (Scylla paramamosain) with phylogenetic consideration. Gene 519(1):120–127. https://doi.org/10.1016/j.gene.2013.01.028
Ma H, Ma C, Li C, Lu J, Zou X, Gong Y, Wang W, Chen W, Ma L, Xia L (2015) First mitochondrial genome for the red crab (Charybdis feriata) with implication of phylogenomics and population genetics. Sci Rep. https://doi.org/10.1038/srep11524
Matzen da Silva J, Creer S, dos Santos A, Costa AC, Cunha MR, Costa FO, Carvalho GR (2011) Systematic and evolutionary insights derived from mtDNA COI barcode diversity in the Decapoda (Crustacea: Malacostraca). PLoS ONE 6(5):e19449. https://doi.org/10.1371/journal.pone.0019449
Montes MM, Castro-Romero R, Martorelli SR (2017) Morphological identification and DNA barcoding of a new species of Parabrachiella (Siphonostomatoida: Lernaeopodidae) with aspects of their intraspecific variation. Acta Trop 173:34–44. https://doi.org/10.1016/j.actatropica.2017.05.025
Moritz C, Cicero C (2004) DNA barcoding: Promise and pitfalls. PLoS Biol 2(10):e354. https://doi.org/10.1371/journal.pbio.0020354
Ocampo EH, Robles R, Terossi M, Nuñez JD, Cledón M, Mantelatto FL (2013) Phylogeny, phylogeography, and systematics of the American pea crab genusCalyptraeotheres Campos, 1990, inferred from molecular markers. Zool J Linn Soc 169(1):27–42. https://doi.org/10.1111/zoj.12045
Okonechnikov K, Golosova O, Fursov M (2012) Unipro ugene: a unified bioinformatics toolkit. Bioinformatics 28(8):1166–1167. https://doi.org/10.1093/bioinformatics/bts091
Palumbi SR, Martin A, Romano S, Mcmillan WV, Stice L, Grabowski G (1991) The Simple Fool’s Guide to PCR. University of Hawaii, Honolulu, p 46
Philippe H, Zhou Y, Brinkmann H, Rodrigue N, Delsuc F (2005) Heterotachy and long-branch attraction in phylogenetics. BMC Evol Biol 5(1):50. https://doi.org/10.1186/1471-2148-5-50
Puckridge M, Andreakis N, Appleyard SA, Ward RD (2013) Cryptic diversity in flathead fishes (Scorpaeniformes: Platycephalidae) across the Indo-West Pacific uncovered by DNA barcoding. Mol Ecol Resour. https://doi.org/10.1111/1755-0998.12022
Ratnasingham S, Hebert PDN (2007) Barcoding: bold: The barcode of life data system. Mol Ecol Notes 7(3):355–364. https://doi.org/10.1111/j.1471-8286.2007.01678.x
Raupach MJ, Barco A, Steinke D, Beermann J, Laakmann S, Mohrbeck I, Neumann H, Kihara TC, Pointner K, Radulovici A, Segelken-Voigt A, Wesse C, Knebelsberger T (2015) The Application of DNA Barcodes for the Identification of Marine Crustaceans from the North Sea and Adjacent Regions. PLoS ONE 10(9):0139421. https://doi.org/10.1371/journal.pone.0139421
Romiguier J, Ranwez V, Douzery EJ, Galtier N (2010) Contrasting GC-content dynamics across 33 mammalian genomes: Relationship with life-history traits and chromosome sizes. Genome Res 20(8):1001–1009. https://doi.org/10.1101/gr.104372.109
Roux C, Fraïsse C, Romiguier J, Anciaux Y, Galtier N, Bierne N (2016) Shedding light on the grey zone of speciation along a continuum of genomic divergence. PLoS Biol 14(12):2000234. https://doi.org/10.1371/journal.pbio.2000234
Roy BJ, Singha NK, Ali SH, Rhaman MG (2012) Exploitation of marine swimming crab in Cox’s Bazar. Bangladesh Bangladesh Journal of Zoology 40(2):241–245. https://doi.org/10.3329/bjz.v40i2.14318
Team R (2015) RStudio: integrated development for R. RStudio, Inc., Boston, MA 42(14):84. http://www.rstudio.com/
Schubart CD, Cuesta JA, Rodríguez A (2001) Molecular phylogeny of the crab genus Brachynotus (Brachyura: Varunidae) based on the 16S rRNA gene. Hydrobiologia 449:41–46
Scott HJ (2004) Evolution, biogeography, and the utility of mitochondrial 16s and COI genes in phylogenetic analysis of the crab genus Austinixa (Decapoda: Pinnotheridae). Mol Phylogenet Evol 30(3):743–754. https://doi.org/10.1016/s1055-7903(03)00250-1
Ševčík J, Kaspřák D, Rulik B (2016) A new species of Docosia Winnertz from Central Europe, with DNA barcoding based on four gene markers (Diptera, Mycetophilidae). Zookeys 549:127–143
Sinniger F, Reimer JD, Pawlowski J (2008) Potential of DNA sequences to identify zoanthids (Cnidaria: Zoantharia). Zoological Sciences 25(12):1253–1260. https://doi.org/10.2108/zsj.25.1253
Spencer CCA (2006) Human polymorphism around recombination hotspots. Biochem Soc Trans 34(Pt 4):535–536. https://doi.org/10.1042/BST0340535
Sun S, Li Q, Kong L, Yu H, Zheng X, Yu R, Dai L, Sun Y, Chen J, Liu J, Ni L, Feng Y, Yu Z, Zou S, Lin J (2016) DNA barcoding reveal patterns of species diversity among Northwestern Pacific molluscs. Sci Rep. https://doi.org/10.1038/srep33367
Van der Meij SET, Berumen ML, Paulay G (2015) A new species of Fizesereneia Takeda & Tamura, 1980 (Crustacea: Brachyura: Cryptochiridae) from the Red Sea and Oman. Zootaxa 3931(4):585. https://doi.org/10.11646/zootaxa.3931.4.8
Vences M, Thomas M, van der Meijden A, Chiari Y, Vieites DR (2005) Comparative performance of the 16S rRNA gene in DNA barcoding of amphibians. Front Zool 2:1–5. https://doi.org/10.1186/1742-9994-2-5
Ward RD (2009) DNA barcode divergence among species and genera of birds and fishes. Mol Ecol Resour 9:1077–1085. https://doi.org/10.1111/j.1755-0998.2009.02541.x
Ward RD, Zemlak TS, Innes BH, Last PR, Hebert PDN (2005) DNA barcoding Australia’s fish species. Philosophical Transactions of the Royal Society B: Biological Sciences 360(1462):1847–1857. https://doi.org/10.1098/rstb.2005.1716
Ward RD, Holmes BH, Yearsley G (2008) DNA barcoding reveals a likely second species of Asian sea bass (barramundi) (Lates calcarifer). J Fish Biol 72(2):458–463. https://doi.org/10.1111/j.1095-8649.2007.01703.x
Xia X (2018) Dambe7: New and improved tools for data analysis in molecular biology and evolution. Mol Biol Evol 35(6):1550–1552. https://doi.org/10.1093/molbev/msy073
Xia X, Xie Z, Salemi M, Chen L, Wang Y (2003) An Index of Substitution Saturation and Its Appl 26(1):1–7. https://doi.org/10.1016/S1055-7903(02)00326-3
Zemlak TS, Ward RD, Connell AD, Holmes BH, Hebert PDN (2009) DNA barcoding reveals overlooked marine fishes. Mol Ecol Res 9:237–242. https://doi.org/10.1111/j.1755-0998.2009.02649.x
Zheng L, He J, Lin Y, Cao W, Zhang W (2013) 16s rRNA is a better choice than COI for DNA barcoding hydrozoans in the coastal waters of China. Acta Oceanol Sin 33(4):55–76. https://doi.org/10.1007/s13131-014-0415-8
Acknowledgements
We sincerely acknowledge the partial financial support from the Ministry of Science and Technology, Government of the Peoples Republic of Bangladesh to the first author as a form of NST Fellowship (2019-2020). We are thankful to Mr. Sujan Kumar Datta and Mr. Tonmoy Saha for their cooperation during sample collection.
Author information
Authors and Affiliations
Contributions
AMS initiated, design and supervised whole experiment; performed the taxonomic analyses and prepared the manuscript. AAA participated experimental design, logistics and manuscript preparation. BA contributed laboratory analyses, participated in generation of sequences and made substantial contribution to the manuscript preparation. AT performed field collection, the taxonomic analyses, laboratory analysis and participated in the writing of the final manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Barua, A., Afrin, T., Akhand, A.A. et al. Molecular characterization and phylogenetic analysis of crabs (Crustacea: Decapoda: Brachyura) based on mitochondrial COI and 16S rRNA genes. Conservation Genet Resour 13, 291–301 (2021). https://doi.org/10.1007/s12686-021-01212-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12686-021-01212-9