
Abstract
Differential gene expression during development is maintained by complex regulatory epigenetic mechanisms that provide the formation of different specialized cell types. Subsequently, a multicellular organism is a mosaic of cells with differing epigenetic characteristics. It seems likely that exceeding the limits of normal epigenetic variability may cause the occurrence of pathological mosaic states in which one part of the cell population has a normal epigenotype, while the other part carries modified epigenetic information. In this review, using the genomic imprinting as a classical epigenetic phenomenon, for the first time, the prevalence of epigenetic mosaicism and the mechanisms of its origin, as well as its role in the etiology of hereditary disorders, determined by the dysfunction of imprinted genomic loci are summarized.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Somatic cells of diploid mammalian organism possess two parental copies of the genes. This provides paternal and maternal loci the potential to be equally active in the genome of the offspring. Genomic imprinting is a phenomenon that violates this biallelic expression of both paternal and maternal genes, leading to their monoallelic expression and haploidization. To date, more than 100 imprinted genes have been identified in mammals [1]. These are mainly protein-coding genes that play a key role in providing normal embryonic development through influencing the expression levels of the genes controlling fetal growth, cell proliferation and differentiation, and other processes concerning fetal development, placenta formation, CNS, and metabolism [2].
Epigenetic mosaicism, like any form of genetic mosaicism, can be defined as the presence of different epigenotypes in different cell populations within an organism developed from a single zygote. However, unlike structural mosaicism of somatic gene mutations or chromosomal abnormalities, epigenetic mosaicism is rather difficult to differentiate from normal epigenetic polymorphism, in which different cells of the body, even within the same cell type, have specific epigenetic characteristics. Obviously, consideration of the issue of the possible pathogenetic value of epigenetic mosaicism (or epimosaicism) should imply the existence of a certain border between normal and pathological epigenetic variability. Concerning imprinted genes, it should be mentioned that some of them have these unique features and can be expressed differently in different tissues. Moreover, some genes are characterized by bidirectional imprinting, when in the cells of some tissues the expression of a maternal allele takes place, while in the others a paternal allele is expressed [3]. The occurrence of tissue-specific mosaicism of these genes suggests that their methylation status can be established not during the formation of gametes, but, possibly, even at different periods of ontogeny. All this leads to the fact that, in a healthy organism, some imprinted genes are active in some cells, while in other cells other genes are active, and some loci are active in the prenatal period, while completely different loci are active during postnatal ontogenesis and in its different periods. The question then arises as to when and how such normal epigenetic variability becomes pathological.
Disturbance of methylation polymorphism of imprinted genes leads to the formation of genomic imprinting disorders that challenge current molecular diagnostics, including clinical and molecular heterogeneity, overlapping clinical features, and multiple epimutations in imprinted genes. Somatic inter- and intratissue epimosaicism becomes another problem in the diagnostics of genomic imprinting disorders, since the presence of a normal cell clone can attenuate or modify clinical features of these syndromes. In this regard, it cannot be excluded that mosaic variants of genomic imprinting disorders may be missed by clinicians. In the present review, we attempted to characterize and differentiate these epigenetic mosaicism cases using the example of imprinted genomic loci.
DIFFERENCES IN THE IMPRINTED GENE EXPRESSION BETWEEN TISSUES
At present, tissue-specific imprinting is known for such imprinted genes as KCNQ1OT1 (11p15.5), UBE3A (15q11), SNURF-SNRPN (15q11), IGF2R (6q25.3), GRB10 (7p21), MEST (7q32.2), and some others (Table 1). These genes violate classical concept of imprinted genes, which show monoallelic expression from only one allele in all tissues. However, it turned out that epigenetic tissue mosaicism is a normal phenomenon in the development of the organism.
For instance, in humans, the IGF2R gene (OMIM 147280, 6q25.3) is characterized by polymorphic imprinting, i.e., monoallelic expression in all tissues, except the brain, where its biallelic expression is observed [5]. At the same time, in mice, monoallelic expression from only maternal allele in all tissues is observed. This is an example of not only tissue-specific but also evolutionary epipolymorphism.
Another gene with monoallelic expression is the gene encoding Bladder Cancer-Associated Protein, BLCAP (OMIM: 613110, 20q11.23; previously BC10), which is a tumor suppressor that limits cell proliferation and stimulates apoptosis. The BLCAP protein is absent from many types of human tumors. This gene is imprinted in mice and humans and demonstrates monoallelic maternal expression in the brain with biallelic expression in the placenta, testicles, heart, lungs, liver, and skeletal muscle tissue [3, 9].
One more gene characterized by variation in allelic expression between tissues is GRB10 (OMIM 601523, 7p12.2). It has a number of isoforms with bidirectional monoallelic expression, i.e., only from paternal allele in human fetal brain and spinal cord, and only from maternal allele in the placenta [4, 10]. All other fetal tissues, including lung, limbs, umbilical cord, skin, kidneys, and organs adrenal, pancreas, liver, and heart, demonstrated biallelic gene expression patterns. Maternal expression of GRB10 in the placenta may be evolutionarily important, presumably, to control fetal growth. Indeed, the loss of maternal allele expression in embryogenesis resulted in fetal and placental overgrowth, which disrupts correct distribution of maternal resources to this process [11]. Garfield et al. [12] showed that disruption of monoallelic expression of Grb10 in the mouse brain resulted in increased social dominance, especially among other aspects of social behavior, as evidenced by the observed increase in allogrooming in the Grb10 deficient males. At the same time, it was demonstrated that, in the knockout Grb10 mice, this gene primarily affected the stability of social behavior, rather than social dominance [13]. The presence of the opposite monoallelic expression in the placenta compared to brain supports the hypothesis that GRB10 imprinting could have evolved to separate different roles of this gene in the mammalian growth and behavior.
The WT1 gene (Wilms tumor 1 gene, OMIM 607102, 11p13) is the imprinted gene with monoallelic expression of only paternal homolog in fibroblasts and lymphocytes, monoallelic maternal expression in the placenta and brain, and biallelic expression in other organs (heart, lung, liver, and intestine) [14].
The UBE3A gene (encodes a ubiquitin E3 ligase, OMIM 601623, 15q11.2) in humans shows imprinted monoallelic expression only in the brain, namely, in the Purkinje cells of the cerebellar cortex and the hippocampal neurons [15].
GNAS (OMIM 139320, 20q13.3) is a complex locus in terms of epigenetic regulation, which encodes one biallelic (Gsα) and four monoallelic (NESP55, GNAS-AS1, XLsα, and A/B) transcripts. XLAS is expressed exclusively from paternal allele, NESP55 is expressed from maternal allele, while Gsα is expressed biallelically. Depending on the tissue, either biallelic or monoallelic expression takes place. For example, in most tissues, both alleles are transcribed with the formation of Gsα. In neuroendocrine tissues of the brain and spinal cord and in the heart, kidney, lung, and muscles, only maternal allele of the GNAS locus is expressed, which encodes the NESP55 transcript [16]. The NESP55 promoter contains a specific differentially methylated region (DMR), which is methylated on paternal allele, because of which expression takes place only from maternal chromosome. NESP55 encodes a chromogranin-like neuroendocrine secretory protein and participates in the regulation of methylation at GNAS DMR, which affects expression of this locus [17]. Gsα is expressed biallelically in most tissues, excluding a small number of tissues, such as renal proximal tubules, thyroid, gonads, and pituitary, where it is monoallelically expressed only from maternal GNAS allele. XLAS is a variant of Gsα, which is expressed exclusively from paternal GNAS allele, primarily in neuroendocrine tissues and the nervous system.
Campbell et al. [18] showed that GNAS was biallelically expressed in a wide range of human fetal tissues. They also showed that Gsα was monoallelically expressed from maternal allele in a normal adult pituitary, while biallelic expression of Gsα was detected in pituitary tumor cells, regardless of GNAS abnormalities. These results pointed to the possible role of the loss of Gsα imprinting in pituitary tumors.
Thus, more and more imprinted genes with polymorphic imprinting have recently been identified and, most likely, it may turn out that such genes are not the exception, but the rule. The determination of imprinted expression of a gene in a certain tissue does not mean that this gene will be imprinted in all other tissues. Conversely, the absence of imprinted status of a gene in some tissues does not exclude its presence in the others. These data support the suggestion that tissue-specific epigenetic gene modification is one of the main mechanisms that ensure differential gene expression in different tissues during development. Disruption of the epigenetic balance of the dosage of imprinted genes leads to the formation of genomic imprinting disorders.
GENOMIC IMPRINTING DISORDERS AND EPIGENETIC MOSAICISM
Epigenetic mosaicism was described in a number of genomic imprinting disorders, including hydatidiform mole, Angelman syndrome (AS), Prader–Willi syndrome (PWS), Silver–Russell syndrome (SRS), Beckwith–Wiedemann syndrome (BWS), Temple syndrome, pseudohypoparathyroidism 1B (PHP1B), and transient neonatal diabetes mellitus (TNDM).
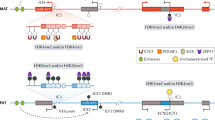
The hydatidiform mole (HM, OMIM 231090) is an abnormal pregnancy accompanied by morphological changes in the chorionic cytotrophoblast. Complete hydatidiform mole (CHM) is characterized by the presence of diploid androgenic trophoblast cells having two haploid genomes of paternal origin. The most frequent mechanism of the CHM formation is the oocyte fertilization by two haploid spermatozoa or one diploid spermatozoon with the subsequent elimination of maternal pronucleus (diandry) (Fig. 1). Sometimes, CHM results from a loss of maternal chromosome set in the oocyte due to abnormal segregation of two haploid sets in the polar body and duplication of haploid paternal genome in the zygote, resulting in so-called homozygous moles. The zygote has 46,ХХ karyotype.

The mechanisms of hydatidiform mole formation. dup, duplication. For the right half of the figure: n, the maternal genome; n, the genome of the first spermatozoon; n, the genome of the second spermatozoon.
Interestingly, in some cases, CHM can develop in the case of normal biparental diploid karyotype of the zygote. This is biparental complete hydatidiform mole (BCHM). Analysis of the epigenetic aspects of BCHM revealed global disruptions in the imprinted gene methylation patterns on chromosomes of maternal origin. It was demonstrated that in a diploid zygote, a number of maternal alleles of imprinted genes acquired paternal epigenotype, and this condition was functionally equivalent to the inheritance of both alleles from the father, as in classical variant of diandrogenetic CHM (Fig. 1). For instance, hypomethylation of the SNRPN, PEG1, PEG3, and KCNQ1OT1 genes imprinted on maternal chromosomes was revealed, which led to their biallelic expression from both parental homologs [19].
In addition to the above-mentioned forms of HM, mosaic cases of this pathology were described. Sunde et al. [20] among 162 cases of diploid hydatidiform mole, identified eight cases of mosaicism, i.e., mixtures of androgenetic diploid and biparental diploid cells with the trophoblast abnormalities developed as a CHM. The authors suggested that mosaic HMs covering the androgenetic cell population were the result of different postzygotic abnormalities, including duplication of paternal pronucleus, asymmetric cytokinesis, and postzygotic diploidization. These findings support the proposal that fertilization of an empty oocyte is not a mandatory event for the creation of androgenic cell population.
Possible mechanisms of formation of HM mosaic variants (Fig. 1) include the following: (a) Fertilization of oocyte (n) with two spermatozoa (n + n). Endoreduplication results in the appearance of cells with paternal and maternal genomes in the population of diploid parental cells and the appearance of cells with two paternal genomes in the population of diploid androgenic cells. (b) Fertilization of oocyte with maternal genome by two spermatozoa, followed by endoreduplication and duplication of paternal genome. This results in the appearance of both androgenetic and biparental diploid cells. (c) Oocyte fertilization by one spermatozoon followed by duplication of paternal pronucleus and the formation of a triploid zygote, followed by endoreduplication and then another duplication of paternal genome. Finally, in the same way as the two above-mentioned cases, diploid cells with diandrogenetic HM and biparental cells with paternal and maternal genomes are formed [20].
Recently, it was demonstrated that during cytokinesis, mammalian zygotes could spontaneously segregate entire parental genomes into distinct blastomeres [21]. The molecular mechanisms underlying the occurrence of blastomeres with different parental genomes during the first mitotic cycle remain unclear. However, the first zygotic metaphase causes asymmetric interactions between the mitotic spindle and parental kinetochores. This leads to the formation of heterogeneous blastomeres with different ploidy levels. For instance, tripolar cytokinesis results in the formation of blastomeres with diploid biparental genomes, haploid paternal genomes, and haploid maternal genomes. All this can lead to the clinical picture of HM. Thus, epigenetic mosaicism at the level of entire haploid chromosome sets can be formed as a result of fertilization, cytokinesis, and chromosomal segregation abnormalities, as well as from the errors in genomic imprinting establishment and maintenance.
Transient neonatal diabetes mellitus (OMIM 601410, chromosome region 6q24, population frequency 1 : 400 000) is characterized by intrauterine growth retardation and neonatal hyperglycemia. The diabetes mellitus typically develops at birth or in adolescence. The patients may have macroglossia and umbilical hernia, and sudden unexpected death in newborns is also possible.
TNDM is associated with hypomethylation of the HYMAI and PLAGL1 genes. The first gene is a tumor suppressor and encodes untranslated RNA. The product of the PLAGL1 tumor suppressor gene is a transcription factor, which, together with the p53 protein, is involved in the regulation of the cell cycle through the control of the G1/S phase transition and the initiation of apoptosis. It inhibits cell growth and is expressed only from the paternal homolog. In this regard, hypomethylation of this gene can lead to a decrease in cell proliferative activity.
Mackay et al. [22] examined 12 patients with TNDM and showed a mosaic hypomethylation spectrum at one or simultaneously at a number of imprinted genes, as well as at genes that in normal conditions are not expressed on maternal homolog, GRB10 (7p12), PEG1 (7q21), KCNQ1OT1 (11p15.5), and PEG3 (7q21), while there was no change in the methylation level of paternal methylated H19 locus (11p15.5). Peripheral blood, skin fibroblasts, and buccal epithelium were examined. It was demonstrated that, in blood lymphocytes of all patients, there was no mosaicism of hypomethylation of these genes, whereas in skin fibroblasts and buccal epithelium, hypomethylation was mosaic. At the same time, the main disease-specific imprinted genes from the 6q24 region (HYMAI and PLAGL1) were completely hypomethylated in all tissues; i.e., they showed no mosaicism of hypomethylation. No changes in the methylation pattern of the studied genes were found in the parents of the probands. The authors found no phenotypic differences in patients with mosaicism of hypomethylation at the imprinted genes. It seems likely that this was associated with the fact that the main genes that determine this syndrome were not involved in mosaicism.
Tissue-specific epigenetic mosaicism, manifested as multilocus methylation defects of the imprinted genes (MLID, multilocus imprinting defects) in TNDM, suggests the presence of some cis- or trans-acting factor. Indeed, in TNDM patients with MLID, mutations in the ZFP57 gene were found, which was indicative of possible involvement of this gene in maintaining maternal and paternal methylation status in the embryonic somatic cells after fertilization [23]. Unfortunately, the authors of [22] did not search for the ZFP57 mutations in patients with mosaic MLID and TNDM.
ZFP57 (OMIM 612192, 6p22.1) encodes a protein that has a KRAB domain associated with Cys2His2 zinc fingers. KRAB is transcriptional repressor that acts through protein-protein interactions with KAP1/TIF1b/Trim28, which can initiate de novo methylation of mouse DNA during embryogenesis. Zfp57 is required to maintain the methylation status of imprinted genes, especially during the period of epigenetic reprogramming during preimplantation development. After fertilization, the paternal genome is actively demethylated, while demethylation of the maternal genome occurs passively. In the zygote, DPPA3 protects the maternal genome and imprinted genes from active demethylation by binding to dimethylated histone H3 lysine 9 (H3K9me2). The ZFP57/TRIM28 complex provides the delivery of DNMT1 DNA methyltransferase to regions of imprinted genes, which ensures the stability of their methylation during reprogramming [24, 25].
Silver–Russell syndrome (OMIM 180860, subchromosomal regions 11p15.5 and 7p11.2, population frequency 1 : 10 000–30 000) combines intrauterine and postnatal growth retardation, low body mass index, and small triangular face. In 50% of patients, skeletal asymmetry, sexual ambiguity, and learning difficulties are observed. Pre- and postnatal growth retardation together with hemihypotrophy represent an opposite phenotype to the overgrowth condition and hemihypertrophy characteristic of BWS.
The absence of the MEST, CDKN1C, IGF2, and KCNQ1OT1 gene products, as well as increased dosage of the H19 gene, can lead to SRS. The MEST gene (7q32) product is involved in the regulation of embryonic and placental growth, and CDKN1C (11p15.5) is a negative regulator of cell proliferation. KCNQ1OT1 (11p15.5) is one of two imprinting centers (IC) in the 11p15 imprinted gene cluster, which encodes a long noncoding RNA that inhibits the activity of adjacent imprinted genes. The IGF2 gene product is a fetal growth factor, and from H19, long noncoding RNA is transcribed that functions as a tumor suppressor. One of the causes of SRS is also the biallelic expression of GRB10, which is the gene for growth factor receptor-bound protein 10. GRB10 belongs to adapter proteins that interact with a number of receptor tyrosine kinases and signaling molecules. The GRB10 protein product interacts with insulin and insulin-like growth factor receptors through the SH2 domain, inhibiting tyrosine kinase activity, which is involved in stimulation of insulin activity and insulin-like growth factors I and II, regulating growth, development, and differentiation of cells and tissues. Thus, GRB10 has a suppressive effect on growth, and increased dosage of this gene may provide suppression of cellular growth [26].
Fuke-Sato et al. [27] described a child with SRS and mosaic uniparental disomy (UPD) of chromosome 7 of maternal origin (mosaic UPD(7)mat). The child had all clinical features of SRS, except for skeletal asymmetry (intrauterine and postnatal growth retardation, low body mass index, small triangular face, and hemihypotrophy). The authors showed that the clone of UPD(7)mat cells constituted 92% in peripheral blood leukocytes, 91% in buccal epithelium cells, and 11% in placental tissue. Similar to epimutations, UPD disrupts the balance of imprinted gene dosage, and therefore, UPD mosaicism can lead to the emergence of epigenetic mosaicism of imprinted genes.
Genetic and epigenetic anomalies involving imprinted genes of the 11p15 region can also cause SRS; i.e., epigenetic and genetic abnormalities in Silver–Russell and Beckwith–Wiedemann syndromes are reciprocal to each other: hypomethylation (SRS) versus hypermethylation (BWS) of the IGF2/H19 imprinting center, maternal duplication of 11p15 region (SRS) versus its paternal duplication (BWS), uniparental disomy of chromosome 11 of maternal origin (SRS) versus paternal UPD of chromosome 11 (BWS).
Fontana et al. [28] revealed mosaic hypomethylation of IGF2/H19 IC (methylation index (MI) ranged from 25 to 36%) in seven SRS patients. Methylation levels were evaluated using the MassArray EpiTYPER system with nucleotide-specific cleavage and subsequent product identification using MALDI-TOF mass spectrometry. In addition, in two patients, hypomethylation of other imprinted loci, MEST (MI 16%) in one patient and MEST (MI 22%) and GNASXL (MI 31%) in the other patient, was observed. One of these patients carried a heterozygous missense mutation in the ZFP42 gene (4q35.2) inherited from the father. In patients with epimosaicism and SRS, no clinical specificities were identified; i.e., their phenotypes correspond to classical SRS.
Hiura et al. [29] described five children with SRS who were born after using assisted reproductive technologies. All patients had hypomethylation of IGF2/H19 IC. In four of these patients, mosaic MLID with the involvement of other imprinted genes, MEST, CDKN1C, IGF2, and KCNQ1OT1, was observed, pointing to the imprinting disruption after fertilization. Whole-genome sequencing in patients with epigenetic mosaicism of MLID to search for genes possibly controlling this pathological process was not carried out in this study. Interestingly, all five patients with epigenetic mosaicism had classical SRS phenotype.
Bullman et al. [30] described a child with SRS and mosaic maternal uniparental isodisomy of chromosome 11. Using the MS-MLPA method, it was demonstrated that the mosaicism level of UPD(11)mat was 18%. The child had typical SRS phenotypes, including intrauterine and postnatal growth retardation, low body mass index at birth (1.76 kg), triangular face, while there was no asymmetry of the extremities. Thus, the presence of even a small clone of cells with UPD, leading to epigenetic disruption of imprinted genes, led to the formation of the clinical picture of this syndrome.
In some cases, in mothers of patients with SRS and MLID, combined with IGF2/H19 hypomethylation, the NLRP5 mutation was observed [31]. NLRP5 (NALP5, OMIM 609658, 19q13.43, homolog of mouse Mater) is a cytosolic protein, a Nod-like receptor of the NALP family, that regulates early embryogenesis and apoptosis. It plays a certain role in early embryogenesis, participating in activation of the embryonic genome, degradation of maternal RNA product, and regulation of the organelle functioning [32]. The corresponding gene can function as trans-acting factor that is caused epigenetic disturbances in SRS. Another gene with similar function can be ZFP42 (OMIM 614572, 4q35.2), which was found in a patient with SRS and epigenetic mosaicism of MLID. The encoded protein belongs to the family of KRAB domain zinc finger proteins and is a DNA-binding transcription factor. Dramatic suppression of gene expression occurs at the beginning of cell differentiation.
Beckwith–Wiedemann syndrome (OMIM 130650, subchromosomal region 11p15.5, population frequency 1 : 13 700) includes clinical features as gigantism, macroglossia, macrosomia, visceromegaly, hemihyperplasia, abdominal wall defects, umbilical hernia, microcephaly, earlobe pits, pigmented nevi, and neonatal hypoglycemia. Wilms tumor is described in 7.5% of cases. Patients with BWS are at risk of developing neuroblastoma, hepatoblastoma, and adrenal tumors. The same molecular alterations as in BWS are found in approximately 30% of patients with isolated hemihyperplasia (OMIM 235000) [33]. There is an increased risk of BWS and KCNQ1OT1 hypomethylation in children born by assisted reproductive technologies [34].
The 11p15.5 region contains at least 12 imprinted genes that are regulated by two separated by unimprinted regions. The imprinted H19 and IGF2 genes located in this chromosomal subregion are regulated in a coordinated manner owing to the competition of their promoters for access to a common enhancer, which plays an important role in regulating the activity of both loci. On the maternally inherited chromosome, the enhancer activates transcription of H19, from which untranslated RNA is read, and the IGF2 gene is in an inactive state. On the chromosome of paternal origin, methylation of the H19 locus results in that the IGF2 gene becomes available for the enhancer and is activated. In the case of SRS, hypomethylation of H19 occurs, resulting in that it completely switches off IGF2 expression, which may cause intrauterine and postnatal growth retardation and low body mass index. In the case of BWS, the opposite pattern is observed; i.e., biallelic IGF2 expression leads to the complete absence of the H19 product, which results in prenatally formed organomegaly.
MLID is detected in 50% of BWS cases. Bliek et al. [35] examined a sample of 149 BWS patients and found KCNQ1OT1 hypomethylation in 17 of them. In addition to hypomethylation of this IC, mosaic hypomethylation of the MEST gene was detected in seven patients; of PLAGL1, in six patients; of IGF2R, in six patients; of GRB10, in four patients; and of GNAS/NESPAS, in ten patients. Epigenotype analysis was performed only in leukocyte DNA. Body weight, frequency of neonatal macrosomia, and hemihypertrophy at birth in patients with multiple hypomethylation at imprinted genes were on average lower than in the subgroup with isolated hypomethylation of KCNQ1OT1. In addition, some patients with hypomethylation of several imprinted genes had some features not associated with BWS, i.e., impaired speech, developmental delay, feeding difficulties at birth, and hearing problems.
The presence of epigenetic mosaicism for MLID supports the hypothesis that trans-acting factors can influence the somatic maintenance of imprinting at several maternal methylated loci. Indeed, in some cases of the presence of multiple epimutations at imprinted genes in BWS patients, in their mothers, mutations in the NLRP2 or NLRP5 genes were detected [31, 36].
Fontana et al. [28] using the MassArray technique in 17 of 21 patients with BWS showed mosaic hypomethylation of IGF2 (methylation index (MI) ranged from 7 to 31%), which was accompanied by hypomethylation at the PEG10 gene (MI from 21 to 41%) in four patients, MEST (MI from 20 to 31%) in three patients, and GNAS (MI from 20 to 34%) in two patients. In three BWS patients, mosaic hypermethylation of IGF2/H19 IC (MI ranged from 59 to 62%) was demonstrated. Changes in the methylation level of other imprinted genes were not detected. In one patient with MLID, a heterozygous mutation in the NLRP2 gene (region 19q13.42), inherited from father, was detected. All patients with epigenetic mosaicism for MLID showed lower weight at birth, as well as the features that were not characteristic of BWS (speech delay, apnea, and feeding difficulties).
Mosaic hypomethylation of the IGF2 gene was detected in children with BWS and nonsyndromic variant, when only Wilms tumor was detected. Analysis of mosaic methylation patterns in kidney and blood specimens of patients with Wilms tumor showed variation of the methylation index in the H19 gene from 7 to 56% and in IGF2 from 6 to 85%. Mosaic biallelic expression of IGF2 and H19 methylation were detected in all kidney tissues adjacent to Wilms tumor, with mosaic hypomethylation at the IGF2 locus. A high proportion of epigenetically modified cells in the normal kidney indicates that epigenetic abnormalities should have occurred at the early stage of development preceding the appearance of Wilms tumor [37]. Therefore, the level and location of mosaicism for either genetic or epigenetic alterations may be responsible for the unusual diagnostic features of BWS.
Itoh et al. [38] described nine patients with BWS and different degrees of mosaicism for paternal UPD of the short arm of chromosome 11 (from 60.9 to 89.5%). All patients had macrosomia at birth, hemihypertrophy, and mild macroglossia. Mosaicism for paternal UPD of chromosome 11 was observed in approximately 20% of BWS patients, most of them had segmental isodisomy of 11p15.5, and only in 8% of the cases was UPD of the entire chromosome 11 of paternal origin detected. The authors suggested that mosaic segmental UPD of 11p15.5 could have resulted from somatic recombination at the early stage of embryonic development.
Romanelli et al. [39] described nine patients with BWS and segmental parental isodisomy of 11p15.5, which encompassed the entire cluster of imprinted genes. The proportion of cells with segmental UPD was 64–70%. The authors showed that Wilms tumor was more frequently found in this pathology.
Ohtsuka et al. [40] on a sample of 32 patients with BWS and paternal UPD of chromosome 11 revealed segmental paternal UPD of 11p15.5 with the mosaicism level of 15 to 70% in 28 of these patients. The minimum size of segmental UPD 11p15.5 was 2.71 Mb and included both imprinting centers, IGF2/H19 and KCNQ1OT1. The authors showed that, in BWS patients with the mosaic variant of segmental UPD 11p15.5, heart abnormalities, hemihyperplasia, and a high risk of developing embryonic tumors, including Wilms and hepatoblastoma tumors, were more common. Macroglossia was also characteristic of the mosaic segmental UPD 11p15.5.
Angelman syndrome (OMIM 105830, syndrome frequency 1 : 16 000). The leading clinical features include severe delay of intellectual and physical development, ataxia and stereotyped puppet movements, specific face with laughter-like grimacing, frequent bouts of laughter, lack of speech, hypotension, convulsions, and microcephaly.
Epimutations at the SNURF-SNRPN locus occur in 2–4% of AS patients, with somatic mosaicism being found in approximately 30% of children [41]. It was demonstrated that imprinting defects occurred at the early stage of embryonic development and the proportion of normal cells ranged from 10 to 40%. Patients with higher percentage of normally methylated cells usually had milder clinical symptoms than patients with a lower percentage of such cells, and the features sometimes overlapped with PWS. For example, hyperphagia and obesity or nonspecific mental retardation were detected. Unlike children with a typical clinical picture of AS, these patients could have a vocabulary of up to 100 words and speak sentences. Ataxia and convulsions could be absent. Most children also had no microcephaly [42].
Buiting et al. [43] in 27 out of 85 patients with AS showed mosaicism for hypomethylation of SNURF-SNRPN IC. It was demonstrated that this epimutation occurred on the chromosome inherited from the grandmother, and was the result of disturbance of demethylation of imprinted genes during spermatogenesis in the father.
Prader–Willi syndrome (OMIM 176270, population frequency 1 : 17 500). In the neonatal period, patients have pronounced muscular hypotonia, a poor sucking reflex, and strong delay in the development of static and locomotor functions. After the first year of life, hyperphagia associated with the damage of the satiety center in the hippocampus appears, so patients have pronounced obesity. Patients are characterized by the presence of hypogonadotropic hypogonadism, acromicria, dolichocephalic skull shape, deformed low-set ears, soft ear cartilage, almond-shaped palpebral fissures, epicanthal folds, hypertelorism, and convergent squint. The intelligence of patients varied within 60–70 IQ. Mosaicism for maternal isodisomy of chromosome 15 and a combination of iso- and heterodisomy of maternal chromosome 15 in another patient with PWS was also described [44]. Newborns were characterized by the presence of mild face dysmorphism and hypotension, as well as delayed physical development; i.e., in these patients, no specific features of the clinical picture of the disorder were found.
Wey et al. [45] described a 20-year-old woman with clinical features of PWS. At the same time, unlike the main clinical picture of the syndrome, in the perinatal period, there were no feeding difficulties. Hyperphagia started later, at the age of 6 years, and epilepsy was also observed. Using the methods of methylation-specific PCR followed by denaturing high-performance liquid chromatography (MSP/DHPLC), methylation-sensitive restriction enzyme analysis, and methylation-specific real-time PCR, it was demonstrated that 50% of blood lymphocytes had hypomethylation of SNURF-SNRPN IC, and 50% of the cells had the normal methylation pattern of the imprinted locus.
The leading clinical features of Temple syndrome (OMIM 616222, chromosome region 14q32) include embryonic and postnatal developmental delay, short height and low weight at birth, neonatal hypotonia with feeding difficulties, retardation of motor and physical development, facial dysmorphia, scoliosis, joint hyperextensibility, and premature sexual development. In infancy, the phenotype of patients may resemble PWS or SRS [46]. Beygo et al. [47] in two of 13 patients with Temple syndrome revealed mosaic hypomethylation of MEG3/DLK1 IC and MEG3. The methylation index constituted 38 and 21% in one patient and 34 and 23.5% in another patient, respectively. The authors suggested that mosaic hypomethylation could have resulted from an error in maintaining the methylation status of MEG3/DLK1 IC after fertilization and, then, of the MEG3 gene, which is under its control.
Pseudohypoparathyroidism 1B (OMIM 603233, 20q13.3) is a rare hereditary disorder of the skeleton that mimics hypoparathyroidism and is characterized by impaired calcium and phosphorus metabolism, often accompanied by mental and physical retardation. Patients have short stature because of shortening of the lower extremities, brachydactyly, round moon-shaped face, and disturbance of intellectual development. The disorder is caused by the lack of tissue receptors for parathyroid hormone in target organs and limited resistance to parathyroid hormone. Maupetit-Mehouas et al. [48], analyzing blood lymphocytes of patients with PHP1B, revealed mosaic decrease in the methylation index at four DMRs of the GNAS gene. In particular, at DMR of NESP55 locus, the methylation index varied from 24 to 68%. Elli et al. [49] in 42% of patients with PHP1B revealed a decrease in the methylation index of different DMRs of the GNAS gene. This was found in 18% of patients at XL DMR, in 7% of patients at AS DMR, and in 10% patients simultaneously at DMRs of XL and AS loci.
Thus, epigenetic mosaicism in genomic imprinting disorders seems to have different contributions to the formation of the clinical picture of the disorder. For instance, it was demonstrated that mosaicism in the case of BWS, PWS, and AS was associated with a milder phenotype and the appearance of clinical features that were not characteristic of the given pathology. At the same time, for TNDM and SRS, even a small clone of epigenetically altered cells forms the classical phenotype of these disorders. In addition, in patients with TNDM, epigenetic mosaicism was observed only in certain tissues. These findings raise the question about the mechanisms underlying the emergence of epigenetic mosaicism.
POSSIBLE MECHANISMS UNDERLYING THE EMERGENCE OF MOSAIC EPIGENETIC ABNORMALITIES
Monoallelic expression of imprinted gene suggests that a mutation in one of the alleles is sufficient for the manifestation of pathological phenotype [50]. The range of such mutations is reduced to four main types as follows:
(1) gene mutations inactivating the only expressed allele;
(2) microdeletions or microduplications of chromosome regions, as well as CNV (copy number variation), containing the imprinted genes;
(3) UPD of chromosomes changing the balance of gene dosages of maternal and paternal origin in the genome;
(4) epimutation, i.e., inherited and noninherited changes in gene expression that are not associated with the disruption of its nucleotide sequence, but caused by epigenetic modifications of DNA or chromatin proteins.
To date, mosaicism in genomic imprinting disorders has been described for UPDs that encompass either the whole chromosome or only a specific region of it and for epimutations mostly affecting several imprinted genes (MLID) responsible for the formation of clinical features of the syndromes and other genes located on different chromosomes [51].
The presence of mosaicism for the methylation patterns of several imprinted genes in different tissues indicates the presence of somatic epimutations which arose after the separation of different germ layers. These epimutations could have occurred in the course of ontogeny during the second wave of reprogramming. In the ontogeny of mammals, two waves of epigenetic reprogramming of the genome are known. One of these occurs during germ cell development and is represented by successive events of total DNA demethylation and remethylation. At the same time, DNA demethylation is not complete, and the average methylation index in male and female primordial germ cells remains at 7.8 and 6.0%, respectively. In mature gametes, de novo hypermethylation occurs, while about 10% of CpG dinucleotides remain in the unmethylated state. Another reprogramming wave occurs immediately after fertilization. At this time, maternal and paternal genomes, with the exception of imprinted genes, undergo demethylation. It was demonstrated that, during fertilization, the average methylation index in mature spermatozoa was 54%, and in oocytes at metaphase II, it constituted 48%. In the zygote, this index value was 41%, decreasing to 32% at the preimplantation stages of development [52]. Thus, somatic hypomethylation of imprinted genes is formed at the preimplantation stage of development and is caused by the disruption of the mechanisms of imprinting maintaining in the somatic embryonic cells. Mosaic hypomethylation could occur after implantation as a result of the inability of supporting DNMT1 methyltransferase to accurately reproduce the imprinted gene status in cell divisions.
Most of the imprinted genes are grouped into clusters and are under general internal regulatory control of IC. In each cluster, these centers in cis-position initiate switching off the imprinted gene transcription through the production of untranslated RNA, and the subsequent DNA methylation fixes this state. In addition, tissue-specific epigenetic mosaicism of multilocus imprinting disruption suggests the presence of some trans-acting factor controlling the processes of imprinting maintaining or, as in the case of GRB10, switching from one imprinting to another. Indeed, in recent years, a new class of genes has been identified in which mutations are accompanied by multiple epimutations in imprinted loci. These include the KHDC3L gene encoding the embryonic stem cell-associated transcript 1 protein; the ZFP57 and ZFP42 genes encoding proteins of a family of zinc finger proteins; the genes for the proteins of NOD-like receptor family, NLRP2, NLRP5, and NLRP7; and the gene for peptidyl arginine deiminase 16, PADI6 [31, 53]. The presence of this class of imprinting control genes indicates the need upon presence of MLID in a patient to conduct a search for mutations in these genes in the patient, as well as in his parents. Accordingly, this raises the question of modification of algorithms for molecular genetic diagnosis of genomic imprinting disorders.
PROBLEMS OF DIAGNOSTICS MOSAIC EPIGENETIC IMPRINTING DISORDERS
At present, 12 genomic imprinting disorders are known with the clinical features affecting growth, physical and intellectual development, metabolism, and the central nervous system. In addition to the well-known SRS, BWS, PWS, and AS, recently this list was supplemented with new Birk–Barel syndrome, Temple syndrome, Kagami–Ogata syndrome, IMAGe syndrome, and Schaaf–Yang syndrome [54]. However, only in half of them was mosaic methylation of imprinted genes responsible for the formation of the clinical picture of these disorders identified so far. These include Silver–Russell, Angelman, Prader–Willi, Beckwith–Wiedemann, and Temple syndromes, as well as pseudohypoparathyroidism and transient neonatal diabetes mellitus.
Mosaic forms of genomic imprinting disorders pose a problem both for making a clinical diagnosis and for molecular genetic analysis, because mosaicism smooths out the typical clinical picture of syndromes unrecognizably and, hence, such patients cannot be sent to appropriate molecular diagnostics. For example, the leading diagnostic features of AS include gait ataxia and absence of speech; whereas patients with SNURF-SNRPN IC mosaicism are able to speak single words, they have no stereotypic movements, and in some cases, there are clinical features overlapping with PWS (obesity and hyperphagia) [42]. Not all children with WBS demonstrate classical phenotype of this disorder because of the heterogeneity of molecular abnormalities and somatic mosaicism. At the same time, the presence of a small MLID clone in the case of TNDM is already able to form a classical clinical picture of this disorder.
Genomic imprinting disorders are usually caused by disturbances of certain imprinted genomic loci. At the same time, somatic epigenetic mosaicism leads to disruption of the methylation pattern of several other imprinted genes simultaneously and, thus, to the difficulties of molecular genetic confirmation of clinical diagnosis [55]. Thus, to determine the etiology of a syndrome, a careful description of patients is necessary, and the molecular diagnosis of genomic imprinting disorders should include the exclusion of all genetic and epigenetic abnormalities described for these syndromes. In addition, to identify a mosaic variant of a syndrome, it is necessary to carry out molecular analysis in several available cell types (for example, in peripheral blood lymphocytes, buccal epithelium, skin fibroblasts) and to use methods of targeted bisulfite mass parallel sequencing and single-cell technologies.
Indeed, the achievements of recent years in the field of molecular and clinical diagnosis of genomic imprinting disorders help to effectively address these issues. For instance, the pyrosequencing method is successfully used to determine the methylation level of individual imprinted genes. It makes it possible to determine the methylation of all CpG dinucleotides in the studied nucleotide sequence, as well as the methylation percentage of each CpG pair in a specimen, which makes it possible to successfully diagnose mosaic forms of imprinting disorders. The MS-MLPA method allows for evaluation of the methylation status of several tens of imprinted loci simultaneously in one reaction. This reduces the number of stages of DNA diagnostics and, thereby, reduces the time of genetic diagnosis. Today, on the basis of this method, specialized MRC-Holland (Amsterdam, Netherlands) kits have been developed. For example, MS-MLPA (ME028-PWS/AS kit), which contains 46 probes, 32 of which are located in the 15q11-q13 region, is used to diagnose PWS and AS. As a control, 14 probes located outside this region are used. Determination of the methylation status of several imprinted loci simultaneously, both in the 15q11-q13 region and beyond, helps to improve testing accuracy. Similar kits are also available for the diagnosis of PWS/SRS (ME030-BWS/RSS kit), TNDM (ME033-TNDM kit), and pseudohypoparathyroidism type 1A and 1B (ME031-GNAS kit).
And finally, special emphasis is placed on the next-generation sequencing (NGS) technology, which allows for massively parallel sequencing of a great number of relatively small DNA fragments, which is the main difference from earlier sequencing methods [56]. NGS generates hundreds of megabases to gigabases of nucleotide sequences in one instrumental run, which makes it possible to obtain an enormous amount of data on the methylation status of the studied genes. This method also facilitates the search for mutations in the genes involved in the regulation of imprinting in the case of a patient having multiple methylation defects in imprinted genes or in the recurrent cases of hydatidiform moles or biparental complete hydatidiform moles.
Thus, in recent years, we have witnessed success in developing protocols for the diagnosis and identification of the genetic and epigenetic bases of many human genomic imprinting disorders. Currently, the existing algorithms are being improved and new ones for the diagnosis of these syndromes are being developed. The ability to identify the methylation status of many imprinted genes with a rather high degree of evaluation of mosaic methylation in one study increases the accuracy and reduces the time frame of molecular genetic diagnosis, and for the genetic counselor, the knowledge of genetic and epigenetic defects in imprinting diseases contributes to more effective family genetic counseling.
REFERENCES
Catalogue of Imprinted Genes. http://igc.otago.ac.nz/IGC.
Tucci, V., Isles, A.R., Kelsey, G., and Ferguson-Smith, A.C., Erice imprinting group: genomic imprinting and physiological processes in mammals, Cell, 2019, vol. 21, no. 176, pp. 952–965. https://doi.org/10.1016/j.cell.2019.01.043
Thamban, T., Sowpati, D., Pai, V., et al., The putative Neuronatin imprint control region is an enhancer that also regulates the Blcap gene, Epigenomics, 2019, vol. 11, no. 3, pp. 251–266. https://doi.org/10.2217/epi-2018-0060
Monk, D., Arnaud, P., Frost, J., et al., Reciprocal imprinting of human GRB10 in placental trophoblast and brain: evolutionary conservation of reversed allelic expression, Hum. Mol. Genet., 2009, vol. 15, no. 18 (16), pp. 3066–3074. https://doi.org/10.1093/hmg/ddp248
Baran, Y., Subramaniam, M., Biton, A., et al., The landscape of genomic imprinting across diverse adult human tissues, Genome Res., 2015, vol. 25, no. 7, pp. 927–936. https://doi.org/10.1101/gr.192278.115
Martins-Taylor, K., Hsiao, J.S., Chen, P.F., et al., Imprinted expression of UBE3A in non-neuronal cells from a Prader—Willi syndrome patient with an atypical deletion, Hum. Mol. Genet., 2014, vol. 23, no. 9, pp. 2364–2373. https://doi.org/10.1093/hmg/ddt628
Bielinska, E., Matiakowska, K., and Haus, O., Heterogeneity of human WT1 gene, Postepy Hig. Meg. Dosw., 2017, vol. 11, no. 71, pp. 595–601.
Bergman, D., Halje, M., Nordin, M., and Engstrom, W., Insulin-like growth factor 2 in development and disease: a mini-review, Gerontology, 2013, vol. 59, no. 3, pp. 240–249. https://doi.org/10.1159/000343995
Schulz, R., McCole, R.B., Woodfine, K., et al., Transcript- and tissue-specific imprinting of a tumour suppressor gene, Hum. Mol. Genet., 2009, vol. 1, no. 18 (1), pp. 118–127. https://doi.org/10.1093/hmg/ddn322
Hsiao, J.S., Germain, N.D., Wilderman, A., et al., A bipartite boundary element restricts UBE3A imprinting to mature neurons, Proc. Natl. Acad. Sci. U.S.A., 2019, vol. 116, no. 6, pp. 2181–2186. https://doi.org/10.1073/pnas.1815279116
Hitchins, M.P., Monk, D., Bell, G.M., et al., Maternal repression of the human GRB10 gene in the developing central nervous system; evaluation of the role for GRB10 in Silver—Russell syndrome, Eur. J. Hum. Genet., 2001, vol. 9, no. 2, pp. 82–90. https://doi.org/10.1038/sj.ejhg.5200583
Garfield, A.S., Cowley, M., Smith, F.M., et al., Distinct physiological and behavioural functions for parental alleles of imprinted Grb10, Nature, 2011, vol. 469, no. 7331, pp. 534–538. https://doi.org/10.1038/nature09651
Rienecker, K.D., Chavasse, A.T., Moorwood, K., et al., Detailed analysis of paternal knockout Grb10 mice suggests effects on stability of social behavior, rather than social dominance, Genes Brain Behav., 2019. e12571. https://doi.org/10.1111/gbb.12571
Nishiwaki, K., Niikawa, N., and Ishikawa, M., Polymorphic and tissue-specific imprinting of the human Wilms tumor gene WT1, Jpn. J. Hum. Genet., 1997, vol. 42, no. 1, pp. 205–211.
Lopez, S.J., Segal, D.J., and La Salle, J.M., UBE3A: An E3 ubiquitin ligase with genome-wide impact in neurodevelopmental disease, Front. Mol. Neurosci., 2019, vol. 11, no. 476. https://doi.org/10.3389/fnmol.2018.00476
Turan, S. and Bastepe, M., The GNAS complex locus and human diseases associated with loss-of-function mutations or epimutations within this imprinted gene, Horm. Res. Paediatr., 2013, vol. 80, no. 4. pp. 229–241. https://doi.org/10.1159/000355384
Chotalia, M., Smallwood, S.A., Ruf, N., et al., Transcription is required for establishment of germline methylation marks at imprinted genes, Genes Dev., 2009, vol. 23, no. 1, pp. 105–117. https://doi.org/10.1101/gad.495809
Campbell, R., Gosden, C.M., and Bonthron, D.T., Parental origin of transcription from the human GNAS1 gene, J. Med. Genet., 1994, vol. 31, no. 8, pp. 607–614. https://doi.org/10.1136/jmg.31.8.607
Hemida, R., van Doorn, H., and Fisher, R., A novel genetic mutation in a patient with recurrent biparental complete hydatidiform mole: a brief report, Int. J. Gynecol. Cancer, 2016, vol. 26, no. 7, pp. 1351–1353.
Sunde, L., Niemann, I., Hansen, E.S., et al., Mosaics and moles, Eur. J. Hum. Genet., 2011, vol. 10, pp. 1026–1031. https://doi.org/10.1038/ejhg.2011.93
Destouni, A. and Vermeesch, J.R., How can zygotes segregate entire parental genomes into distinct blastomeres? The zygote metaphase revisited, Bioessays, 2017, vol. 39, no. 4. https://doi.org/10.1002/bies.201600226
Mackay, D.J., Boonen, S.E., Clayton-Smith, J., et al., A maternal hypomethylation syndrome presenting as transient neonatal diabetes mellitus, Hum. Genet., 2006, vol. 120, pp. 262–269. https://doi.org/10.1007/s00439-006-0205-2
Boonen, S.E., Mackay, D.J., Hahnemann, J.M., et al., Transient neonatal diabetes, ZFP57, and hypomethylation of multiple imprinted loci: a detailed follow-up, Diabetes Care, 2013, vol. 36, pp. 505–512. https://doi.org/10.2337/dc12-0700
Messerschmidt, D.M., de Vries, W., Ito, M., et al., Trim is required for epigenetic stability during mouse oocyte to embryo transition, Science, 2012, vol. 335, no. 6075, pp. 1499–1502. https://doi.org/10.1126/science.1216154
Smith, Z.D. and Meissner, A., DNA methylation: roles in mammalian development, Nat. Rev. Genet., 2013, vol. 14, no. 3, pp. 204–220. https://doi.org/10.1038/nrg3354
Plasschaert, R.N. and Bartolomei, M.S., Tissue-specific regulation and function of Grb10 during growth and neuronal commitment, Proc. Natl. Acad. Sci. U.S.A., 2015, vol. 112, no. 22, pp. 6841–6847. https://doi.org/10.1073/pnas.1411254111
Fuke-Sato, T., Yamazawa, K., Nakabayashi, K., et al., Mosaic upd(7)mat in a patient with Silver—Russell syndrome, Am. J. Med. Genet. Part A., 2012, vol. 158, no. 2, pp. 465–468. https://doi.org/10.1002/ajmg.a.34404
Fontana, L., Bedeschi, M.F., Maitz, S., et al., Characterization of multi-locus imprinting disturbances and underlying genetic defects in patients with chromosome 11p15.5 related imprinting disorders, Epigenetics, 2018, vol. 13, no. 9, pp. 897–909. https://doi.org/10.1080/15592294.2018.1514230
Hiura, H., Okae, H., Chiba, H., et al., Imprinting methylation errors in ART, Reprod. Med. Biol., 2014, vol. 13, no. 4, pp. 193–202.
Bullman, H., Lever, M., Robinson, D.O., et al., Mosaic maternal uniparental disomy of chromosome 11 in a patient with Silver—Russell syndrome, J. Med. Genet., 2008, vol. 45, no. 6, pp. 396–399.
Docherty, L.E., Rezwan, F.I., Poole, R.L., et al., Mutations in NLRP5 are associated with reproductive wastage and multilocus imprinting disorders in humans, Nat. Commun., 2015, vol. 1, no. 6. e8086. https://doi.org/10.1038/ncomms9086
Mahadevan, S., Wen, S., Wan, Y.W., et al., NLRP7 affects trophoblast lineage differentiation, binds to overexpressed YY1 and alters CpG methylation, Hum. Mol. Genet., 2014, vol. 23. pp. 706—716.
Weksberg, R., Shuman, C., and Beckwith, J.B., Beckwith—Wiedemann syndrome, Eur. J. Hum. Genet., 2010, vol. 18, no. 1, pp. 8–14. https://doi.org/10.1038/ejhg.2009.106
Choufani, S., Shuman, C., and Weksberg, R., Molecular findings in Beckwith—Wiedemann syndrome, Am. J. Med. Genet., Part C, 2013, vol. 163, no. 2, pp. 131–140.
Bliek, J., Verde, G., Callaway, J., et al., Hypomethylation at multiple maternally methylated imprinted regions including PLAGL1 and GNAS loci in Beckwith—Wiedemann syndrome, Eur. J. Hum. Genet., 2009, vol. 17, no. 5, pp. 611–619. https://doi.org/10.1038/ejhg.2008.233
Meyer, E., Lim, D., Pasha, S., et al., Germline mutation in NLRP2 (NALP2) in a familial imprinting disorder (Beckwith—Wiedemann syndrome), PLoS Genet., 2009, vol. 5, no. 3. e1000423
Okamoto, K., Morison, I.M., Taniguchi, T., and Reeve, A.E., Epigenetic changes at the insulin-like growth factor II/H19 locus in developing kidney is an early event in Wilms tumorigenesis, Proc. Natl. Acad. Sci. U.S.A., 1997, vol. 94, no. 10, pp. 5367–5371. https://doi.org/10.1073/pnas.94.10.5367
Itoh, N., Becroft, D.M., Reeve, A.E., and Morison, I.M., Proportion of cells with paternal 11p15 uniparental disomy correlates with organ enlargement in Wiedemann—Beckwith syndrome, Am. J. Med. Genet. 2000, vol. 92, no. 2, pp. 111–116.
Romanelli, V., Meneses, H.N., Fernández, L., Martínez-Glez, V., et al., Beckwith–Wiedemann syndrome and uniparental disomy 11p: fine mapping of the recombination breakpoints and evaluation of several techniques, Eur. J. Hum. Genet., 2011, vol. 19, no. 4, pp. 416–421. https://doi.org/10.1038/ejhg.2010.236
Ohtsuka, Y., Higashimoto, K., Sasaki, K., et al., Autosomal recessive cystinuria caused by genome-wide paternal uniparental isodisomy in a patient with Beckwith—Wiedemann syndrome, Clin. Genet., 2015, vol. 88, pp. 261–266.
Fairbrother, L.C., Cytrynbaum, C., Boutis, P., et al., Mild Angelman syndrome phenotype due to a mosaic methylation imprinting defect, Am. J. Med. Genet., Part A, 2015, vol. 167, no. 7, pp. 1565–1569. https://doi.org/10.1002/ajmg.a.37058
Le Fevre, A., Beygo, J., Silveira, C., Kamien, B., et al., Atypical Angelman syndrome due to a mosaic imprinting defect: case reports and review of the literature, Am. J. Med. Genet., Part A, 2017, vol. 173, no. 3, pp. 753–757. https://doi.org/10.1002/ajmg.a.38072
Buiting, K., Gross, S., Lich, C., et al., Epimutations in Prader—Willi and Angelman syndromes: a molecular study of 136 patients with an imprinting defect, Am. J. Hum. Genet., 2003, vol. 72, no. 3, pp. 571—577.
Izumi, K., Santani, A.B., Deardorff, M.A., and Feret, H.A., Mosaic maternal uniparental disomy of chromosome 15 in Prader—Willi syndrome: utility of genome-wide SNP array, Am. J. Med. Genet., Part A, 2013, vol. 161, no. 1, pp. 166–171.
Wey, E., Bartholdi, D., Riegel, M., et al., Mosaic imprinting defect in a patient with an almost typical expression of the Prader—Willi syndrome, Eur. J. Hum. Genet., 2005, vol. 13, no. 3, pp. 273–277.
Kagami, M., Kato, F., Matsubara, K., et al., Relative frequency of underlying genetic causes for the development of UPD(14)pat-like phenotype, Eur. J. Hum. Genet., 2012, vol. 20, no. 9, pp. 928–932. https://doi.org/10.1038/ejhg.2012.26
Beygo, J., Mertel, C., Kaya, S., et al., The origin of imprinting defects in Temple syndrome and comparison with other imprinting disorders, Epigenetics, 2018, vol. 13, no. 8, pp. 822–828. https://doi.org/10.1080/15592294.2018.1514233
Maupetit-Mehouas, S., Mariot, V., Reynes, C., et al., Quantification of the methylation at the GNAS locus identifies subtypes of sporadic pseudohypoparathyroidism type, Ib. J. Med. Genet., 2011, vol. 48, no. 1, pp. 55–63. https://doi.org/10.1136/jmg.2010.081356
Elli, F.M., Bordogna, P., Arosio, M., and Spada, A., Mosaicism for GNAS methylation defects associated with pseudohypoparathyroidism type 1B arose in early post-zygotic phases, Clin Epigenet., 2018, vol. 6, no. 10. e16. https://doi.org/10.1186/s13148-018-0449-4
Soellner, L., Begemann, M., Mackay, D.J., et al., Recent advances in imprinting disorders, Clin. Genet., 2017, vol. 91, pp. 3–13. https://doi.org/10.1111/cge.12827
Monk, D., Mackay, D.J., Eggermann, T., et al., Genomic imprinting disorders: lessons on how genome, epigenome and environment interact, Nat. Rev. Genet., 2019, vol. 20, no. 4, pp. 235–248. https://doi.org/10.1038/s41576-018-0092-0
Guo, F., Yan, L., Guo, H., et al., The transcriptome and DNA methylome landscapes of human primordial germ cells, Cell, 2015, vol. 161, no. 6, pp. 1437–1452. https://doi.org/10.1016/j.cell.2015.05.015
Qian, J., Nguyen, N.M., Rezaei, M., et al., Biallelic PADI6 variants linking infertility, miscarriages, and hydatidiform moles, Eur. J. Hum. Genet., 2018, vol. 26, no. 7, pp. 1007–1013. https://doi.org/10.1038/s41431-018-0141-3
Elhamamsy, A.R., Role of DNA methylation in imprinting disorders: an updated review, J. Assist. Reprod. Genet., 2017, vol. 9, pp. 1–14. https://doi.org/10.1007/s10815-017-0895-5
Grafodatskaya, D., Choufani, S., Basran, R., et al., An update on molecular diagnostic testing of human imprinting disorders, J. Pediatr. Genet., 2017, vol. 6, no. 1, pp. 3–17. https://doi.org/10.1055/s-0036-1593840
Kim, J., Kim, D., Lim, J.S., et al., The use of technical replication for detection of low-level somatic mutations in next-generation sequencing, Nat. Commun., 2019, vol. 10, no. 1. e.1047. https://doi.org/10.1038/s41467-019-09026-y
Funding
This study was supported by the state contract with the Research Institute of Medical Genetics, Tomsk National Research Medical Center, Russian Academy of Sciences, Tomsk (state registration no. AAAA-A19-119020890005-5).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflict of interest. This article does not contain any studies involving animals or human participants performed by any of the authors.
Additional information
Translated by N. Maleeva
Rights and permissions
About this article

Cite this article
Sazhenova, E.A., Lebedev, I.N. Epigenetic Mosaicism in Genomic Imprinting Disorders. Russ J Genet 55, 1196–1207 (2019). https://doi.org/10.1134/S1022795419100119
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1022795419100119