
Abstract—
Epigenetic regulation is hereditary and non-hereditary changes in the expression of a particular gene without any corresponding structural changes in its nucleotide sequence. Genomic imprinting is an epigenetic mechanism for regulating the expression of homologous genes depending on parental origin, i.e., they are expressed monoallelically in the mammalian diploid cell. Being genetically imprinted, only the maternal or only the paternal genome is unable to ensure normal embryonic development. The most studied epigenetic modification, which plays one of the main roles in the maintenance of imprinting processes, is the specific methylation of cytosine in CpG-dinucleotides. All known imprinted genes contain differential DNA methylation regions on homologous parent chromosomes, which are necessary for their monoallelic expression. However, it is now known that not only DNA methylation, but chromatin remodeling, histone modifications, and non-coding RNAs also ensure the proper functioning of imprinted genes in the human body. Structural and functional disturbances of epigenetic mechanisms lead to imprinting diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Genomic imprinting is an epigenetic mechanism that allows a gene to be expressed in a parent-of-origin specific manner without altering the DNA sequence. The differential expression of an imprinted gene depends on its parental origin, i.e., diploid cells that contain two parental copies of all genes will express only one parental copy of an imprinted gene and silence the other. Being genetically imprinted, only the maternal or only the paternal genome is unable to ensure normal embryonic development, since paternal epigenetic imprints determine parent-of-origin expression of genes fundamental to placental development, and maternal epigenetic imprints contribute to the early development of embryonic structures [1, 2]. Genomic imprinting has been identified not only in marsupial and placental mammals, but also in flowering plants and some groups of insects, which indicates the evolutionary conservation of genomic imprinting by convergent evolution [3].
Allele-specific DNA methylation is the main mechanism of genomic imprinting, which introduces parental allele-specific epigenetic marks, leading to their monoallelic expression. It is the most studied epigenetic modification and has a major role in imprinting processes. All known imprinted genes contain differentially methylated regions (DMRs) on the two parent chromosomes, and these differences are essential for their monoallelic expression [4]. DMRs are commonly regulatory imprinting centers (ICs). The functional activity of imprinted genes depends not only on methylation, but is also significantly associated with chromatin structure [5, 6]. Almost all differentially methylated imprinted regions share overlapping DNA regions transcribed into non-coding and antisense RNAs with regulatory function [7–10].
The main mechanisms leading to aberrant function of imprinted genes and regions include: (1) gene mutations; (2) structural rearrangement of chromosomes, mainly deletion and duplication; (3) uniparental disomy (UPD) on certain chromosomes or their regions; (4) imprinting anomalies due to epimutations [11], and (5) functional anomalies of the epitranscriptome due to malfunction of non-coding RNAs [10].
At present, approximately 150 imprinted genes with tissue-specific monoallelic expression have been identified in humans, but the number of imprinted genes is likely more than 200 [4, 12, 13]. Herein, approximately 17 syndromes are associated with abnormal genomic imprinting. Multi-locus imprinting disturbances (MLID) are of particular interest. MLID are non-syndrome conditions associated with multi-locus methylation anomalies of imprinted regions and genes. Apparently, a system of reciprocal control operates in the epitranscriptome between non-coding RNAs at imprinted loci, which in some cases explains the clinical overlap between imprinting disorders [9, 14]. In addition, complete or segmental chromosomal maternal or paternal UPD is often detected in patients with non-syndrome pathology [15, 16]. In a range of cases, phenotypic overlap is associated with MLID caused by mutations in genes that establish or maintain specific allele methylation in regulatory regions subject to imprinting [17, 18].
Almost every human chromosome contains genes or regions that are differentially methylated and expressed monoallelically in a tissue and/or during a particular period of ontogenesis. It is known that gene methylation in gametes, placenta, and at the earliest stages of embryogenesis significantly differs from methylation and expression in the tissues of an adult organism. This review focuses on differentially methylated and monoallelically expressed imprinted genes and chromosomal regions, disorders in which (both structural and functional) cause pathological conditions called syndromes or imprinting diseases.
IMPRINTED CHROMOSOMAL REGIONS AND GENES, WHOSE PATHOLOGY LEADS TO IMPRINTING DISEASES
Imprinted Chromosomal Region 6q24.2 and Transient Neonatal Diabetes Mellitus
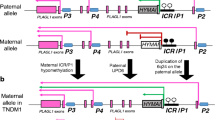
Transient neonatal diabetes mellitus (OMIM #601410) is a rare type of diabetes that occurs in infants and has a range of specific clinical features (Table 1) [19]. Transient neonatal diabetes mellitus type 1 is caused by aberrant expression of the PLAGL1 imprinted gene on chromosome 6q24.2. Several sporadic cases of transient neonatal diabetes mellitus with paternal UPD of chromosome 6 suggest either a double dose of a paternally expressed imprinted gene, or deficiency of a maternally expressed one. In a small number of familial cases, the transmission is always paternal and is associated with duplications in 6q24 [20]. The smallest size of duplications identified by molecular exploration of such families was 500 kb [21].
The PLAGL1 gene regulates transcription and is a tumor suppressor gene [22]. The PLAGL1 protein has a zinc finger motif and induces transcription of the PACAP1-R (ADCYAP1R1) gene, which encodes pituitary adenylate cyclase-activating polypeptide, induces insulin secretion, and regulates pancreatic β-cell function. Overexpression of PLAGL1 impaired β-cell function [23]. PLAGL1 includes 12 exons and is expressed from four different promoters, resulting in the formation of four isoforms of the protein. Transcripts from promo-ters 2, 3, and 4 are produced via biallelic expression, only the PLAGL1 coding transcript that overlaps DMR and the imprinting center (IC) is paternally expressed from promoter 1 (Р1) [24]. In addition, PLAGL1 regulates a range of imprinted and non-imprinted genes, including IGF2, H19, SLC2A4, CDKN1C, and PPARγ1, involved in cell growth and metabolism [25].
HYMAI is another gene in the PLAGL1 region that encodes a long non-coding RNA (lncRNA). The HYMAI gene consists of one exon and has a transcription start site that overlaps the PLAGL1 gene transcript in IC/P1. Hence, the IC/P1 methylation status regulates both the expression of HYMAI and certain PLAGL1 transcripts. DMR/IC, which is methylated on the maternal chromosome and unmethylated on the paternal chromosome, regulates both imprinted genes, i.e., PLAGL1 and HYMAI, which are abundantly expressed in the pancreas [26].
The paternal IC/P1 is commonly hypomethylated, and the maternal IC/P1 is hypermethylated; hence, the HYMAI and PLAGL1 transcripts regulated by IC/P1 are expressed from the paternal allele. Consequently, transient neonatal diabetes mellitus type 1 results from paternal UPD on chromosome 6 in 35–40% of cases, 6q24 duplication—in 35–40% of cases, and aberrant IC methylation (epimutation) of the maternal allele in 6q24 in 20–28% of cases. The loss of methylation at the maternal allele in patients with this type of diabetes leads to an elevated body mass index [19].
Another cause of transient neonatal diabetes mellitus is associated with mutations in the ZFP57 gene that is located on chromosome 6p22.1 and encodes a transcription factor involved in maintaining correct methylation at imprinted loci; these mutations are rare and have been described in 14 cases of transient neonatal diabetes mellitus [27]. We discuss the functions and role of ZFP57 in more detail in the Multi-Locus Imprinting Disturbances (MLID) Affecting Methylation in Regulatory Regions section.
No more than 20 cases of maternal UPD on chromosome 6 have been reported. These UPDs were not accompanied by any characteristic pathological features, except for intrauterine growth restriction [28].
Imprinted PEG13–KCNK9 Locus in Chromosome Region 8q24.3 and Birk–Barel Syndrome
The PEG13–KCNK9 imprinted cluster that harbors two imprinted brain-specific genes, PEG13 and KCNK9, has been mapped to chromosome region 8q24.3. The PEG13 gene located in an intron within the TRAPPC9 non-imprinted gene is paternally expressed to form a long non-coding PEG13 transcript that arises from a maternally methylated DMR. This DMR binds a CTCF-cohesin complex that acts as a methylation-sensitive enhancer-blocker on the unmethylated paternal allele [29]. The KCNK9/TASK3 gene is expressed from the maternal allele and encodes a member of the two pore-domain potassium channel subfamily. KCNK9 potassium channels are found throughout the body, and are especially abundant in the brain, where they play a role in the migration of cortical pyramidal neurons [30]. The reciprocal expression of KCNK9/TASK3 genes is regulated by maternal DMR located within the PEG13 transcript. In addition, chromatin loops with classic histone modification (H3K4me1, H3K27ac) for the enhancer regions were also found between the enhancer domain located in intron 17 of the TRAPPC9 gene and the KCNK9 and PEG13 promoter regions in brain tissues, indicating mutual regulation of the allelic expression of the PEG13 and KCNK9 transcripts [29, 31].
Birk–Barel syndrome (OMIM #612292) is characterized by multiple features of dysembriogenesis, delayed physical development and mental retardation (Table 1) [32]. This disease is usually caused by a maternal specific missense mutation (c.770G>A or 770G>С, p.Gly236Arg) [33]. A novel variant c.710C > A: p.Ala237Asp has also been described in the maternal copy of the KCNK9 gene [34]. In addition, complete loss of the maternal allele of the gene has been reported that arises due to complex structural rearrangement of chromosome 8 involving the imprinted region [35].
Molecular Organization of Imprinted Chromosome Region 11p15.5 and the Genes Associated with Beckwith–Wiedemann, Russell–Silver, and IMAGe Syndromes
The 11р15.5 chromosomal region contains a cluster of imprinted genes that appear intermittently with non-imprinted loci. Several imprinted genes and their regulatory regions make a major contribution to the phenotypes of Beckwith–Wiedemann and Russell–Silver syndromes.
The CDKN1C/Р57KIP2 gene encodes a cyclin-dependent kinase inhibitor of the CIP family factors. The gene is a negative regulator of cell proliferation, it is expressed from the maternal chromosome, and its overexpression may promote cell cycle arrest at the G1 border of cell cycle [36, 37].
The KCNQ1 gene encodes a potassium channel. It is expressed in almost all tissues from the maternal allele and has biallelic expression only in the heart muscle. The KCNQ1 human gene is known to have four transcript isoforms. The first form is transcribed only from the maternal allele (in most tissues), the second is transcribed biallelically in the heart muscle, and the third and fourth isoforms are not translated [38].
The KCNQ1OT1/LIT1 gene is localized in intron 10 of the KCNQ1 gene. The paternally expressed lncRNA emerges from an intron of the KCNQ1 gene in the antisense direction [39]. This lncRNA may interact with chromatin enriched with trimethylated lysine residues at positions 9 and 27 (H3K9 and H3K27), as well as with H3K9- and H3K27-specific histone methyltransferases G9a, EHMT2 and the polycomb-2 repressor complex. KCNQ1OT1 may cause specific transcription silencing of neighboring genes by recruiting chromatin remodeling complexes and maintain this state for several cell divisions [40, 41].
The IGF2 gene encodes fetal growth factor, which is a mitogen for almost all cell types, but specifically modulates muscle cell growth and proliferation. IGF2 contains 9 exons, the coding region is confined to only exons 7–9. IGF2 has five promoter regions (P1, P0, P2, P3, and P4) located in exons 1, 2, 4, 5, and 6, respectively, and four CpG-islands, the final CpG island maps to coding exon 9. In both embryonic and mature tissues, it is expressed only from the paternal chromosome [42].
The H19 gene is expressed from the maternal chromosome and encodes lncRNA, which modulates growth suppression, placental growth retardation, cell cycle regulation, carcinogenesis, and metastasis [43]. The study of gene methylation during embryogenesis revealed that H19 biallelic expression is confined to the placenta until 10 weeks of gestation, after which it becomes exclusively maternal and does not affect allele-specificity or levels of IGF2 expression [44].
Structural and Functional Organization of Imprinting Centers IC1 and IC2 in Chromosome Region 11р15.5
The H19 and IGF2 genes are tightly linked, and their expression is regulated by a common enhancer located in the 3'-region of H19 [45]. The common enhancer regulates the transcription of H19 lncRNA, intragenic microRNA miR-675 on the maternal chromosome, and IGF2 and intragenic miR-483 on the paternal chromosome. The IC1 domain, also known as the H19–IGF2 intergenic DMR, contains tandem repeats and binds CTCF, POU5F1, and SOX2 transcription factors, which maintain the maternal allele in an unmethylated state, which silences IGF2 expression. At the same time, ZFP57 maintains the methylated state of the IC1 paternal allele and, hence, is not able to bind CTCF and does not prevent the possible activation of IGF2 by an enhancer on the paternal chromosome [4, 45]. The CTCF protein blocks communication between enhancers and the promoter regions of genes, it is necessary for the normal functioning of epigenetic marks [46]. In addition, IC1 demonstrates different states of chromatin on the parent chromosomes: the presence of the repressive marks of histone 3 and 4 (H3K9me2, H3K9me3 and H4K20me3) on the methylated allele, and the activated state of chromatin with di- and tri-methylated histone 3 lysine residue 4 (H3K4me2 and H3K4me3) associated with the non-methylated maternal allele [47]. The secondary DMRs: H19 promoter, IGF2 DMR0, and IGF2 DMR2, are methylated on the paternal chromosome [4, 42]. IGF2 DMR0 is methylated on the paternal chromosome and has a silencer function, it downregulates the expression of IGF2 on the maternal chromosome in mesodermal tissues, except for muscles. Maternal deletion is associated with a lack of the IGF2 silencing effect: the gene is expressed biallelically, with loss of imprinting. When a IGF2 deletion is transmitted paternally, the paternal IGF2 allele is transcribed. The region that contains miR-483 in IGF2 includes another regulatory element that has a silencer function—DMR2. Its deletion on the maternal chromosome causes biallelic expression of IGF2, robustly in the muscles, especially in the muscles of the tongue, which leads to macroglossia. Deletion of DMR2 on the paternal chromosome does not perturb IGF2 expression [48].
A second DMR within the IC2 region is located in intron 10 of the KCNQ1 gene prior to the transcriptional start site for KCNQ1OT1/LIT1, a paternally expressed antisense transcript [49]. It was shown that KCNQ1OT1 lncRNA represses the expression of coding genes in cis in this region, i.e., it downregulates expression of the KCNQ1 and Р57KIP genes, which are expressed from the maternal allele [50]. Methylation of IC2 and silencing of the KCNQ1OT1 promoter are maintained through interaction with ZFP57 on the maternal chromosome [4]. Epimutation results in aberrant expression of KCNQ1OT1 from the maternal chromosome, which leads to biallelic silencing of KCNQ1 and Р57KIP and a pathological phenotype [51]. Hence, the imprinted region of chromosome 11р15.5 contains two ICs located at a distance of about 600 kb. Their dysfunction leads to Beckwith–Wiedemann or Russell–Silver syndromes.
Beckwith–Wiedemann syndrome (OMIM #130650) is a common disease with a characteristic phenotype and a prevalence of 1 : 10 000–15 000 live births (Table 1) [51, 52].
Molecular-genetic and epigenetic anomalies may be detected in 80–85% of patients with Beckwith–Wiedemann syndrome. Approximately 50% of the cases are caused by loss of methylation (LOM) at the maternal IC2 allele (IC2-LOM), leading to reduced expression of CDKN1C and KCNQ1. IC2-LOM is usually a sporadic primary epigenetic defect; however, rare familial cases carrying genetic mutations causing secondary hypomethylation have been described [51, 53, 54]. Recently, a growing proportion of patients with IC2-LOM that have impaired methylation at other imprinted loci has been noticed, which is associated with the appearance of additional phenotypic features [55] (see the Multi-Locus Imprinting Disturbances (MLID) Affecting Methylation in Regulatory Regions section).
Mosaic segmental paternal UPD of chromosome 11 accounts for 20% of cases and leads to altered expression at both gene clusters: IC2-LOM and gain of methylation at IC1 (IC1-GOM), which inactivates Н19 on the maternal chromosome. Complete UPD at chromosome 11 is quite frequent and associated with higher cancer risk [54, 56].
IC1-GOM results in biallelic expression of the IGF2 gene, which is normally expressed by the paternal allele only, and a lack of H19 gene expression from the maternal allele. IC1-GOM is found in 5–10% of cases and in a subset of patients is caused by microdeletions encompassing the OCT4/SOX2 binding site localized inside IC1, leading to a maternally transmitted Beckwith–Wiedemann phenotype [51, 53, 54]. Mice with H19 deletion display epigenetic lesions at the IGF2 gene that lead to overgrowth and exomphalos [57, 58].
Maternal CDKN1C loss-of-function mutations responsible for the maternal transmission of Beckwith–Wiedemann syndrome account for 5–10% of all cases of this disease. Mutations of this gene are found in almost 40% of familial cases and in less than 5% of sporadic cases [53, 59].
Approximately 1% of Beckwith–Wiedemann cases are caused by chromosomal rearrangements (duplications, translocations, inversions, deletions) involving the 11p15.5 chromosomal region, which leads to secondary IC2-LOM and IC1-GOM [60]. Approximately 15% of individuals with a clinical diagnosis of Beckwith–Wiedemann syndrome have no detectable molecular defect when investigated using commonly employed diagnostic molecular techniques. However, low somatic mosaicism of the above mentioned defects is increasingly found by using novel sequencing techniques and analyzing tissues other than blood (buccal epithelium, skin fibroblasts) [53].
Molecular subtypes of Beckwith–Wiedemann syndrome are characterized by a gradient in cancer development probability and display different histotypes allowing differentiation of tumor surveillance protocols according to the epigenotype. This facilitates the early detection of the relevant associated tumors [51]. Biallelic expression of IGF2 is often detected in nephroblastoma, rhabdomyosarcoma, adrenal cortical cancer and other embryonal tumors characteristic of Beckwith–Wiedemann syndrome. Overexpression of this gene in transgenic mice causes overgrowth, macroglossia, organomegaly, and tumors [57, 61]. Indeed, it was found that 20% of tumors show UPD and aberrant IC1 and IC2 methylation. Isolated IC1-GOM Н19 was observed in 7% of patients, and isolated IC2-LOM KCNQ1OT1 was reported in 55% of cases. In the latter group of patients, no tumors were detected, whereas individuals with IC1-GOM or paternal UPD had neoplasms characteristic of the syndrome in 33% of cases [54].
Russell–Silver syndrome (OMIM #180860) is a relatively common inherited condition. Its prevalence in various populations is 1 : 75 000–1 : 100 000 live births. The main phenotypic features of Russell–Silver syndrome include postnatal growth failure, relative macrocephaly at birth, signs of dysmorphogenesis, among others (Table 1) [62, 63].
Since Russell–Silver syndrome displays an array of dysembriogenic features, and delayed mental and physical development, chromosomal analysis was the first stage in identification of its causes. Indeed, several cases were linked to various nonspecific chromosomal anomalies: translocations and inversions, partial trisomies of the long arm of chromosome 1q42, deletion of 8(q11–q12), 13(q22–q32), 18р, and others [64]. Several cases of ring chromosome 15 with deletion of region 15q26.3 encompassing the IGF1R gene, have been reported. Isolated deletions of this gene lead to severe growth delay, triangular face, clinodactyly, micrognathia, microcephaly and mental retardation, but no mutations of the gene were found in Russell–Silver syndrome [65]. In patients with a phenotype similar to Russell–Silver syndrome, balanced de novo translocations were detected corresponding to a breakpoint in chromosome 17(q24–q25). This region contains a cluster of growth hormone genes; their deletions were found in patients without overt syndromic disorders [64, 66].
It had not been expected that alterations in this region may cause another syndrome until two maternal microduplications of chromosomal region 11p15.5 with contrasting phenotypes were described. In the first case, an inverted duplication of the 11p15 maternal chromosome with a size of 1.2 Mb led to Russell–Silver syndrome. The duplication spanned the entire cluster of 11p15.5 imprinted genes, and CpG hypermethylation was observed in the IC2 region. In the second case, a maternally inherited inverted duplication with a size of 160 kb included only IC2 and 20 kb of the 5′-KCNQ1OT1 region, which led to the Beckwith–Wiedemann phenotype in five people over two generations [67].
A nuanced molecular analysis revealed hypomethylation at the paternal allele of the Н19 gene in a range of patients due to epimutation, i.e., the gene switches to biallelic expression. This occurs due to the fact that IC1 is not methylated at the paternal chromosome (found in 40–60% of patients) and binds the CTCF protein, which leads to biallelic silencing of IGF2 [53, 62]. For example, it was shown that DMR in the 5'-region of IGF2 on the paternal chromosome is also hypomethylated in these patients. Since Beckwith–Wiedemann syndrome and Russell–Silver syndrome are imprinting disorders with mirror opposite alterations at the genomic loci in 11p15.5, IC2 (KCNQ1OT1) contributed to the Russell–Silver syndrome phenotype, but insignificantly. Several cases of de novo IC2 methylation on the paternal chromosome have been described [53, 57, 68].
In a range of patients with Russell–Silver syndrome, aberrant Н19 methylation occurs with a frequency of 0–35%. This means that Н19 demethylation on the paternal chromosome occurs only in some cells [69]. The mosaic etiology of imprinting anomalies shows that the lesions arise during the first rounds of cell division, indicating that most cases of the disease are sporadic.
The molecular cause is detected in approximately 60% of patients with a clinical diagnosis of Russell–Silver syndrome. Maternal UPD on chromosome 7 is the second most common etiological factor, which accounts for 5–10% of cases [70, 71]. A systematic analysis of differential methylation at chromosome 7 in maternal and paternal UPD showed that 65% of DMRs on the maternal chromosome are hypomethylated [72]. The short arm region on chromosome 7 (р11.2–р13) contains an imprinted GRB10 gene encoding a cytoplasmic adapter protein that acts as a negative regulator of tyrosine kinase receptor signaling. Paternal expression of this gene is observed in the brain and spinal cord, maternal—in skeletal muscles, and in all other tissues the gene is expressed biallelically. No mutations or aberrant methylation were identified in the regulatory region of the GRB10 gene in patients with Russell–Silver syndrome [73]. The genes in the imprinted loci on the long arm of chromosome 7 (SGCE/PEG10 and PEG/MEST) also did not have any structural and functional disturbances that could cause the development of Russell–Silver syndrome [74]. Therefore, no gene has been conclusively implicated in Russell–Silver syndrome [62, 75]. In addition, paternal UPD for chromosome 7 has no apparent phenotypic effect [72].
CDKN1C mutations of maternal origin have been described in patients with Russell–Silver syndrome in several families [76–78] and paternal inactivating IGF2 mutations in other cases [79, 80]. In addition, variants of two non-imprinted genes were identified in Russell–Silver syndrome: HMGA2 variants were described for three families and three sporadic cases [81], and two families and one sporadic case with PLAG1 alterations have been reported [53, 70, 82].
The PLAG1 transcription factor contains seven canonical zinc finger domains for DNA binding and a C-end enriched with serine residues that can activate transcription. PLAG1 binds the P3 IGF2 promoter, thereby increasing its expression, which is necessary for normal embryonic growth. Silencing of the gene may lead to prenatal and postnatal growth retardation and delayed development [83].
Furthermore, multi-locus methylation imprinting disturbances (MLID) in imprinted regions of other chromosomes are possible in Russell–Silver syndrome which leads to a partial phenotypic overlap. For example, when structural and functional disorders of chromosome 11р15 and maternal UPD on chromosome 7 are not detected, maternal UPD on chromosomes 6, 16, 20, as well as the methylation status of two DMR on chromosome 14 need to be investigated [53, 84].
IMAGe syndrome (OMIM #614732) is characterized by intrauterine growth restriction, and dysembriogenic features (Table 1). It is caused by missense mutations in the CDKN1C gene, which inhibits cell-cycle progression [85]. All known single-nucleotide variants associated with IMAGe syndrome are located in a highly conserved hotspot in the PCNA-binding domain of CDKN1C between codons 272–279 and disrupt the binding of PCNA (proliferating cell nuclear antigen). The mutations most often detected are p.Asp274Asn, p.Lys278Glu or p.Arg279Leu. These mutations may increase protein stability and enhance its function, thereby preventing growth [86, 87]. The mechanism has not been established, but it may be associated with decreased degradation of CDKN1C, allowing prolonged cell cycle repression and delayed S-phase progression. CDKN1C is expressed only from the maternal allele, hence only maternal transmission of the mutation results in IMAGe syndrome [88].
Imprinted Region of the RB1 Gene in Chromosome Locus 13q14.1
It has long been known that the tumor suppressor gene would be silenced in various types of tumors via methylation within the promoter regions. One or both RB1 alleles are functionally inactivated frequently in the tumor [89], and inactivation of the gene due to germline epimutation is a rare event. Mosaic methylation at CpG 106 encompassing the promoter region of RB1 has been described in a patient with retinoblastoma, indicating that epimutation occurred in the maternal allele at an early stage of embryonic development [90].
A more nuanced molecular-genetic analysis of the promoter region and CpG islands of the RB1 gene revealed a range of features. The RB1 gene contains an imprinted site (1.2 kb)—CpG 85, a CpG island within the second intron of the gene, which shows parent-of-origin specific methylation—it is methylated on the maternal chromosome and unmethylated on the paternal chromosome. In addition, RB1 contains two more CpG islands: CpG 106 is associated with the promoter and exon 1, it is unmethylated and provides biallelic expression of the main transcript encoding the RB protein; and CpG 42 is located in intron 2 of the gene, it is methylated on both chromosomes and has no regulatory activity [91].
It was found that CpG 85 is part of a 5'-truncated processed pseudogene derived from the KIAA0649 protein-coding gene on chromosome 9 that integrated into RB1 in reverse orientation. CpG 85 acts as a promoter for an alternative RB1 transcript, which is expressed only from the unmethylated paternal chromosome [91]. Despite the expected higher overall expression level of mRNA transcripts from the paternal allele (compared to the maternal allele), the level of expression from the paternal allele is twice or three times lower due to transcriptional interference by combined expression of regular and alternative transcripts. Transcription interference is a mechanism whereby transcription of one gene suppresses the transcription of another gene. For example, the transcription complex of the alternative transcript of the RB1 gene, which binds to the unmethylated CpG 85 island on the paternal chromosome, acts as a block for the transcription complex of the regular transcript on the same allele, which decreases the expression on the paternal chromosome [91, 92]. This indicates an epigenetic parent-of-origin-specific regulation of RB1 gene expression.
In inherited retinoblastoma (ОMIM #180200) (Table 1), which accounts for 40% of retinoblastoma tumors, a germline mutation on one allele of the RB1 gene causes predisposition for retinoblastoma and its familial transmission. The tumor is initiated by a somatic mutation on the other allele of the RB1 gene in a retinal cell, which occurs in utero or early in childhood. In some families with retinoblastoma with two or more carriers of the same germline mutation, the mutations cause a milder phenotype with reduced disease penetrance, where some carriers of the germline mutation do not develop retinoblastoma, or with variable expressivity, and the same mutation in different family members manifests as unilateral or bilateral disease. The phenotypic manifestation of inherited retinoblastoma depends on the functional type of germline mutation in the RB1 gene [93]. In turn, the molecular mechanisms underlying the variable phenotypic expression of the same mutation in different members of the same family are explained by the parent-of-origin impact of the RB1 mutation. Herein, paternally inherited low-penetrance germline RB1 mutations more frequently lead to retinoblastoma and cause a more severe form than maternally inherited mutations. It is believed that when the mutation is inherited from the maternal side, pRB retains sufficient tumor suppressor activity to prevent retinoblastoma development. In contrast, when the mutation is paternally transmitted, the low residual activity would mimic a null mutation and subsequently lead to retinoblastoma [94, 95].
Imprinted Chromosome Region 14q32.2 and Uniparental Disomy at Chromosome 14
The 14q32.2 chromosome region contains a cluster of imprinted genes: some are expressed from the paternal chromosome—DLK1, RTL1 and DIO3, and others are expressed from the maternal chromosome—lncRNA genes MEG3/GTL2, MEG8, RTL1as, snoRNAs, and miRNAs [96]. DLK1, preadipocyte factor 1, or fetal antigen 1, encodes an EGF-like membrane-bound protein involved in Notch signaling and regulation of preadipocyte differentiation. It is expressed in neuroendocrine tissues, especially in the adrenal cortex [97]. A range of structural alterations of the gene were detected in patients with central precocious puberty (Table 1). At the same time, the metabolic phenotype, including an increased incidence of obesity, early glucose intolerance, type 2 diabetes mellitus, and hyperlipidemia, was more common in patients with DLK1 mutations than in patients with mutations in other genes [98].
The retrotransposon-like RTL1 gene is expressed in the placenta at the late fetal stage. RTL1 is responsible for fetal muscle defects characteristic of Kagami–Ogata and Temple syndromes. It also plays an important role in the central nervous system, since its irregular expression leads to impaired innervation of motor neurons to skeletal muscles as well as malfunction of the hippocampus-amygdala complex [99]. The strong expression of DIO3 in the developing and adult brain inactivates thyroid hormones and plays an important role in preventing neurological abnormalities caused by inappropriate levels of these hormones. Altered exposure to thyroid hormones during development may lead to some phenotypic features associated with chromosome 14 UPD [100]. MEG3 encodes lncRNA, which orchestrates a wide range of different cellular processes that require epigenetic regulation of genes and interaction with key signaling proteins. Its intronic DMR contains two CTCF-binding domains and thus has regulatory functions [101]. It is possible that CTCF binding upregulates all genes expressed from the maternal chromosome, which represents a single transcriptional unit and represses all paternal genes by impairing histone acetylation [102]. The functions of RTL1as and MEG8, other than regulatory roles, are unknown. Parent-of-origin patterns of expression are regulated by the germline intergenic DMRs (MEG3/DLK1 DMR), and DMR that is acquired after fertilization (MEG3-DMR). Both DMRs are normally methylated on the paternal chromosome [102, 103]. Loss of DLK1 expression in conjunction with RTL1 and DIO3, due to maternal UPD, leads to Temple syndrome [104].
Temple syndrome (OMIM #616222) has overlapping clinical features with Prader–Willi and Russell–Silver syndromes (Table 1) [9, 62].
The mechanisms that impair hemizygosity of the imprinted genes in the 14q32 region and the clinical phenotypic anomalies include: (1) maternal UPD on chromosome 14 (72–78%); (2) isolated loss of methylation (epimutation) in the MEG3-DMR region (12–20%); and (3) paternal origin of the 14q32 deletion (10%) [104, 105].
Kagami–Ogata syndrome (OMIM #608149) is an imprinting disorder with a range of dysmorphic features and developmental defects (Table 1). The pathognomonic feature is a bell-shaped thorax with coat-hanger configuration of the ribs. Some non-specific phenotypic features are similar to the manifestations of Beckwith–Wiedemann syndrome [106].
The syndrome may be caused by three different molecular events: (1) paternal UPD at chromosome 14 (65% of cases); (2) maternal microdeletions on chromosome 14q32.2 (20%); (3) hypermethylation of MEG3-DMR on the maternal chromosome (15%) [107]. While paternal UPD 14 and MEG3 hypermethylation occur sporadically, microdeletions may lead to a maternal inheritance. It has been shown that deletions of the imprinted region do not necessarily include DMR, hence a normal methylation pattern does not exclude the possibility of the syndrome [108].
Hypomethylation of the DLK1/MEG3 domain decreases the expression of imprinted genes such as IGF2, SNURF, and IPW, as well as several other non-imprinted genes involved in growth stimulation. Alterations in expression may reflect, directly or indirectly, the participation of MEG3 and MEG8 lncRNAs in this process; lncRNAs regulate gene expression both in cis, as well as in trans by recruiting chromatin modifiers [7]. Overexpression or inactivation of MEG3 and MEG8 expression in normal fibroblast cultures may be associated with impaired regulation of imprinted genes in the 11p15.5 and (15q11–q13) regions. For example, it was found that: (1) MEG3 overexpression is associated with lower levels of IPW transcripts; (2) overexpression of MEG8 is associated with a lower level of the SNURF transcript; and (3) concomitant overexpression of MEG3 and MEG8 was associated with lower levels of IGF2 transcripts. This indicates that MEG3 and MEG8 can regulate, in trans, the expression of other imprinted genes [9]. It has been found that IPW overexpression can lead to the downregulation of MEG3 expression [109]. Hence, it is likely that there is a reciprocal control system in the epitranscriptome between lncRNAs within imprinted regions, and this, in turn, may contribute to the overlapping phenotypes of the Prader–Willi, Beckwith–Wiedemann, and Russell–Silver syndromes [14].
The molecular organization of the imprinted chromosomal region 15(q11.2–q13) and the genes linked to Prader–Willi, Angelman, Schaaf–Young syndromes, and central precocious puberty. Chromosome region 15(q11.2–q13) includes one of the extended imprinted regions containing a cluster of imprinted genes. The structural and functional defects in these genes cause the well-known Prader–Willi (OMIM #176270) and Angelman syndromes (OMIM #105830) as well as the rare Schaaf–Yang syndrome (OMIM #615547) and central precocious puberty type 2 (OMIM #615346). Phenotypic features, disease rates, and variants of molecular pathology are shown in Table 1.
The critical region of chromosome 15(q11.2–q13) is the most susceptible region to deletions (5–7 Mb) in the human genome. It may be divided into four fragments: (1) the proximal region containing non-imprinted genes (TUBGCP5, CYPFIP1, NIPA2 and NIPA1); (2) an imprinted region containing genes expressed monoallelically only from the paternal chromosome: protein-coding genes (MKRN3/ZNF127, MAGEL2, NECDIN, NPAP1/C15orf2, SNURF-SNRPN), genes encoding untranslated RNAs (MKRN3-AS/ZNF127-AS, IPW, UBE3A-ATS), and a cluster of genes encoding snoRNAs (SNORD116: 29 copies, SNORD115: 48 copies, SNORD64, SNORD107, SNORD108, SNORD109A and SNORD109B); (3) a region with monoallelic expression of the UBE3A protein-coding gene from the maternal chromosome and biallelic expression of ATP10C; (4) the distal region contains biallelically expressed genes—OCA2 responsible for albinism type 2, the HERC2 gene and three gamma-aminobutyric acid (GABA) receptor genes [110–112].
Deletions arise due to non-homologous recombination between domains of highly homologous low-copy repeats at break points (BP) BP1–BP5. ВР1 and ВР2 are localized proximally to MKRN3, and ВР3, 4 and 5 are located in the telomeric area of the imprinted region. There are two classes of deletions: class 1 deletions constitute 40% and they are located in ВР1–ВР3; class 2 deletions are present in 50% of deletion cases and extend from BP2 to BP3. In rare cases (less than 10%), the position of a deletion may not coincide with regular break points, but may be much smaller or extend beyond ВР4 and ВР5 [113–117]. The distal cluster of break points has been mapped telomeric to the HERC2 gene. Such deletions include a cluster of GABA receptor genes—GABRB3, GABRA5 and GABRG3. The most frequent break points BP1, BP2 and BP3 are flanked by low-copy repeats originating from duplicated fragments of the HERC2 gene. The original copy of the HERC2 gene is located in ВР3. ВР4 and ВР5 also contain low-copy repeats, but they do not have homology with HERC2. Deletions are caused by intra- or interchromosomal crossover, occur de novo, due to the formation of a large (4–5 Mb) loop, and only in rare cases are they caused by structural rearrangement [114, 118]. In addition, in a large number of cases, the region 15(q11.2–q13) was involved not only in deletions, but also in inverted duplications (tetrasomy), duplications (trisomy), unbalanced (monosomy) and balanced translocations, as well as inversions.
The SNURF/SNRPN gene, the SNORD non-coding gene cluster, and the lncRNA UBE3A-ATS are central to the imprinted region and are expressed from the non-methylated paternal allele. This locus is sophisticated complex genetic locus SNHG14 (Small Nucleolar RNA Host Gene 14) that spans 465–600 kb and contains at least 148 exons subject to alternative splicing [119, 120]. SNURF-SNRPN is a bicistronic gene encoding two different proteins. Its 5'-region contains a CpG island, including the promoter, the first exon and the PWS-IC intron-regulatory region, with a length of 4.3 kb. The SNRPN minimal promoter region includes 71 bp of 5′-UTR and the first 51 bp of SNURF-SNRPN exon 1. This region represents an essential part of IC [121, 122] and is crucial for the regulation of the entire imprinted region [123]. Imprinting is associated with parental allele-specific methylation of CpG residues, and the methylation imprints are established during or after gametogenesis and maintained during embryogenesis. The PWS-IC region is unmethylated on the paternal chromosome and methylated on the maternal one [124].
The main function of PWS-IC is to upregulate the transcription of paternal allele genes, including MKRN3, MAGEL2, NECDIN, NPAP1, SNURF-SNRPN, IPW and UBE3A-ATS. The expression of UBE3A-ATS downregulates UBE3A on the paternal allele by means of transcriptional interference [125].
IC is a bipartite imprinting center, i.e., it contains PWS-IC and AS-IC [126]. AS-IC is located 35 kb more proximal, consists of 880 bp, and contains an alternative 5'-non-coding SNRPN exon, which is expressed only in oocytes [127]. The oocyte-specific transcription leads to methylation and transcriptional inactivation of the maternal allele, but not the paternal allele. AS-IC is crucial for silencing imprinted genes on the maternal allele. The lack of UBE3A-ATS expression on the maternal allele is necessary for normal UBE3A expression [128, 129]. The structural pathology of AS-IC erases methylation imprints on the maternal promoter of SNRPN and upregulates the imprinted genes on the maternal allele [130].
The PWS-IC functions in omatic tissue to activate paternally expressed genes during gametogenesis and guarantee paternal gene expression. The inheritance of a structural pathology or epimutation in PWS-IC leads to Prader–Willi syndrome, biallelic expression of UBE3A and, consequently, silences UBE3A-ATS on the paternal chromosome. During oogenesis, the AS-IC functions to negatively regulate the PWS-IC and suppresses the processes modulated by PWS-IC during spermatogenesis. Hence, a deletion or epimutation in the maternal AS-IC allele interferes with silencing of the paternal genes on the maternal chromosome, including UBE3A-ATS, which leads to biallelic UBE3A gene inactivation [131–133].
Mutations or epimutations affecting the function of PWS-IC alter the methylation profile of secondary DMR genes NDN and MKRN3 and lead to loss of allelic expression from the entire imprinted region, indicating that PWS-IC is the main regulator of imprinting within this region in somatic tissues [134].
The first three exons of the SNURF-SNRPN transcript give rise to the small nuclear SNURF protein of unknown function [135], exons 4–10 correspond to the SNRPN portion and encode the SmN spliceosomal protein necessary for the formation of spliceosomes, which are responsible for alternative splicing of various mRNAs [136]. Six snoRNA genes extend more distally and are expressed as a single long transcript; they are regulated by SNURF-SNRPN expression and do not encode proteins [120, 137, 138]. These genes contain five one-copy snoRNA genes (SNORD64, SNORD107, SNORD108, SNORD109A and SNORD109B) and two genes, SNORD115 and SNORD116, which include clusters of repeat sequences that encode C/D-box snoRNAs. These snoRNAs are located in the introns of the SNHG14 “maternal” locus, which encodes an untranslated brain-specific transcript expressed from the paternal chromosome [139]. Ribonucleoprotein complexes between C/D-box snoRNAs guide 2′-O-methylation at specific rRNA sites in association with fibrillarin [140].
In humans, SNORD115 is only expressed in neurons and participates in RNA editing of the 5HTR2C receptor [141]. SNORD116 is located in the nucleolus and may participate in splicing, RNA modifications, and post-transcriptional regulation, although its exact role in the genome still remains to be determined [112, 142]. Isolated deletions of SNORD116 in both humans and mice lead to Prader–Willi syndrome [110, 112, 143]. The snoRNAs processed from the SNORD116 and SNORD115 introns, referred to as 116HG and 115HG, are localized in the form of RNA clouds in the regions of their own transcription in the nucleus of neurons and regulate the functioning of many genes [112, 144, 145].
UBE3A-ATS is part of the lncRNA SNHG14 and is expressed from the SNRPN promoter on the paternal chromosome [119]. SNHG14 can be divided into two functional units based on tissue-specific transcription patterns in humans [119]. The proximal part of the SNHG14/SNRPN transcript includes two mRNAs encoding the SNURF and SNRPN proteins; two lncRNAs with snoRNAs at their 5'-ends and polyadenylated at 3'-ends, i.e., SPA1 and SPA2 that are involved in protein binding and alternative mRNA splicing [146, 147]; the non-coding “maternal” gene of several C/D-box snoRNAs (SNORD109A, SNORD107, SNORD108 and SNORD116) [119], and IPW encoding polyadenylated lncRNAs [148] represent exons in the proximal part of SNHG14, which is transcribed in all tissues [149, 150].
It has been shown that SPA2 and IPW lncRNAs regulate the expression of other genes. For example, lncRNA IPW controls the DLK1-DIO3 imprinted region and expression of the MEG3 imprinted gene on chromosome 14. Some clinical features of Prader–Willi syndrome may be caused by aberrant expression of maternal alleles within the DLK1-DIO3 region [109].
The transcription from the distal part of SNHG14, which includes another “maternal” non-coding gene containing snoRNA (SNORD115 and SNORD109B), and the non-coding UBE3A-ATS, is restricted almost exclusively to the brain [119, 120, 151]. The region separating the expressed proximal portion of the SNHG14 from the repressed distal portion includes a stretch of weak polyadenylation sites and conserved sequences within the last IPW exon and a cluster of CTCF binding sites in and around the exon of SNHG14 annotated as PWAR1/PAR1. Although both elements contribute to boundary function, IPW plays a larger role and is required to completely stop transcription in nonneuronal cells [152]. This confirms that UBE3A-ATS silences the paternal UBE3A allele by means of transcriptional interference.
The imprinted genes in the proximal region (MKRN3, MAGEL2, NECDIN, PWRN1, and NPAP1/C15orf2) are methylated on the maternal chromosome and expressed from the paternal chromosome.
The MKRN3 gene includes one exon. It is expressed in all tissues and encodes a zinc finger containing protein (for more information, see the Central Precocious Puberty Type 2 section).
The MAGEL2 gene is also a single-exon gene. It encodes MAGEL2, a member of the MAGE-protein family, whose mutations cause a phenotype similar to Prader–Willi syndrome (for more information, see the Schaaf–Young Syndrome section). MAGE proteins interact with E3-ubiquitin ligases containing the RING-zinc finger motif, with ubiquitin-specific proteases (deubiquitinases) and affect the ubiquitination of substrate proteins [153, 154].
The NDN gene contains one exon. It encodes the NECDIN protein that also belongs to the MAGE family and participates in neuronal differentiation and maturation, and it suppresses the growth of almost all postmitotic neurons in the brain [155].
The PWRN1 gene (Prader–Willi Region Non-Protein Coding RNA 1) encodes lncRNA with biallelic expression in the testes and kidneys and monoallelic expression in the brain. PWRN1 represents an alternative 5′-part of SNURF-SNRPN. It is assumed that PWRN1 may function to maintain the open chromatin conformation on the paternal allele and provide access to transcription factors [156].
The NPAP1/C15orf2 gene encodes a nuclear pore complex associated protein; it is part of the nuclear pore complex that mediates the transport of macromolecules between the nucleus and the cytoplasm and regulates gene expression, mRNA biogenesis, and the cell cycle. The gene contains one exon, is biallelically expressed in adult testis, and monoallelically expressed in the fetal brain, including the hypothalamus, which is related to several endocrine features of Prader–Willi syndrome [157, 158].
The only imprinted gene in the distal part of the imprinted region is UBE3A. The UBE3A gene is monoallelically expressed from the maternal allele in the brain, while the paternal allele is imprinted. The paternal UBE3A allele is silenced by the neuron-specific antisense transcript UBE3A-ATS on the paternal allele [119, 159].
The UBE3A gene, consisting of 16 exons, encodes E6-AP-ubiquitin-protein-ligase [160]. This gene is biallelically expressed in all tissues, and in several brain structures UBE3A is active only on the maternal chromosome [161]. In mice, the Ube3a gene is monoallelically expressed in the Purkinje cells of the cerebellar cortex, hippocampal neurons, and in the olfactory bulb [162]. The silencing of the maternal allele for UBE3A leads to Angelman syndrome.
The gene encoding ATPase phospholipid transporting 10A-ATP10A/ATP10C is located 200 kb more distally to the UBE3A gene. Initially, it was assumed that it is mainly expressed from the maternal allele in fibroblasts and various brain structures and functions to maintain contacts between cell membranes and communicate signals to the central nervous system, but it was revealed that this gene is not imprinted [163]. It is likely that the lack of gene expression in the brain in patients with Angelman syndrome caused by deletions may lead to more severe symptoms related to severe autism.
The pathogenesis of epileptic seizures in Angelman syndrome is related to a cluster of GABA-receptor genes that are subject to frequent deletions. One such gene, GABRB3, encodes the GABA receptor β3 subunit [164]. Experimental mice with a homozygous deletion of the Gabrb3 gene display memory disorders, learning disability, convulsive seizures, and motor disorders similar to the symptoms of Angelman syndrome. In animals heterozygous for this deletion, neurological disorders are less expressed [165]. Consequently, in the case of an extended deletion, the clinical manifestations of Angelman syndrome may be due to GABRB3 haploinsufficiency [166].
The OCA2 gene encoding melanosomal transmembrane protein is also subject to deletions in Angelman and Prader–Willi syndromes. The loss of both alleles of this gene leads to albinism, and heterozygous carriers of the mutation demonstrate a slight decrease in pigmentation. Indeed, skin hypopigmentation in Prader–Willi and Angelman syndromes occurs only due to extended deletions of the critical area, but is absent in patients with UPD, IC mutations, or point mutations in the UBE3A gene [118, 167].
Molecular Pathology that Causes Prader–Willi and Angelman Syndromes
The most common cause of Prader–Willi and Angelman syndromes is an extended deletion in the 15(q11.2–q13) critical region. This deletion occurs in 65–75% of patients, and in the population its prevalence is 0.67–1.0 per 10 000 live births. Deletion in the critical region on paternal chromosome 15 causes Prader–Willi syndrome, whereas Angelman syndrome is caused by deletion of the same region on its maternal homologue [168–170]. Several patients diagnosed with Prader–Willi syndrome had 400 kb microdeletions of the SNORD116 cluster [110, 171]. The smallest microdeletion is restricted to the SNORD116 cluster, a portion of IPW, and the SPA2 transcript. These microdeletions show that a region between SNRPN and UBE3A, the SNORD116 cluster in particular, may be critical to the key clinical presentations of Prader–Willi syndrome [172]. Since this imprinted region has a large homology with the corresponding mouse region, several models with SNORD116 deletion have been created. Mice with the deletion demonstrated phenotypic features characteristic of Prader–Willi syndrome, such as difficulties in motor learning, poor memory, hyperphagia, growth delay, and elevated anxiety [110].
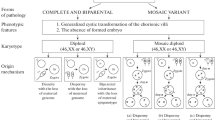
UPD is the second most common cause of Prader–Willi and Angelman syndromes. Maternal UPD accounts for 20–30% of Prader–Willi cases. Heterodisomy occurs in most patients with Prader–Willi syndrome and UPD due to nondisjunction of maternal chromosomes during the first meiotic division. Isodisomy due to nondisjunction of maternal chromosomes in the second meiotic division or segmental disomy because of crossing-over errors occur less frequently [170, 173]. This may be explained by the higher survival rate from zygotes with chromosome 15 trisomy and the early death of monosomic zygotes. Paternal UPD accounts for 3–7% of Angelman syndrome cases; UPD in this syndrome is commonly manifested by isodisomy due to the nondisjunction of paternal chromosomes during the second meiotic division and occurs due to corrections of monosomy to disomy or, more likely, as a result of postzygotic events [111, 173, 174]. Since zygotes carrying mono- or trisomy on chromosome 15 are not viable, the doubling of a single chromosome in monosomy and loss of an extra chromosome in trisomy may be regarded as an emergency measure, after which further development of the embryo becomes possible. The loss of an extra chromosome in a trisomic germ cell appears a more probable event than chromosomal doubling in a monosomal cell at an early stage of development, which explains the different frequency of UPD in Prader–Willi and Angelman syndromes.
The imprinting pathology in Prader–Willi and Angelman syndromes accounts for no more than 1 and 3%, respectively, and may involve a difficult-to-determine structural pathology in the IC or epigenetic alterations affecting methylation and gene expression in the entire imprinted region [175]. In both cases, patients inherit one copy of chromosome 15 from each parent. Loss of methylation at PWS-IC on the maternal chromosome leads to biallelic expression of SNHG14 and silencing of the maternal UBE3A. The imprinting pathology in 10–15% of cases is associated with a small deletion affecting the AS-IC, which regulates the establishment and maintenance of imprinting on the maternal chromosome [176]. More than 85% of the imprinting pathology in Angelman syndrome is caused by epimutation without an underlying change in the DNA sequence. In such cases, the maternal chromosome carrying an epimutation, and an aberrant paternal epigenotype/imprint (modifications of DNA and histone proteins that mark parental alleles and provide a monoallelic expression of imprinted genes) may be inherited from a maternal grandmother or maternal grandfather. The imprinting pathology is caused by an error in the establishment/switching of epigenetic imprints during gametogenesis or results from failure to maintain epigenetic imprint during the early stages of embryo [131]. Patients with Angelman syndrome quite frequently have somatic mosaicism including epimutation, which indicates that mosaic mutations may be acquired after fertilization during early embryogenesis [177].
The imprinting pathology in Prader–Willi syndrome is represented by abnormal methylation of PWS-IC, which downregulates the expression of imprinted genes on the paternal chromosome; 10–15% of cases include PWS-IC microdeletions, both inherited and arising during spermatogenesis or after fertilization. Most cases are associated with epimutations, i.e., accidental errors that occur during the establishment of paternal imprint or due to disruption of the maternal imprinting switch to paternal imprint during spermatogenesis or imprint erasure; if it occurs in early embryogenesis, then somatic mosaicism will result [131, 175].
Mutations in the UBE3A gene occur in 10–15% of cases of Angelman syndrome and represent mutations of the maternal allele with a premature stop during translation. Most of the known mutations alter the HECT-ligase domain. About 29% of mutations are inherited maternally and 71% occur de novo. Frameshift mutations and nonsense mutations dominate. Apparently, missense mutations do not significantly alter the protein function, and the phenotype of such patients differs from the phenotype characteristic of Angelman syndrome. Mutations in the UBE3A gene on the paternal chromosome are not phenotypically manifested [169, 178–181]. Atypical UBE3A deletions or rare types of structural rearrangements that alter the critical area have been reported in only a small number of patients with Angelman syndrome [182, 183].
Deficient expression of the maternal copy of the UBE3A gene in Purkinje cells may explain ataxia and tremor in Angelman syndrome, while epileptic seizures and learning inability may be associated with the lack of expression of this gene in hippocampal neurons [162].
Schaaf–Young syndrome (OMIM #615547), which is similar to Prader–Willi syndrome (Table 1), is caused by nonsense and missense mutations in the MAGEL2 gene, which is imprinted on the maternal chromosome and is expressed from the paternal chromosome [184, 185]. The phenotypic expressions range from severe fetal akinesia to mild akinesia, and include mental retardation and finger contractures [186].
MAGEL2 regulates ubiquitination, which is necessary for protein decay [187, 188]. Surprisingly, mutations that results in a shortened protein of the MAGEL2 gene cause Schaaf–Young syndrome, while deletion of the entire gene leads only to an insignificant clinical phenotype or to a complete lack of symptoms. Since MAGEL2 contains only one exon, mutations that result in a truncated protein may be explained by dominant-negative effects. It is likely that deletion of the entire paternal copy of the gene, including the promoter, may partly reverse the expression of the maternal methylated allele [189, 190].
Another disease is central precocious puberty type 2 (OMIM #615346), also known as gonadotropin-dependent precocious puberty (Table 1) [191, 192].
Loss-of-function mutations in the MKRN3 gene are detected most frequently in this disease. The MKRN3 gene encodes a 507-amino acid protein with a RING (C3HC4) zinc finger motif and multiple C3H zinc finger motifs, which predict MKRN3 RNA binding function. The gene contains one exon; it is expressed ubiquitously to yield a 3-kb transcript. The entire coding sequence and 5'-CpG island overlap a second gene, ZNF127AS, which is transcribed from the antisense strand with a different transcript size and pattern of expression. The antisense ZNF127AS RNA of unknown function probably regulates MKRN3 expression. Allele-specific analysis showed that the MKRN3 gene is expressed only from the paternal allele, and the maternal allele is imprinted [193]. Therefore, all affected patients with familial Schaaf–Young syndrome inherit MKRN3 mutations from their fathers [194].
Multiple mutations have been identified in the coding region of the MKNR3 gene, including deletions, nonsense, missense mutations, and frameshift loss-of-function mutations [195, 196]. In addition, some researchers reported mutations in the promoter region and at the transcription start site that alter normal MKNR3 gene expression [197].
MKRN3 can reduce pulsatile GnRH secretion and regulate puberty initiation. The level of MKRN3 decreases during puberty and negatively correlates with gonadotropin secretion in prepubertal girls. Its level in the blood decreases during puberty in healthy boys, but the exact mechanism of its action remains to be determined [198].
Another cause of central precocious puberty type 2 is mutations in the DLK1 gene that resides in the imprinted region of chromosome 14q32.2. Only the paternal allele of the DLK1 gene is expressed (similar to the MKRN3 gene), while the maternal allele is imprinted (for more information, see the Imprinted Chromosome Region 14q32.2 section). It is assumed that the KCNK9 imprinted gene (paternal allele), whose polymorphic variants were described in association with the early age of menarche, may participate in the pathogenesis of central precocious puberty type 2 [199].
Finally, we mention duplication of the imprinted region on chromosome 15q(11.2–q13.1) (OMIM #608636). Carriers of maternal duplication at the imprinted area have hypotonia and delayed motor and physical development and mental retardation, seizures, autism, and other behavioral anomalies, such as psychoses. Furthermore, the grade of these symptoms varies significantly even in individuals with the same genotype. Patients with an isodicentric chromosome of maternal origin (tetrasomy of the imprinted region) are commonly affected more seriously than patients with interstitial duplication. Such pathology of paternal origin is not phenotypically manifested [200–203].
Imprinted Chromosome Locus 20q13.2 and a Range of Diseases Associated with Defects in the GNAS gene
GNAS is a complex imprinting locus resulting in maternally, paternally, or biallelically expressed transcripts in differentially imprinted tissues. Gsα A (the stimulatory G protein alpha-subunit) encoded by exons 1–13 has no DMR and is biallelically expressed in most tissues except for the proximal renal tubules, neonatal brown adipose tissue, thyroid gland, gonads, paraventricular nucleus of the hypothalamus, and pituitary gland, where it is expressed from the maternal allele, but the promoter of the paternal allele is not methylated [204, 205].
The three alternative first exons of this locus (А/B, XLas and NESP55) are spliced with exons 2–13 to form different transcripts. DMRs are located in close proximity to alternative exons, which leads to the expression of NESP55 only from the maternal chromosome (its DMR is methylated on the paternal chromosome), whereas XLas, exon А/В, and NESP55АS/GNAS-AS1 antisense transcript are expressed from the paternal chromosome, since the maternal DMRs are methylated [205–207].
XLas, a longer version of Gsα, is synthesized mainly in neuroendocrine tissues and the nervous system; truncated neural transcripts of Gsα and XLas, referred to as GsαN1 and XLN1, are also known, and prematurely terminate prior to exon 4. Both proteins differ only by the N-terminal region. NESP55, another product of the GNAS gene, is a chromogranin-like neuroendocrine secretory protein. Similar to XLas, the alternative exon is spliced with exons 2–13, but NESP55 has no homology with the Gsα protein, since it contains a termination codon. The other two transcripts, A/B and AS1, which have their own exons, do not overlap with any of the other exons, and they are expressed in all tissues from the paternal chromosome, but are not translated [206–208].
The GNAS locus has two different IC regions. The first is located within the STX16 gene and controls the establishment of differential imprinting at the alternative GNAS A/B promoter, while the second, encompassing the antisense GNAS-AS1 transcript in exons 3–4, controls the establishment of imprinting over the entire GNAS locus [209].
The STX16 gene encoding syntaxin 16, which mediates intracellular interactions, is mapped 220 kb centromeric of the GNAS locus. Evidently, this gene cannot be involved in the pathogenesis of the disease, but all patients with maternal STX16 deletions demonstrate A/B loss of methylation, which impairs a cis-acting element regulating the imprinting of the maternal GNAS allele [205]. In most cases, two variants of deletions are detected: a 3-kb deletion with the loss of exons 4–6 or a 4.4-kb deletion with the loss of exons 2–4. The smallest region of deletion overlap contains a CpG-site that is not subject to differential methylation. STX16 deletions are not restricted only to this region, large deletions and deletions of the entire gene have been reported [210, 211].
In several cases of GNAS locus imprinting pathology, maternal deletions in exons 3 and 4, or 40 and 33-bp microdeletions in introns 4 and 3 of GNAS-AS1 were detected, which led to a loss of methylation in four DMRs and the appearance of biallelic DMR methylation of the NESP55 exon. These deletions lead to complete loss of the maternal imprint in the GNAS locus, resulting in biallelic expression of XL, A/B and antisense transcript, thus pointing to another regulatory element within antisense exons 3 and 4 necessary for full methylation of the maternal GNAS allele [207, 212, 213].
Molecular-genetic and epigenetic alterations in the GNAS locus cause a heterogeneous group of rare endocrine disorders called pseudohypoparathyroidism. It is mainly characterized by renal resistance to parathyroid hormone, which stimulates hypocalcemia, increased phosphate levels, and an increased secretion of parathyroid hormone. In addition to the increased level of parathyroid hormone, resistance to other hormones, such as thyrotropic hormone, whose activities are mediated by Gsα-subunit receptors, has also been described. Depending on the molecular anomalies, pseudohypoparathyroidism includes other endocrine disorders associated with resistance to a range of hormones and some non-endocrine features. The estimated prevalence of pseudohypoparathyroidism is 1.1 per 100 000. The occurrence of clinical heterogeneous phenotypes is related to structural and functional changes in the GNAS gene [209].
Pseudohypoparathyroidism type 1A (OMIM #103580) caused by loss-of-function mutations in the maternal allele of the GNAS gene has characteristic clinical features (Table 1) [206, 214].
Loss of Gsα function on the paternal allele causes pseudopseudohypoparathyroidism (OMIM #612463). The renal tubular cells express mainly the maternal GNAS allele, hence a paternally inherited mutation leads to a normal response of the kidney to the parathyroid hormone (Table 1) [215]. Paternal loss-of-function mutations may cause progressive osseous heteroplasia (OMIM #166350) (Table 1) [216]. In pseudopseudohypoparathyroidism and progressive osseous heteroplasia, Gsα expression in red blood cells is reduced twofold, although GNAS is normally biallelically expressed. The phenotype of Albright hereditary osteodystrophy can be caused by Gsα haploinsufficiency in tissues where GNAS is expressed from both alleles.
In contrast, pseudohypoparathyroidism type 1B (OMIM #603233) is characterized by isolated renal resistance to parathyroid hormone, and in some cases, resistance to thyrotropic hormone. In such patients, Albright hereditary osteodystrophy is rarely detected (Table 1) [209, 215, 217].
All patients with pseudohypoparathyroidism type 1B have at least loss of methylation at GNAS A/B DMR on the maternal allele, which leads to biallelic expression of the А/В-transcript and decreased expression of the GNAS-Gsα transcript in tissues subject to imprinting; hormone resistance occurs due to loss of methylation on the maternal allele [204]. The majority of sporadic pseudohypoparathyroidism type 1B involves disturbances of imprinting at both А/В exon DMR and all DMRs of the GNAS locus. In the absence of deletions within STX16 or NESP55 DMR and paternal UPD, molecular disorders in other distant regulatory regions may be suggested that still need to be determined. While mutations in GNAS are commonly detected in patients with pseudohypoparathyroidism type 1A, such mutations have not yet been detected in patients with pseudohypoparathyroidism type 1B. Since Gsα expression is disrupted in the disease, the correct methylation of exon А/В on the maternal chromosome located closely to the Gsα promoter is necessary for the expression of this protein, at least in the proximal renal tubules [209]. Approximately 20% of pseudohypoparathyroidism type 1B cases are inherited and caused by deletions within IC, while the other 80% are sporadic and are linked to methylation anomalies over the entire GNAS locus [211, 215].
Complete or segmented paternal UPD on chromosome 20 can reach 24% and leads to sporadic pseudohypoparathyroidism type 1B [218].
Maternal UPD on chromosome 20 (Table 1) commonly results from trisomy reduction to disomy, which occurred upon chromosomal nondisjunction in the second meiotic division, and causes Mulchandani–Bhoj–Conlin syndrome (ОМIМ #617352) [219, 220].
Multi-Locus Imprinting Disturbances (MLID) Affecting Methylation in Regulatory Regions
New multi-locus imprinting disturbances (MLID) caused by maternal hypomethylation of various imprinted loci have been in discussion since 2006 [221]. In a family (closely-related marriage), two daughters had phenotypic features of transient neonatal diabetes mellitus with some symptoms of Beckwith–Wiedemann syndrome. The investigation of methylation status at the imprinted regions showed that loss of methylation occurred not only in the imprinted PLAGL1 (6q24) region, but also within KCNQ1OT1 (11p15.5), GRB10 (7p11.2-р12), PEG3 (19q13), PEG1/MEST (7q32) and NESP55AS (20q13). It was hypothesized that the family either carried an autosomal recessive defect that disturbed methylation in children, or establishment of imprints in oocytes was disrupted [222].
The investigation of patients with transient neonatal diabetes mellitus and MLID revealed mutations in the ZFP57 gene [223]. Fourteen families with missense and nonsense mutations in ZFP57 have been described. All patients had very similar methylation patterns at imprinted DMRs: complete hypomethylation at DMR PLAGL1 and combinations of mosaic hypomethylation at GRB10, PEG3, MEST, NAP1L5 and GNAS [27].
ZFP57 encodes a transcription repressor that contains the Krüppel associated box (KRAB) domain encoded by exons 4 and 5 and seven zinc finger motifs in exon 6. The main function of the protein is to maintain DNA methylation in germline DMRs by binding a methylated hexanucleotide TGCmetCGC motif [224]. ZFP57 forms a complex with the KAP1 corepressor protein (KRAB-associated protein-1). KAP1 acts as a scaffold protein for the formation of an inactivating complex comprising histone lysine methyltransferase (SETDB1), the nucleosome remodeling and deacetylation complex (NuRD), heterochromatin protein 1 (HP-1), DNMT1 and UHRF1, which are necessary for DNA methylation maintenance. Proteins containing zinc finger motifs and the KRAB-domain act as transcription repressors by inducing KAP1 heterochromatin and DNA methylation in early embryonic cells [225]. Therefore, this protein complex plays a significant role in the regulation and maintenance of DNA methylation in various imprinted DMRs [226]. Consequently, heterozygous ZFP57 mutations, leading to loss or appearance of a defective protein, perturb methylation at various ICs, resulting in loss of imprinting [27, 227, 228].
ZFP57 binds methylated ICs in the preimplantation embryo, protects these ICs from demethylation and conserves their parental identity. The ZFP57 binding site was found in 17 out of 31 imprinted DMRs, and mutations in it most frequently impair methylation at PEG3, PLAGL1, INPPF5, NAP1L5 and GRB10 [228].
The expression profile of another member of the zinc finger motif protein family, ZNF445, its resistance to loss-of-function mutations, and its ability to bind to KAP1 and form heterochromatin in IC regions indicates its important role in methylation maintenance at the early stages of embryo development. Knockdown of the gene impairs KAP1 binding and H3K9me3 methylation, and, gene expression increases and loss of methylation in IC, including Н19, is detected. All this confirms that ZNF445, similar to ZFP57, can bind to IC, recruit KAP1 and trigger H3K9me3 methylation [229]. At the same time, these two genes are not expressed simultaneously: ZNF445 is expressed first and then ZFP57, and they recruit KAP1 one after another. ZNF445 binds to 13 imprinted DMRs: DIRAS3, ZDBF2, MEST, PEG 13, H19, KCNQ1OT1, MEG3-DLK1, MEG3, NET, GNAS-NESP55, GNAS-AS1, GNAS-XL, and SNU13. These two proteins work in tune to preserve imprints (methylation) on DMRs during early embryonic development [228].
Another gene, ZNF202, binds only four imprinted DMRs (FAM50B, PLAGL1, KCNQ1OT1, and L3MBTL1), but it is thought that it performs a similar function [228].
ZFP42, a zinc finger motif protein, is a marker of stem cells and is abundantly expressed in the preimplantation embryo. It protects the normally unmethylated alleles at imprinted DMR from methylation, Peg3 and Gnas in particular [230]. One patient with Russell–Silver syndrome and MLID with loss of methylation at H19/IGF2 (11p15.5) and MEST (7q32.2) was found to carry a paternally inherited mutation of this gene [55].
Mutations in the maternal genes encoding proteins of the subcortical oocyte complex may cause reproductive disturbances at the epigenetic level [231]. The subcortical oocyte complex plays an essential role at the early stage of embryogenesis and contains at least seven proteins (NLRP2, NLRP5, NLRP7, PADI6, KHDC3L, TLE6, and OOEP). These proteins are expressed only by the maternal genome in oocytes and at the early stages of embryo development, and they are then inactivated when the embryonic genome begins to function independently [231]. The NLRP2, NLRP5, NLRP7 genes encoding a small subfamily of cytoplasmic proteins containing pirin domains are actively expressed in growing oocytes and are necessary for oocyte maturation, regulation of methylation at the early stages of embryogenesis, and maintenance of ploidy in the early embryo [231]. Pathogenic variants of these proteins were found in mothers of children with MLID. These women had reproductive abnormalities (miscarriage and molar pregnancy). For example, the NLRP2 maternal mutation was found in two children with Beckwith–Wiedemann syndrome and MLID. The NLRP5 mutations led to loss of methylation at both maternal and paternal imprinted DMRs, resulting in MLID. NLRP7 is involved in the establishment of oocyte-specific methylation, and its mutations lead to recurrent molar pregnancy with extended loss of the maternal methylated imprint, while the paternal methylated DMRs are not altered [17, 232]. Mutations in the NLRP2, NLRP5, NLRP7 genes lead to Beckwith–Wiedemann syndrome, Russell–Silver syndrome, and transient neonatal diabetes mellitus; OOEP gene mutation cause transient neonatal diabetes mellitus, UHRF1 is associated with Russell–Silver syndrome, and ZAR1—Beckwith–Wiedemann syndrome [17]. Mutations in the PADI6 gene cause loss of methylation at imprinted DMRs: KCNQ1OT1, PLAGL1, GRB10, MEST, H19/IGF2, GNAS-AS1, GNAS-XL, MEG3, SNURF and may cause Russell–Silver, Beckwith–Wiedemann, and Temple syndromes [17, 233].
Other genes of the subcortical oocyte complex, i.e., KHDC3L, TLE6, OOEP, UHRF1, and ZAR1, also contribute (not significantly) to MLID [17, 232, 233].
Molecular-genetic studies have made it possible to detect MLID in some patients with imprinting diseases caused by epimutations. The frequency of MLID contribution to the phenotypic manifestations of imprinting diseases varies significantly in different studies. For example, transient neonatal diabetes mellitus with loss of PLAGL1-DMR methylation due to MLID is estimated to be 30–70%; Russell–Silver syndrome with loss of methylation at H19-DMR is caused by MLID in 7–30% of cases; Beckwith–Wiedemann syndrome with loss of methylation at KCNQ1OT1 or biallelic methylation at H19-DMR due to MLID may constitute 20–50%. MLID accounts for 6.3–12.5% of pseudohypoparathyroidism type 1B cases caused by loss of methylation in GNAS-A/B DMR. MLID-linked methylation disorders occur very rarely in Prader–Willi and Angelman syndromes [4, 17, 18, 234].
Multi-locus DNA methylation disturbances in imprinted DMRs of the genome have already been detected in a significant number of patients with imprinting disorders. Patients with MLID demonstrate a specific classical phenotype of a certain imprinting disease, but some of them develop a complex of symptoms characteristic of different syndromes caused by imprinting anomalies, as shown above. In addition, MLID are often mosaic, since the pathology occurs in a limited number of cells at the earliest stages of embryo development. Therefore, it is very difficult to determine specific correlations between epi-/genotype and the phenotypic manifestations in patients with MLID.
CONCLUSIONS
Over the past decade, significant progress has been achieved in the study of epigenetic regulation of gene expression. Investigation of the structural organization, specific allelic methylation and a certain allelic structure of chromatin in regulatory regions, cis- and trans-interaction of lncRNA with primary/germline (PLAGL1, H19/IGF2, PEG13, IGF2-DMR0, KCNQ1OT1, RB1, MEG3/DLK1, SNURF, GNAS-AS1, GNAS-XL, GNAS A/B) and secondary/somatic (IGF2-DMR2, DLK1, GTL2, MEG8, MAGEL2, NDN, SNRPN, SNORD116, GNAS-NESP) regulatory regions (DMRs) at various stages of ontogenesis allows us to understand how the epigenome functions in normal and pathological conditions. Disturbances of concerted interactions between these structural and functional components of the genome may cause pathological conditions, such as imprinting diseases, non-syndromic forms of mental retardation and delayed physical development, and multi-factor diseases, including oncological, autoimmune, and others.
Genomic imprinting plays a key role in a range of ontogenetic stages. It is controlled by a complex of genes that are monoallelically expressed in certain tissues or cell types. Therefore, the role of imprinted genes in development processes represents the most investigated area of epigenetics. Modern methods of molecular genetic analysis allow us to determine complete and segmental chromosomal UPD, where imprinted genes can be located [235–237], to conduct a whole-genome analysis of methylation [238, 239] or to study epitranscriptome and monoallelic expression, including in individual cells [15, 240]. These investigations reveal an increasing quantity of imprinted genes and DMRs. The next stage will clarify the role of these genes in ontogenesis and the contribution of methylation pathology to phenotypic manifestations. It is reasonable to assume that the cause of a number of syndromic conditions and non-specific forms of intrauterine/postnatal delayed development, where a molecular defect is still unknown, are epigenetic disorders represented by abnormal DNA methylation, impaired chromatin structure, and changes in lncRNA expression. In addition, one needs to take into account structural disorders of the genes involved in the establishment and maintenance of monoallelic gene expression both in germline cells and at the early stages of embryo development. Mutations in such genes may lead to MLID, and this, in turn, will require the elaboration of diagnostic protocols to determine molecular genetic pathology and prevent phenotypic disturbances in the offspring. Modern achievements in molecular biology and genetics inspire optimism that in the next decade, regularities of the functioning and interaction between the genome and epigenome will allow not only conduction of high-tech molecular genetic diagnostics of diseases based on epigenetic regulation disorders, but also development of effective ways to treat such diseases.
REFERENCES
McGrath J., Solter D. 1984. Completion of mouse embryogenesis requires both the maternal and paternal genomes. Cell. 37, 179–183.
Surani M.A., Barton S.C., Norris M.L. 1984. Development of reconstituted mouse eggs suggests imprinting of the genome during gametogenesis. Nature. 308, 548–550.
Sazhenova E.A., Lebedev I.N. 2021. Evolutionary aspects of genomic imprinting. Mol. Biol. (Moscow). 55 (1), 1–16.
Monk D., Mackay D.J.G., Eggermann T., Maher E.R., Riccio A. 2019. Genomic imprinting disorders: Lessons on how genome, epigenome and environment interact. Nat. Rev. Genet. 20, 235–248.
Singh P., Wu X., Lee D.-H., Li A.X., Rauch T.A., Pfeifer G.P., Mann J.R., Szabó P.E. 2011. Chromosome-wide analysis of parental allele-specific chromatin and DNA methylation. Mol. Cell. Biol. 31, 1757–1770.
Sanli I., Feil R. 2015. Chromatin mechanisms in the developmental control of imprinted gene expression. Int. J. Biochem. Cell. Biol. 67, 139–147.
Kota S.K., Llères D., Bouschet T., Hirasawa R., Marchand A., Begon-Pescia C., Sanli I., Arnaud P., Journot L., Girardot M., Feil R. 2014. ICR noncoding RNA expression controls imprinting and DNA replication at the Dlk1–Dio3 domain. Dev. Cell. 31, 19–33.
Kanduri C. 2016. Long noncoding RNAs: Lessons from genomic imprinting. Biochim. Biophys. Acta. 1859, 102–111.
Abi Habib W., Brioude F., Azzi S., Rossignol S., Linglart A., Sobrier M-L., Giabicani É., Steunou V., Harbison M.D., Le Bouc Y., Netchine I. 2019. Transcriptional profiling at the DLK1/MEG3 domain explains clinical overlap between imprinting disorders. Sci. Adv. 5, eaau9425.
MacDonald W.A., Mann M.R.W. 2020. Long noncoding RNA functionality in imprinted domain regulation. PLoS Genet. 16, e1008930.
Horsthemke B. 2014. In brief: genomic imprinting and imprinting diseases. J. Pathol. 232, 485–487.
Barlow D.P., Bartolomei M.S. 2014. Genomic imprinting in mammals. Cold Spring Harb. Perspect. Biol. 6 (2), a018382. https://doi.org/10.1101/cshperspect.a018382
Tucci V., Isles A.R., Kelsey G., Ferguson-Smith A.C., Erice Imprinting Group. 2019. Genomic imprinting and physiological processes in mammals. Cell. 176, 952–965.
Patten M.M., Cowley M., Oakey R.J., Feil R. 2016. Regulatory links between imprinted genes: Evolutionary predictions and consequences. Proc. Biol. Sci. 283 (1824), 20152760. https://doi.org/10.1098/rspb.2015.2760
Jadhav B., Monajemi R., Gagalova K.K., Ho D., Draisma H.H.M., van de Wiel M.A., Franke L., Heijmans B.T., van Meurs J., Jansen R., GoNL Consortium, BIOS Consortium, ’t Hoen PAC, Sharp A.J., Kiełbasa S.M. 2019. RNA-Seq in 296 phased trios provides a high-resolution map of genomic imprinting. BMC Biol. 17, 50.
Chaves T.F., Oliveira L.F., Ocampos M., Barbato I.T., de Luca G.R., Barbato Filho J.H., de Camargo Pinto L.L., Bernardi P., Maris A.F. 2019. Long contiguous stretches of homozygosity detected by chromosomal microarrays (CMA) in patients with neurodevelopmental disorders in the South of Brazil. BMC Med. Genomics. 12, 50.
Elbracht M., Mackay D., Begemann M., Kagan K.O., Eggermann T. 2020. Disturbed genomic imprinting and its relevance for human reproduction: Causes and clinical consequences. Hum. Reprod. Update. 26, 197–213.
Cerrato F., Sparago A., Ariani F., Brugnoletti F., Calzari L., Coppedè F., De Luca A., Gervasini C., Giardina E., Gurrieri F., Lo Nigro C., Merla G., Miozzo M., Russo S., Sangiorgi E., et al. 2020. DNA methylation in the diagnosis of monogenic diseases. Genes (Basel). 11 (4), 355. https://doi.org/10.3390/genes11040355
Temple I.K., Mackay D.J. 1993. Diabetes mellitus, 6q24-related transient neonatal. In: GeneReviews. Eds Adam M.P., Ardinger H.H., Pagon R.A., Wallace S.E., Bean L.J., Mirzaa G., Amemiya A. Seattle (WA): Univ. of Washington, 1993–2021. https://www.ncbi. nlm.nih.gov/books/NBK1534/
Temple I.K., Gardner R.J., Robinson D.O., Kibirige M.S., Ferguson A.W., Baum J.D., Barber J.C., James R.S., Shield J.P. 1996. Further evidence for an imprinted gene for neonatal diabetes localised to chromosome 6q22–q23. Hum. Mol. Genet. 5, 1117–1121.
Gardner R.J., Mackay D.J., Mungall A.J., Polychronakos C., Siebert R., Shield J.P., Temple I.K., Robinson D.O. 2000. An imprinted locus associated with transient neonatal diabetes mellitus. Hum. Mol. Genet. 9, 589–596.
Su H.-C., Wu S.-C., Yen L.-C., Chiao L.-K., Wang J.-K., Chiu Y.-L., Ho C.-L., Huang S.-M. 2020. Gene expression profiling identifies the role of Zac1 in cervical cancer metastasis. Sci. Rep. 10, 11837.
Hoffmann A., Spengler D. 2015. Role of ZAC1 in transient neonatal diabetes mellitus and glucose metabolism. World J. Biol. Chem. 6, 95–109.
Iglesias-Platas I., Court F., Camprubi C., Sparago A., Guillaumet-Adkins A., Martin-Trujillo A., Riccio A., Moore G.E., Monk D. 2013. Imprinting at the PLAGL1 domain is contained within a 70-kb CTCF/cohesin-mediated non-allelic chromatin loop. Nucleic Acids Res. 41, 2171–2179.
Varrault A., Dantec C., Le Digarcher A., Chotard L., Bilanges B., Parrinello H., Dubois E., Rialle S., Severac D., Bouschet T., Journot L. 2017. Identification of Plagl1/Zac1 binding sites and target genes establishes its role in the regulation of extracellular matrix genes and the imprinted gene network. Nucleic Acids Res. 45, 10466–10480.
Mackay D.J.G., Coupe A.-M., Shield J.P.H., Storr J.N.P., Temple I.K., Robinson D.O. 2002. Relaxation of imprinted expression of ZAC and HYMAI in a patient with transient neonatal diabetes mellitus. Hum. Genet. 110, 139–144.
Touati A., Errea-Dorronsoro J., Nouri S., Halleb Y., Pereda A., Mahdhaoui N., Ghith A., Saad A., Perez de Nanclares G., H′mida Ben Brahim D. 2019. Transient neonatal diabetes mellitus and hypomethylation at additional imprinted loci: Novel ZFP57 mutation and review on the literature. Acta Diabetol. 56, 301–307.
Kerr E.R., Stuhlmiller G.M., Maha G.C., Ladd M.A., Mikhail F.M., Koester R.P., Hurst A.C.E. 2018. Maternal uniparental isodisomy for chromosome 6 discovered by paternity testing: a case report. Mol. Cytogenet. 11, 60.
Court F., Camprubi C., Garcia C.V., Guillaumet-Adkins A., Sparago A., Seruggia D., Sandoval J., Esteller M., Martin-Trujillo A., Riccio A., Montoliu L., Monk D. 2014. The PEG13-DMR and brain-specific enhancers dictate imprinted expression within the 8q24 intellectual disability risk locus. Epigenetics Chromatin. 7, 5.
Ruf N., Bähring S., Galetzka D., Pliushch G., Luft F.C., Nürnberg P., Haaf T., Kelsey G., Zechner U. 2007. Sequence-based bioinformatic prediction and QUASEP identify genomic imprinting of the KCNK9 potassium channel gene in mouse and human. Hum. Mol. Genet. 16, 2591–2599.
Liang Z.S., Cimino I., Yalcin B., Raghupathy N., Vancollie V.E., Ibarra-Soria X., Firth H.V., Rimmington D., Farooqi I.S., Lelliott C.J., Munger S.C., O’Rahilly S., Ferguson-Smith A.C., Coll A.P., Logan D.W. 2020. Trappc9 deficiency causes parent-of-origin dependent microcephaly and obesity. PLoS Genet. 16, e1008916.
Zadeh N., Graham J.M. 1993. KCNK9 imprinting syndrome. In: GeneReviews. Eds. Adam M.P., Ardinger H.H., Pagon R.A., Wallace S.E., Bean L.J., Mirzaa G., Amemiya A. Seattle (WA): Univ. of Washington, 1993–2021. https://www.ncbi.nlm.nih.gov/ books/NBK425128/
Graham J.M., Zadeh N., Kelley M., Tan E.S., Liew W., Tan V., Deardorff M.A., Wilson G.N., Sagi-Dain L., Shalev S.A. 2016. KCNK9 imprinting syndrome-further delineation of a possible treatable disorder. Am. J. Med. Genet. A. 170, 2632–2637.
Šedivá M., Laššuthová P., Zámečník J., Sedláčková L., Seeman P., Haberlová J. 2020. Novel variant in the KCNK9 gene in a girl with Birk Barel syndrome. Eur. J. Med. Genet. 63, 103619.
Kashevarova A.A., Nikitina T.V., Mikhailik L.I., Belyaeva E.O., Vasilyev S.A., Lopatkina M.E., Fedotov D.A., Fonova E.A., Zarubin A.A., Sivtsev A.A., Skryabin N.A., Nazarenko L.P., Lebedev I.N. 2020. 46,XY,r(8)/45,XY,-8 mosaicism as a possible mechanism of the imprinted Birk–Barel syndrome: A case study. Genes (Basel). 11(12), 1473. https://doi.org/10.3390/genes11121473
Besson A., Dowdy S.F., Roberts J.M. 2008. CDK inhibitors: Cell cycle regulators and beyond. Dev. Cell. 14, 159–169.
Creff J., Besson A. 2020. Functional versatility of the CDK inhibitor p57Kip2. Front. Cell. Dev. Biol. 8, 584590.
Neyroud N., Richard P., Vignier N., Donger C., Denjoy I., Demay L., Shkolnikova M., Pesce R., Chevalier P., Hainque B., Coumel P., Schwartz K., Guicheney P. 1999. Genomic organization of the KCNQ1 K+ channel gene and identification of C-terminal mutations in the long-QT syndrome. Circ. Res. 84, 290–297.
Mitsuya K., Meguro M., Lee M.P., Katoh M., Schulz T.C., Kugoh H., Yoshida M.A., Niikawa N., Feinberg A.P., Oshimura M. 1999. LIT1, an imprinted antisense RNA in the human KvLQT1 locus identified by screening for differentially expressed transcripts using monochromosomal hybrids. Hum. Mol. Genet. 8, 1209–1217.
Pandey R.R., Mondal T., Mohammad F., Enroth S., Redrup L., Komorowski J., Nagano T., Mancini-Dinardo D., Kanduri C. 2008. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol. Cell. 32, 232–246.
Kanduri C. 2011. Kcnq1ot1: A chromatin regulatory RNA. Semin. Cell Dev. Biol. 22, 343–350.
Monk D., Sanches R., Arnaud P., Apostolidou S., Hills F.A., Abu-Amero S., Murrell A., Friess H., Reik W., Stanier P., Constância M., Moore G.E. 2006. Imprinting of IGF2 P0 transcript and novel alternatively spliced INS-IGF2 isoforms show differences between mouse and human. Hum. Mol. Genet. 15, 1259–1269.
Ghafouri-Fard S., Esmaeili M., Taheri M. 2020. H19 lncRNA: Roles in tumorigenesis. Biomed. Pharmacother. 123, 109774.
Jinno Y., Ikeda Y., Yun K., Maw M., Masuzaki H., Fukuda H., Inuzuka K., Fujishita A., Ohtani Y., Okimoto T. 1995. Establishment of functional imprinting of the H19 gene in human developing placentae. Nat. Genet. 10, 318–324.
Higashimoto K., Jozaki K., Kosho T., Matsubara K., Fuke T., Yamada D., Yatsuki H., Maeda T., Ohtsuka Y., Nishioka K., Joh K., Koseki H., Ogata T., Soejima H. 2014. A novel de novo point mutation of the OCT-binding site in the IGF2/H19-imprinting control region in a Beckwith–Wiedemann syndrome patient. Clin. Genet. 86, 539–544.
Hark A.T., Schoenherr C.J., Katz D.J., Ingram R.S., Levorse J.M., Tilghman S.M. 2000. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature. 405, 486–489.
Nativio R., Sparago A., Ito Y., Weksberg R., Riccio A., Murrell A. 2011. Disruption of genomic neighbourhood at the imprinted IGF2-H19 locus in Beckwith–Wiedemann syndrome and Silver–Russell syndrome. Hum. Mol. Genet. 20, 1363–1374.
Lopes S., Lewis A., Hajkova P., Dean W., Oswald J., Forné T., Murrell A., Constância M., Bartolomei M., Walter J., Reik W. 2003. Epigenetic modifications in an imprinting cluster are controlled by a hierarchy of DMRs suggesting long-range chromatin interactions. Hum. Mol. Genet. 12, 295–305.
Lee M.P., DeBaun M.R., Mitsuya K., Galonek H.L., Brandenburg S., Oshimura M., Feinberg A.P. 1999. Loss of imprinting of a paternally expressed transcript, with antisense orientation to KVLQT1, occurs frequently in Beckwith–Wiedemann syndrome and is independent of insulin-like growth factor II imprinting. Proc. Natl. Acad. Sci. U. S. A. 96, 5203–5208.
Mancini-Dinardo D., Steele S.J.S., Levorse J.M., Ingram R.S., Tilghman S.M. 2006. Elongation of the Kcnq1ot1 transcript is required for genomic imprinting of neighboring genes. Genes Dev. 20, 1268–1282.
Ferrero G.B., Boonen S.E., Cole T., Baker R., Bertoletti M., Cocchi G., Coze C., De Pellegrin M., Hussain K., Ibrahim A., et al. 2018. Expert consensus document: Clinical and molecular diagnosis, screening and management of Beckwith–Wiedemann syndrome: An international consensus statement. Nat. Rev. Endocrinol. 14, 229–249.
Wang K.H., Kupa J., Duffy K.A., Kalish J.M. 2019. Diagnosis and management of Beckwith–Wiedemann syndrome. Front. Pediatr. 7, 562.
Eggermann T., Brück J., Knopp C., Fekete G., Kratz C., Tasic V., Kurth I., Elbracht M., Eggermann K., Begemann M. 2020. Need for a precise molecular diagnosis in Beckwith–Wiedemann and Silver–Russell syndrome: What has to be considered and why it is important. J. Mol. Med. (Berl.). 98, 1447–1455.
Papulino C., Chianese U., Nicoletti M.M., Benedetti R., Altucci L. 2020. Preclinical and clinical epigenetic-based reconsideration of Beckwith–Wiedemann syndrome. Front. Genet. 11, 563718.
Fontana L., Bedeschi M.F., Maitz S., Cereda A., Faré C., Motta S., Seresini A., D′Ursi P., Orro A., Pecile V., Calvello M., Selicorni A., Lalatta F., Milani D., Sirchia S.M., et al. 2018. Characterization of multi-locus imprinting disturbances and underlying genetic defects in patients with chromosome 11p15.5 related imprinting disorders. Epigenetics. 13, 897–909.
Nemtsova M.V., Strel’nikov V.V., Babenko S.V., Zemlyakova V.V., Zaletaev D.V. 2005. Molecular diagnosis of epigenetic disorders in Beckwith–Wiedemann syndrome. Med. Genet. 4, 33–38.
Chang S., Bartolomei M.S. 2020. Modeling human epigenetic disorders in mice: Beckwith–Wiedemann syndrome and Silver–Russell syndrome. Dis. Model. Mech. 13 (5), dmm044123. https://doi.org/10.1242/dmm.044123
Yamaguchi Y., Tayama C., Tomikawa J., Akaishi R., Kamura H., Matsuoka K., Wake N., Minakami H., Kato K., Yamada T., Nakabayashi K., Hata K. 2019. Placenta-specific epimutation at H19-DMR among common pregnancy complications: Its frequency and effect on the expression patterns of H19 and IGF2. Clin. Epigenetics. 11, 113.
Brioude F., Netchine I., Praz F., Le Jule M., Calmel C., Lacombe D., Edery P., Catala M., Odent S., Isidor B., Lyonnet S., Sigaudy S., Leheup B., Audebert-Bellanger S., Burglen L., et al. 2015. Mutations of the imprinted CDKN1C gene as a cause of the overgrowth Beckwith–Wiedemann syndrome: Clinical spectrum and functional characterization. Hum. Mutat. 36, 894–902.
Eggermann T., Begemann M., Pfeiffer L. 2021. Unusual deletion of the maternal 11p15 allele in Beckwith–Wiedemann syndrome with an impact on both imprinting domains. Clin. Epigenetics. 13, 30.
Sun F.L., Dean W.L., Kelsey G., Allen N.D., Reik W. 1997. Transactivation of Igf2 in a mouse model of Beckwith–Wiedemann syndrome. Nature. 389, 809–815.
Wakeling E.L., Brioude F., Lokulo-Sodipe O., O’Connell S.M., Salem J., Bliek J., Canton A.P.M., Chrzanowska K.H., Davies J.H., Dias R.P., Dubern B., Elbracht M., Giabicani E., Grimberg A., Grønskov K., et al. 2017. Diagnosis and management of Silver–Russell syndrome: First international consensus statement. Nat. Rev. Endocrinol. 13, 105–124.
Lokulo-Sodipe O., Ballard L., Child J., Inskip H.M., Byrne C.D., Ishida M., Moore G.E., Wakeling E.L., Fenwick A., Mackay D.J.G., Davies J.H., Temple I.K. 2020. Phenotype of genetically confirmed Silver–Russell syndrome beyond childhood. J. Med. Genet. 57, 683–691.
Tümer Z., López-Hernández J.A., Netchine I., Elbracht M., Grønskov K., Gede L.B., Sachwitz J., den Dunnen J.T., Eggermann T. 2018. Structural and sequence variants in patients with Silver–Russell syndrome or similar features – curation of a disease database. Hum. Mutat. 39, 345–364.
Forbes B.E., Blyth A.J., Wit J.M. 2020. Disorders of IGFs and IGF-1R signaling pathways. Mol. Cell. Endocrinol. 518, 111035.
Dörr S., Midro A.T., Färber C., Giannakudis J., Hansmann I. 2001. Construction of a detailed physical and transcript map of the candidate region for Russell–Silver syndrome on chromosome 17q23–q24. Genomics. 71, 174–181.
Chiesa N., De Crescenzo A., Mishra K., Perone L., Carella M., Palumbo O., Mussa A., Sparago A., Cerrato F., Russo S., Lapi E., Cubellis M.V., Kanduri C., Cirillo Silengo M., Riccio A., Ferrero G.B. 2012. The KCNQ1OT1 imprinting control region and non-coding RNA: New properties derived from the study of Beckwith–Wiedemann syndrome and Silver–Russell syndrome cases. Hum. Mol. Genet. 21, 10–25.
Cytrynbaum C., Chong K., Hannig V., Choufani S., Shuman C., Steele L., Morgan T., Scherer S.W., Stavropoulos D.J., Basran R.K., Weksberg R. 2016. Genomic imbalance in the centromeric 11p15 imprinting center in three families: Further evidence of a role for IC2 as a cause of Russell–Silver syndrome. Am. J. Med. Genet A. 170, 2731–2739.
Gicquel C., Rossignol S., Cabrol S., Houang M., Steunou V., Barbu V., Danton F., Thibaud N., Le Merrer M., Burglen L., Bertrand A.-M., Netchine I., Le Bouc Y. 2005. Epimutation of the telomeric imprinting center region on chromosome 11p15 in Silver–Russell syndrome. Nat. Genet. 37, 1003–1007.
Inoue T., Nakamura A., Iwahashi-Odano M., Tanase-Nakao K., Matsubara K., Nishioka J., Maruo Y., Hasegawa Y., Suzumura H., Sato S., Kobayashi Y., Murakami N., Nakabayashi K., Yamazawa K., Fuke T., et al. 2020. Contribution of gene mutations to Silver–Russell syndrome phenotype: Multigene sequencing analysis in 92 etiology-unknown patients. Clin. Epigenet. 12, 86.
Saal H.M., Harbison M.D., Netchine I. 1993. Silver–Russell syndrome. In: GeneReviews. Eds Adam M.P., Ardinger H.H., Pagon R.A., Wallace S.E., Bean L.J., Mirzaa G., Amemiya A. Seattle (WA): Univ. of Washington, 1993–2021. https://www.ncbi.nlm.nih.gov/ books/NBK1324/
Hannula-Jouppi K., Muurinen M., Lipsanen-Nyman M., Reinius L.E., Ezer S., Greco D., Kere J. 2014. Differentially methylated regions in maternal and paternal uniparental disomy for chromosome 7. Epigenetics. 9, 351–365.
Hitchins M.P., Monk D., Bell G.M., Ali Z., Preece M.A., Stanier P., Moore G.E. 2001. Maternal repression of the human GRB10 gene in the developing central nervous system; evaluation of the role for GRB10 in Silver–Russell syndrome. Eur. J. Hum. Genet. 9, 82–90.
Schöherr N., Jäger S., Ranke M.B., Wollmann H.A., Binder G., Eggermann T. 2008. No evidence for isolated imprinting mutations in the PEG1/MEST locus in Silver–Russell patients. Eur. J. Med. Genet. 51, 322–324.
Su J., Wang J., Fan X., Fu C., Zhang S., Zhang Y., Qin Z., Li H., Luo J., Li C., Jiang T., Shen Y. 2017. Mosaic UPD(7q)mat in a patient with Silver–Russell syndrome. Mol. Cytogenet. 10, 36.
Brioude F., Oliver-Petit I., Blaise A., Praz F., Rossignol S., Jule M.L., Thibaud N., Faussat A.-M., Tauber M., Bouc Y.L., Netchine I. 2013. CDKN1C mutation affecting the PCNA-binding domain as a cause of familial Russell–Silver syndrome. J. Med. Genet. 50, 823–830.
Sabir A.H., Ryan G., Mohammed Z., Kirk J., Kiely N., Thyagarajan M., Cole T. 2019. Familial Russell–Silver syndrome like phenotype in the PCNA domain of the CDKN1C gene, a further case. Case Rep. Genet. 2019, 1398250. https://doi.org/10.1155/2019/1398250
Binder G., Ziegler J., Schweizer R., Habhab W., Haack T.B., Heinrich T., Eggermann T. 2020. Novel mutation points to a hot spot in CDKN1C causing Silver–Russell syndrome. Clin. Epigenetics. 12, 152.
Rockstroh D., Pfäffle H., Le Duc D., Rößler F., Schlensog-Schuster F., Heiker J.T., Kratzsch J., Kiess W., Lemke J.R., Abou Jamra R., Pfäffle R. 2019. A new p.(Ile66Serfs*93) IGF2 variant is associated with pre- and postnatal growth retardation. Eur. J. Endocrinol. 180, K1–13.
Masunaga Y., Inoue T., Yamoto K., Fujisawa Y., Sato Y., Kawashima-Sonoyama Y., Morisada N., Iijima K., Ohata Y., Namba N., Suzumura H., Kuribayashi R., Yamaguchi Y., Yoshihashi H., Fukami M., et al. 2020. IGF2 mutations. J. Clin. Endocrinol. Metabolism. 105, 116–125.
Hübner C.T., Meyer R., Kenawy A., Ambrozaityte L., Matuleviciene A., Kraft F., Begemann M., Elbracht M., Eggermann T. 2020. HMGA2 variants in Silver–Russell syndrome: Homozygous and heterozygous occurrence. J. Clin. Endocrinol. Metab. 105, 2401–2407.
Vado Y., Pereda A., Llano-Rivas I., Gorria-Redondo N., Díez I., Perez de Nanclares G. 2020. Novel variant in PLAG1 in a familial case with Silver–Russell syndrome suspicion. Genes. 11, 1461.
Akhtar M., Holmgren C., Göndör A., Vesterlund M., Kanduri C., Larsson C., Ekström T.J. 2012. Cell type and context-specific function of PLAG1 for IGF2 P3 promoter activity. Int. J. Oncol. 41, 1959–1966.
Hara-Isono K., Matsubara K., Fuke T., Yamazawa K., Satou K., Murakami N., Saitoh S., Nakabayashi K., Hata K., Ogata T., Fukami M., Kagami M. 2020. Genome-wide methylation analysis in Silver–Russell syndrome, Temple syndrome, and Prader–Willi syndrome. Clin. Epigenetics. 12, 159.
Vilain E., Le Merrer M., Lecointre C., Desangles F., Kay M.A., Maroteaux P., McCabe E.R. 1999. IMAGe, a new clinical association of intrauterine growth retardation, metaphyseal dysplasia, adrenal hypoplasia congenita, and genital anomalies. J. Clin. Endocrinol. Metab. 84, 4335–4340.
Borges K.S., Arboleda V.A., Vilain E. 2015. Mutations in the PCNA-binding site of CDKN1C inhibit cell proliferation by impairing the entry into S phase. Cell Div. 10, 2.
Suntharalingham J.P., Ishida M., Buonocore F., Del Valle I., Solanky N., Demetriou C., Regan L., Moore G.E., Achermann J.C. 2019. Analysis of CDKN1C in fetal growth restriction and pregnancy loss. F1000Res. 8, 90.
Eggermann T., Binder G., Brioude F., Maher E.R., Lapunzina P., Cubellis M.V., Bergadá I., Prawitt D., Begemann M. 2014. CDKN1C mutations: Two sides of the same coin. Trends Mol Med. 20, 614–622.
Babenko O.V., Zemlyakova V.V., Saakyan S.V., Brovkina A.F., Strelnikov V.V., Zaletaev D.V., Nemtsova M.V. 2002. RB1 and CDKN2A functional defects resulting in retinoblastoma. Mol. Biol. (Moscow) 36 (5), 625–630.
Gelli E., Pinto A.M., Somma S., Imperatore V., Cannone M.G., Hadjistilianou T., De Francesco S., Galimberti D., Currò A., Bruttini M., Mari F., Renieri A., Ariani F. 2019. Evidence of predisposing epimutation in retinoblastoma. Hum. Mutat. 40, 201–206.
Kanber D., Berulava T., Ammerpohl O., Mitter D., Richter J., Siebert R., Horsthemke B., Lohmann D., Buiting K. 2009. The human retinoblastoma gene is imprinted. PLoS Genet. 5 (12), e1000790. https://doi.org/10.1371/journal.pgen.1000790
Buiting K., Kanber D., Horsthemke B., Lohmann D. 2010. Imprinting of RB1 (the new kid on the block). Brief Funct. Genomics. 9, 347–353.
Taylor M., Dehainault C., Desjardins L., Doz F., Levy C., Sastre X., Couturier J., Stoppa-Lyonnet D., Houdayer C., Gauthier-Villars M. 2007. Genotype–phenotype correlations in hereditary familial retinoblastoma. Hum. Mutat. 28, 284–293.
Eloy P., Dehainault C., Sefta M., Aerts I., Doz F., Cassoux N., Lumbroso le Rouic L., Stoppa-Lyonnet D., Radvanyi F., Millot G.A., Gauthier-Villars M., Houdayer C. 2016. A parent-of-origin effect impacts the phenotype in low penetrance retinoblastoma families segregating the c.1981C>T/p.Arg661Trp mutation of RB1. PLoS Genet. 12, e1005888.
Alekseeva E.A., Babenko O.V., Kozlova V.M., Ushakova T.L., Kazubskaya T.P., Saakyan S.V., Tanas A.S., Zaletaev D.V., Strelnikov V.V. 2019. The effect of parental origin of RB1 mutations in hereditary retinoblastoma with low penetrance. Med. Genet. 18 (8), 21–28.
Kagami M., Sekita Y., Nishimura G., Irie M., Kato F., Okada M., Yamamori S., Kishimoto H., Nakayama M., Tanaka Y., Matsuoka K., Takahashi T., Noguchi M., Tanaka Y., Masumoto K., et al. 2008. Deletions and epimutations affecting the human 14q32.2 imprinted region in individuals with paternal and maternal upd(14)-like phenotypes. Nat. Genet. 40, 237–242.
Falix F.A., Aronson D.C., Lamers W.H., Gaemers I.C. 2012. Possible roles of DLK1 in the Notch pathway during development and disease. Biochim. Biophys. Acta – Mol. Basis Disease. 1822, 988–995.
Gomes L.G., Cunha-Silva M., Crespo R.P., Ramos C.O., Montenegro L.R., Canton A., Lees M., Spoudeas H., Dauber A., Macedo D.B., Bessa D.S., Maciel G.A., Baracat E.C., Jorge A.A.L., Mendonca B.B., et al. 2019. DLK1 is a novel link between reproduction and metabolism. J. Clin. Endocrinol. Metab. 104, 2112–2120.
Kitazawa M., Sutani A., Kaneko-Ishino T., Ishino F. 2021. The role of eutherian-specific RTL1 in the nervous system and its implications for the Kagami–Ogata and Temple syndromes. Genes Cells. 26, 165–179.
Martinez M.E., Cox D.F., Youth B.P., Hernandez A. 2016. Genomic imprinting of DIO3, a candidate gene for the syndrome associated with human uniparental disomy of chromosome 14. Eur. J. Hum. Genet. 24, 1617–1621.
Hamilton S., de Cabo R., Bernier M. 2020. Maternally expressed gene 3 in metabolic programming. Biochim. Biophys. Acta – Gene Regul. Mech. 1863, 194396.
Kagami M., O′Sullivan M.J., Green A.J., Watabe Y., Arisaka O., Masawa N., Matsuoka K., Fukami M., Matsubara K., Kato F., Ferguson-Smith A.C., Ogata T. 2010. The IG-DMR and the MEG3-DMR at human chromosome 14q32.2: Hierarchical interaction and distinct functional properties as imprinting control centers. PLoS Genet. 6, e1000992.
da Rocha S.T., Edwards C.A., Ito M., Ogata T., Ferguson-Smith A.C. 2008. Genomic imprinting at the mammalian Dlk1-Dio3 domain. Trends Genet. 24, 306–316.
Ioannides Y., Lokulo-Sodipe K., Mackay D.J.G., Davies J.H., Temple I.K. 2014. Temple syndrome: Improving the recognition of an underdiagnosed chromosome 14 imprinting disorder: An analysis of 51 published cases. J. Med. Genet. 51, 495–501.
Kagami M., Nagasaki K., Kosaki R., Horikawa R., Naiki Y., Saitoh S., Tajima T., Yorifuji T., Numakura C., Mizuno S., Nakamura A., Matsubara K., Fukami M., Ogata T. 2017. Temple syndrome: Comprehensive molecular and clinical findings in 32 Japanese patients. Genet. Med. 19, 1356–1366.
Ogata T., Kagami M. 2016. Kagami–Ogata syndrome: A clinically recognizable upd(14)pat and related disorder affecting the chromosome 14q32.2 imprinted region. J. Hum. Genet. 61, 87–94.
Soellner L., Begemann M., Mackay D.J.G., Grønskov K., Tümer Z., Maher E.R., Temple I.K., Monk D., Riccio A., Linglart A., Netchine I., Eggermann T. 2017. Recent advances in imprinting disorders. Clin. Genet. 91, 3–13.
van der Werf I.M., Buiting K., Czeschik C., Reyniers E., Vandeweyer G., Vanhaesebrouck P., Lüdecke H.-J., Wieczorek D., Horsthemke B., Mortier G., Leroy J.G., Kooy R.F. 2016. Novel microdeletions on chromosome 14q32.2 suggest a potential role for non-coding RNAs in Kagami–Ogata syndrome. Eur. J. Hum. Genet. 24, 1724–1729.
Stelzer Y., Sagi I., Yanuka O., Eiges R., Benvenisty N. 2014. The noncoding RNA IPW regulates the imprinted DLK1–DIO3 locus in an induced pluripotent stem cell model of Prader–Willi syndrome. Nat. Genet. 46, 551–557.
Cavaillé J. 2017. Box C/D small nucleolar RNA genes and the Prader–Willi syndrome: A complex interplay. Wiley Interdisc. Rev. RNA. 8 (4). https://doi.org/10.1002/wrna.1417
Wang T.-S., Tsai W.-H., Tsai L.-P., Wong S.-B. 2020. Clinical characteristics and epilepsy in genomic imprinting disorders: Angelman syndrome and Prader–-Willi syndrome. Ci Ji Yi Xue Za Zhi. 32, 137–144.
Mendiola A.J.P., LaSalle J.M. 2021. Epigenetics in Prader–Willi syndrome. Front. Genet. 12, 624581.
Christian S. 1999. Large genomic duplicons map to sites of instability in the Prader–Willi/Angelman syndrome chromosome region (15q11–q13). Hum. Mol. Genet. 8, 1025–1037.
Nicholls R.D., Knepper J.L. 2001. Genome organization, function, and imprinting in Prader–Willi and Angelman syndromes. Annu. Rev. Genomics Hum. Genet. 2, 153–175.
Kim S.-J., Miller J.L., Kuipers P.J., German J.R., Beaudet A.L., Sahoo T., Driscoll D.J. 2012. Unique and atypical deletions in Prader–Willi syndrome reveal distinct phenotypes. Eur. J. Hum. Genet. 20, 283–290.
Chung M.S., Langouët M., Chamberlain S.J., Carmichael G.G. 2020. Prader–Willi syndrome: Reflections on seminal studies and future therapies. Open Biol. 10, 200195.
Butler M.G. 2020. Imprinting disorders in humans: A review. Curr. Opin. Pediatr. 32, 719–729.
Cheon C.K. 2016. Genetics of Prader–Willi syndrome and Prader–Will-like syndrome. Ann. Pediatr. Endocrinol. Metab. 21, 126–135.
Runte M., Hüttenhofer A., Gross S., Kiefmann M., Horsthemke B., Buiting K. 2001. The IC-SNURF-SNRPN transcript serves as a host for multiple small nucleolar RNA species and as an antisense RNA for UBE3A. Hum. Mol. Genet. 10, 2687–2700.
Galiveti C.R., Raabe C.A., Konthur Z., Rozhdestvensky T.S. 2014. Differential regulation of non-protein coding RNAs from Prader–Willi syndrome locus. Sci. Rep. 4, 6445.
Maina E.N., Webb T., Soni S., Whittington J., Boer H., Clarke D., Holland A. 2007. Analysis of candidate imprinted genes in PWS subjects with atypical genetics: A possible inactivating mutation in the SNURF/SNRPN minimal promoter. J. Hum. Genet. 52, 297–307.
Green Finberg Y., Kantor B., Hershko A.Y., Razin A. 2003. Characterization of the human SNRPN minimal promoter and cis elements within it. Gene. 304, 201–206.
Cassidy S.B., Schwartz S., Miller J.L., Driscoll D.J. 2012. Prader–Willi syndrome. Genet. Med. 14, 10–26.
Geuns E., De Rycke M., Van Steirteghem A., Liebaers I. 2003. Methylation imprints of the imprint control region of the SNRPN-gene in human gametes and preimplantation embryos. Hum. Mol. Genet. 12, 2873–2879.
Meng L., Person R.E., Huang W., Zhu P.J., Costa-Mattioli M., Beaudet A.L. 2013. Truncation of Ube3a-ATS unsilences paternal Ube3a and ameliorates behavioral defects in the Angelman syndrome mouse model. PLoS Genet. 9, e1004039.
Smith E.Y., Futtner C.R., Chamberlain S.J., Johnstone K.A., Resnick J.L. 2011. Transcription Is required to establish maternal imprinting at the Prader–Willi syndrome and Angelman syndrome locus. PLoS Genet. 7 (12), e1002422. https://doi.org/10.1371/journal.pgen.1002422
Buiting K., Lich C., Cottrell S., Barnicoat A., Horsthemke B. 1999. A 5-kb imprinting center deletion in a family with Angelman syndrome reduces the shortest region of deletion overlap to 880 bp. Hum. Genet. 105, 665–666.
Lewis M.W., Brant J.O., Kramer J.M., Moss J.I., Yang T.P., Hansen P.J., Williams R.S., Resnick J.L. 2015. Angelman syndrome imprinting center encodes a transcriptional promoter. Proc. Natl. Acad. Sci. U. S. A. 112, 6871–6875.
Lewis M.W., Vargas-Franco D., Morse D.A., Resnick J.L. 2019. A mouse model of Angelman syndrome imprinting defects. Hum. Mol. Genet. 28, 220–229.
Wu M.-Y., Tsai T.-F., Beaudet A.L. 2006. Deficiency of Rbbp1/Arid4a and Rbbp1l1/Arid4b alters epigenetic modifications and suppresses an imprinting defect in the PWS/AS domain. Genes Dev. 20, 2859–2870.
Buiting K., Gross S., Lich C., Gillessen-Kaesbach G., el-Maarri O., Horsthemke B. 2003. Epimutations in Prader–Willi and Angelman syndromes: a molecular study of 136 patients with an imprinting defect. Am. J. Hum. Genet. 72, 571–577.
Ohta T., Gray T.A., Rogan P.K., Buiting K., Gabriel J.M., Saitoh S., Muralidhar B., Bilienska B., Krajewska-Walasek M., Driscoll D.J., Horsthemke B., Butler M.G., Nicholls R.D. 1999. Imprinting-mutation mechanisms in Prader–Willi syndrome. Am. J. Hum. Genet. 64, 397–413.
Saitoh S., Buiting K., Cassidy S.B., Conroy J.M., Driscoll D.J., Gabriel J.M., Gillessen-Kaesbach G., Glenn C.C., Greenswag L.R., Horsthemke B., Kondo I., Kuwajima K., Niikawa N., Rogan P.K., Schwartz S., et al. 1997. Clinical spectrum and molecular diagnosis of Angelman and Prader–Willi syndrome patients with an imprinting mutation. Am. J. Med. Genet. 68, 195–206.
Bielinska B., Blaydes S.M., Buiting K., Yang T., Krajewska-Walasek M., Horsthemke B., Brannan C.I. 2000. De novo deletions of SNRPN exon 1 in early human and mouse embryos result in a paternal to maternal imprint switch. Nat. Genet. 25, 74–78.
Gray T.A., Saitoh S., Nicholls R.D. 1999. An imprinted, mammalian bicistronic transcript encodes two independent proteins. Proc. Natl. Acad. Sci. U. S. A. 96, 5616–5621.
Glenn C.C., Saitoh S., Jong M.T., Filbrandt M.M., Surti U., Driscoll D.J., Nicholls R.D. 1996. Gene structure, DNA methylation, and imprinted expression of the human SNRPN gene. Am. J. Hum. Genet. 58, 335–346.
de los Santos T., Schweizer J., Rees C.A., Francke U. 2000. Small evolutionarily conserved RNA, resembling C/D box small nucleolar RNA, is transcribed from PWCR1, a novel imprinted gene in the Prader–Willi deletion region, which Is highly expressed in brain. Am. J. Hum. Genet. 67, 1067–1082.
Gallagher R.C., Pils B., Albalwi M., Francke U. 2002. Evidence for the role of PWCR1/HBII-85 C/D box small nucleolar RNAs in Prader–Willi syndrome. Am. J. Hum. Genet. 71, 669–678.
Bortolin-Cavaille M.-L., Cavaille J. 2012. The SNORD115 (H/MBII-52) and SNORD116 (H/MBII-85) gene clusters at the imprinted Prader–Willi locus generate canonical box C/D snoRNAs. Nucleic Acids Res. 40, 6800–6807.
Bratkovič T., Božič J., Rogelj B. 2020. Functional diversity of small nucleolar RNAs. Nucleic Acids Res. 48, 1627–1651.
Raabe C.A., Voss R., Kummerfeld D-M., Brosius J., Galiveti C.R., Wolters A., Seggewiss J., Huge A., Skryabin B.V., Rozhdestvensky T.S. 2019. Ectopic expression of Snord115 in choroid plexus interferes with editing but not splicing of 5-Ht2c receptor pre-mRNA in mice. Sci. Rep. 9, 4300.
Leung K.N., Vallero R.O., DuBose A.J., Resnick J.L., LaSalle J.M. 2009. Imprinting regulates mammalian snoRNA-encoding chromatin decondensation and neuronal nucleolar size. Hum. Mol. Genet. 18, 4227–4238.
Bieth E., Eddiry S., Gaston V., Lorenzini F., Buffet A., Conte Auriol F., Molinas C., Cailley D., Rooryck C., Arveiler B., Cavaillé J., Salles J.P., Tauber M. 2015. Highly restricted deletion of the SNORD116 region is implicated in Prader–Willi syndrome. Eur. J. Hum Genet. 23, 252–255.
Powell W.T., Coulson R.L., Crary F.K., Wong S.S., Ach R.A., Tsang P., Alice Yamada N., Yasui D.H., Lasalle J.M. 2013. A Prader–Willi locus lncRNA cloud modulates diurnal genes and energy expenditure. Hum. Mol. Genet. 22, 4318–4328.
Coulson R.L., Yasui D.H., Dunaway K.W., Laufer B.I., Vogel Ciernia A., Zhu Y., Mordaunt C.E., Totah T.S., LaSalle J.M. 2018. Snord116-dependent diurnal rhythm of DNA methylation in mouse cortex. Nat. Commun. 9, 1616.
Wu H., Yin Q.-F., Luo Z., Yao R.-W., Zheng C.-C., Zhang J., Xiang J.-F., Yang L., Chen L.-L. 2016. Unusual processing generates SPA lncRNAs that sequester multiple RNA binding proteins. Mol. Cell. 64, 534–548.
Yin Q.-F., Yang L., Zhang Y., Xiang J.-F., Wu Y.-W., Carmichael G.G., Chen L.-L. 2012. Long noncoding RNAs with snoRNA ends. Mol. Cell. 48, 219–230.
Wevrick R., Kerns J.A., Francke U. 1994. Identification of a novel paternally expressed gene in the Prader–Willi syndrome region. Hum. Mol. Genet. 3, 1877–1882.
Cavaillé J., Buiting K., Kiefmann M., Lalande M., Brannan C.I., Horsthemke B., Bachellerie J.P., Brosius J., Hüttenhofer A. 2000. Identification of brain-specific and imprinted small nucleolar RNA genes exhibiting an unusual genomic organization. Proc. Natl. Acad. Sci. U. S. A. 97, 14311–14316.
Castle J.C., Armour C.D., Löwer M., Haynor D., Biery M., Bouzek H., Chen R., Jackson S., Johnson J.M., Rohl C.A., Raymond C.K. 2010. Digital genome-wide ncRNA expression, including snoRNAs, across 11 human tissues using polyA-neutral amplification. PLoS One. 5, e11779.
Chamberlain S.J., Chen P.-F., Ng K.Y., Bourgois-Rocha F., Lemtiri-Chlieh F., Levine E.S., Lalande M. 2010. Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader–Willi syndromes. Proc. Natl. Acad. Sci. U. S. A. 107, 17668–17673.
Hsiao J.S., Germain N.D., Wilderman A., Stoddard C., Wojenski L.A., Villafano G.J., Core L., Cotney J., Chamberlain S.J. 2019. A bipartite boundary element restricts UBE3A imprinting to mature neurons. Proc. Natl. Acad. Sci. U. S. A. 116, 2181–2186.
Wijesuriya T.M., De Ceuninck L., Masschaele D., Sanderson M.R., Carias K.V., Tavernier J., Wevrick R. 2017. The Prader–Willi syndrome proteins MAGEL2 and necdin regulate leptin receptor cell surface abundance through ubiquitination pathways. Hum. Mol. Genet. 26, 4215–4230.
Tacer K.F., Potts P.R. 2017. Cellular and disease functions of the Prader–Willi syndrome gene MAGEL2. Biochem. J. 474, 2177–2190.
Pagliardini S., Ren J., Wevrick R., Greer J.J. 2005. Developmental abnormalities of neuronal structure and function in prenatal mice lacking the Prader–Willi syndrome gene necdin. Am. J. Pathol. 167, 175–191.
Wawrzik M., Spiess A.-N., Herrmann R., Buiting K., Horsthemke B. 2009. Expression of SNURF-SNRPN upstream transcripts and epigenetic regulatory genes during human spermatogenesis. Eur. J. Hum. Genet. 17, 1463–1470.
Buiting K., Nazlican H., Galetzka D., Wawrzik M., Groß S., Horsthemke B. 2007. C15orf2 and a novel noncoding transcript from the Prader–Willi/Angelman syndrome region show monoallelic expression in fetal brain. Genomics. 89, 588–595.
Neumann L.C., Markaki Y., Mladenov E., Hoffmann D., Buiting K., Horsthemke B. 2012. The imprinted NPAP1/C15orf2 gene in the Prader–Willi syndrome region encodes a nuclear pore complex associated protein. Hum. Mol. Genet. 21, 4038–4048.
Rougeulle C., Cardoso C., Fontés M., Colleaux L., Lalande M. 1998. An imprinted antisense RNA overlaps UBE3A and a second maternally expressed transcript. Nat. Genet. 19, 15–16.
Kishino T., Wagstaff J. 1998. Genomic organization of the UBE3A/E6-AP gene and related pseudogenes. Genomics. 47, 101–107.
Rougeulle C., Glatt H., Lalande M. 1997. The Angelman syndrome candidate gene, UBE3A/E6-AP, is imprinted in brain. Nat. Genet. 17, 14–15.
Dindot S.V., Antalffy B.A., Bhattacharjee M.B., Beaudet A.L. 2008. The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Hum. Mol. Genet. 17, 111–118.
DuBose A.J., Johnstone K.A., Smith E.Y., Hallett R.A.E., Resnick J.L. 2010. Atp10a, a gene adjacent to the PWS/AS gene cluster, is not imprinted in mouse and is insensitive to the PWS-IC. Neurogenetics. 11, 145–151.
Mohamad F.H., Has A.T.C. 2019. The α5-containing GABAA receptors-a. Brief summary. J. Mol. Neurosci. 67, 343–351.
DeLorey T.M., Sahbaie P., Hashemi E., Homanics G.E., Clark J.D. 2008. Gabrb3 gene deficient mice exhibit impaired social and exploratory behaviors, deficits in non-selective attention and hypoplasia of cerebellar vermal lobules: a potential model of autism spectrum disorder. Behav. Brain Res. 187, 207–220.
Delahanty R.J., Zhang Y., Bichell T.J., Shen W., Verdier K., Macdonald R.L., Xu L., Boyd K., Williams J., Kang J.-Q. 2016. Beyond epilepsy and autism: Disruption of GABRB3 causes ocular hypopigmentation. Cell Repts. 17, 3115–3124.
Buiting K., Williams C., Horsthemke B. 2016. Angelman syndrome: Insights into a rare neurogenetic disorder. Nat. Rev. Neurol. 12, 584–593.
Zemlyakova V.V., Ermakova M.A., Zaletaev D.V., Nemtsova M.V. 2009. Molecular diagnosis of Prader–Willi and Angelman syndromes. Med. Genet. 8, 16–20.
Bird L.M. 2014. Angelman syndrome: Review of clinical and molecular aspects. Appl. Clin. Genet. 7, 93–104.
Butler M.G., Miller J.L., Forster J.L. 2019. Prader–Willi syndrome: Clinical genetics, diagnosis and treatment approaches: an update. Curr. Pediatr. Rev. 15, 207–244.
Anderlid B.-M., Lundin J., Malmgren H., Lehtihet M., Nordgren A. 2014. Small mosaic deletion encompassing the snoRNAs and SNURF-SNRPN results in an atypical Prader–Willi syndrome phenotype. Am. J. Med. Genet. A. 164A, 425–431.
de Smith A.J., Purmann C., Walters R.G., Ellis R.J., Holder S.E., Van Haelst M.M., Brady A.F., Fairbrother U.L., Dattani M., Keogh J.M., Henning E., Yeo G.S.H., O’Rahilly S., Froguel P., Farooqi I.S., Blakemore A.I.F. 2009. A deletion of the HBII-85 class of small nucleolar RNAs (snoRNAs) is associated with hyperphagia, obesity and hypogonadism. Hum. Mol. Genet. 18, 3257–3265.
Fridman C., Koiffmann C.P. 2000. Origin of uniparental disomy 15 in patients with Prader–Willi or Angelman syndrome. Am. J. Med. Genet. 94, 249–253.
Robinson W.P., Christian S.L., Kuchinka B.D., Peñaherrera M.S., Das S., Schuffenhauer S., Malcolm S., Schinzel A.A., Hassold T.J., Ledbetter D.H. 2000. Somatic segregation errors predominantly contribute to the gain or loss of a paternal chromosome leading to uniparental disomy for chromosome 15. Clin. Genet. 57, 349–358.
Beygo J., Buiting K., Ramsden S.C., Ellis R., Clayton-Smith J., Kanber D. 2019. Update of the EMQN/ACGS best practice guidelines for molecular analysis of Prader–Willi and Angelman syndromes. Eur. J. Hum. Genet. 27, 1326–1340.
Beygo J., Grosser C., Kaya S., Mertel C., Buiting K., Horsthemke B. 2020. Common genetic variation in the Angelman syndrome imprinting centre affects the imprinting of chromosome 15. Eur. J. Hum. Genet. 28, 835–839.
Le Fevre A., Beygo J., Silveira C., Kamien B., Clayton-Smith J., Colley A., Buiting K., Dudding-Byth T. 2017. Atypical Angelman syndrome due to a mosaic imprinting defect: case reports and review of the literature. Am. J. Med. Genet. A. 173, 753–757.
Ermakova M.A., Babenko O.V., Zaletaev D.V., Nemtsova M.V. 2010. Analysis of UBE3A gene mutations in patients with Angelman syndrome. Med. Genet. 9 (5), 18–23.
Eggermann T., Perez de Nanclares G., Maher E.R., Temple I.K., Tümer Z., Monk D., Mackay D.J.G., Grønskov K., Riccio A., Linglart A., Netchine I. 2015. Imprinting disorders: A group of congenital disorders with overlapping patterns of molecular changes affecting imprinted loci. Clin. Epigenetics. 7, 123.
Buiting K., Clayton-Smith J., Driscoll D.J., Gillessen-Kaesbach G., Kanber D., Schwinger E., Williams C., Horsthemke B. 2015. Clinical utility gene card for Angelman syndrome. Eur. J. Hum. Genet. 23 (2). https://doi.org/10.1038/ejhg.2014.93
Beasley S.A., Kellum C.E., Orlomoski R.J., Idrizi F., Spratt D.E. 2020. An Angelman syndrome substitution in the HECT E3 ubiquitin ligase C-terminal lobe of E6AP affects protein stability and activity. PLoS One. 15, e0235925.
Aguilera C., Viñas-Jornet M., Baena N., Gabau E., Fernández C., Capdevila N., Cirkovic S., Sarajlija A., Miskovic M., Radivojevic D., Ruiz A., Guitart M. 2017. Novel intragenic deletions within the UBE3A gene in two unrelated patients with Angelman syndrome: Case report and review of the literature. BMC Med. Genet. 18, 137.
Bossuyt S.N.V., Punt A.M., de Graaf I.J., van den Burg J., Williams M.G., Heussler H., Elgersma Y., Distel B. 2021. Loss of nuclear UBE3A activity is the predominant cause of Angelman syndrome in individuals carrying UBE3A missense mutations. Hum. Mol. Genet. 30 (6), 430–442. https://doi.org/10.1093/hmg/ddab050
McCarthy J.M., McCann-Crosby B.M., Rech M.E., Yin J., Chen C.-A., Ali M.A., Nguyen H.N., Miller J.L., Schaaf C.P. 2018. Hormonal, metabolic and skeletal phenotype of Schaaf–Yang syndrome: A comparison to Prader–Willi syndrome. J. Med. Genet. 55, 307–315.
Negishi Y., Ieda D., Hori I., Nozaki Y., Yamagata T., Komaki H., Tohyama J., Nagasaki K., Tada H., Saitoh S. 2019. Schaaf–Yang syndrome shows a Prader–Willi syndrome-like phenotype during infancy. Orphanet. J. Rare Dis. 14, 277.
Chen X., Ma X., Zou C. 2020. Phenotypic spectrum and genetic analysis in the fatal cases of Schaaf–Yang syndrome: Two case reports and literature review. Medicine. 99, e20574.
Schaaf C.P., Gonzalez-Garay M.L., Xia F., Potocki L., Gripp K.W., Zhang B., Peters B.A., McElwain M.A., Drmanac R., Beaudet A.L., Caskey C.T., Yang Y. 2013. Truncating mutations of MAGEL2 cause Prader–Willi phenotypes and autism. Nat. Genet. 45, 1405–1408.
Carias K.V., Zoeteman M., Seewald A., Sanderson M.R., Bischof J.M., Wevrick R. 2020. A MAGEL2-deubiquitinase complex modulates the ubiquitination of circadian rhythm protein CRY1. PLoS One. 15, e0230874.
Patak J., Gilfert J., Byler M., Neerukonda V., Thiffault I., Cross L., Amudhavalli S., Pacio-Miguez M., Palomares-Bralo M., Garcia-Minaur S., Santos-Simarro F., Powis Z., Alcaraz W., Tang S., Jurgens J., et al. 2019. MAGEL2-related disorders: A study and case series. Clin. Genet. 96, 493–505.
Ahn H., Seo G.H., Oh A., Lee Y., Keum C., Heo S.H., Kim T., Choi J., Kim G.-H., Ko T.-S., Yum M.-S., Lee B.H., Choi I.H. 2020. Diagnosis of Schaaf–Yang syndrome in Korean children with developmental delay and hypotonia. Medicine (Baltimore). 99, e23864.
Roberts S.A., Kaiser U.B. 2020. Genetics in endocrinology: Genetic etiologies of central precocious puberty and the role of imprinted genes. Eur. J. Endocrinol. 183, R107–R117.
Seraphim C.E., Canton A.P.M., Montenegro L., Piovesan M.R., Macedo D.B., Cunha M., Guimaraes A., Ramos C.O., Benedetti A.F.F., de Castro Leal A., Gagliardi P.C., Antonini S.R., Gryngarten M., Arcari A.J., Abreu A.P., et al. 2021. Genotype–phenotype correlations in central precocious puberty caused by MKRN3 mutations. J. Clin. Endocrinol. Metab. 106, 1041–1050.
Jong M.T.C., Gray T.A., Ji Y., Glenn C.C., Saitoh S., Driscoll D.J., Nicholls R.D. 1999. A novel imprinted gene, encoding a ring zinc-finger protein, and overlapping antisense transcript in the Prader–Willi syndrome critical region. Hum. Mol. Genet. 8, 783–793.
Latronico A.C., Brito V.N., Carel J.-C. 2016. Causes, diagnosis, and treatment of central precocious puberty. Lancet Diabetes Endocrinol. 4, 265–274.
Valadares L.P., Meireles C.G., De Toledo I.P., Santarem de Oliveira R., Gonçalves de Castro L.C., Abreu A.P., Carroll R.S., Latronico A.C., Kaiser U.B., Guerra E.N.S., Lofrano-Porto A. 2019. MKRN3 mutations in central precocious puberty: A systematic review and meta-analysis. J. Endocrine Soc. 3, 979–995.
Maione L., Naulé L., Kaiser U.B. 2020. Makorin RING finger protein 3 and central precocious puberty. Curr. Opin. Endocrine Metab. Res. 14, 152–159.
Fanis P., Skordis N., Toumba M., Papaioannou N., Makris A., Kyriakou A., Neocleous V., Phylactou L.A. 2019. Central precocious puberty caused by novel mutations in the promoter and 5′-UTR region of the imprinted MKRN3 gene. Front. Endocrinol. 10, 677.
Abreu A.P., Macedo D.B., Brito V.N., Kaiser U.B., Latronico A.C. 2015. A new pathway in the control of the initiation of puberty: the MKRN3 gene. J. Mol. Endocrinol. 54, R131–139.
Perry J., Day F., Elks C., et Collaborators. 2014. Parent-of-origin-specific allelic associations among 106 genomic loci for age at menarche. Nature. 514, 92–97. https://doi.org/10.1038/nature13545
Li H., Du J., Li W., Cheng D., He W., Yi D., Xiong B., Yuan S., Tu C., Meng L., Luo A., Lin G., Lu G., Tan Y.-Q. 2018. Rare partial octosomy and hexasomy of 15q11–q13 associated with intellectual impairment and development delay: report of two cases and review of literature. Mol. Cytogenet. 11, 15.
Lu Y., Liang Y., Ning S., Deng G., Xie Y., Song J., Zuo N., Feng C., Qin Y. 2020. Rare partial trisomy and tetrasomy of 15q11–q13 associated with developmental delay and autism spectrum disorder. Mol. Cytogenet. 13, 21.
Yang L., Zhan G.D., Ding J.J., Wang H.J., Ma D., Huang G.Y., Zhou W.H. 2013. Psychiatric illness and intellectual disability in the Prader–Willi syndrome with different molecular defects – a meta analysis. PLoS One. 8 (8), e72640. https://doi.org/10.1371/journal.pone.0072640
Dykens E.M., Roof E., Hunt-Hawkins H., Dankner N., Lee E.B., Shivers C.M., Daniell C., Kim S.-J. 2017. Diagnoses and characteristics of autism spectrum disorders in children with Prader–Willi syndrome. J. Neurodev. Disord. 9, 18.
Linglart A., Maupetit-Méhouas S., Silve C. 2013. GNAS-related loss-of-function disorders and the role of imprinting. Horm. Res. Paediatr. 79, 119–129.
Turan S., Bastepe M. 2013. The GNAS complex locus and human diseases associated with loss-of-function mutations or epimutations within this imprinted gene. Horm. Res. Paediatr. 80, 229–241.
Lemos M.C., Thakker R.V. 2015. GNAS mutations in pseudohypoparathyroidism type 1a and related disorders. Hum. Mutat. 36, 11–19.
Mantovani G., Elli F.M. 2019. Inactivating PTH/PTHrP signaling disorders. Front. Horm. Res. 51, 147–159.
Turan S., Bastepe M. 2013. The GNAS complex locus and human diseases associated with loss-of-function mutations or epimutations within this imprinted gene. Horm. Res. Paediatr. 80, 229–241.
Mantovani G., Bastepe M., Monk D., de Sanctis L., Thiele S., Usardi A., Ahmed S.F., Bufo R., Choplin T., De Filippo G., Devernois G., Eggermann T., Elli F.M., Freson K., García Ramirez A., et al. 2018. Diagnosis and management of pseudohypoparathyroidism and related disorders: First International Consensus Statement. Nat. Rev. Endocrinol. 14, 476–500.
Yang Y., Chu X., Nie M., Song A., Jiang Y., Li M., Xia W., Xing X., Wang O. 2020. A novel long-range deletion spanning STX16 and NPEPL1 causing imprinting defects of the GNAS locus discovered in a patient with autosomal-dominant pseudohypoparathyroidism type 1B. Endocrine. 69, 212–219.
Kiuchi Z., Reyes M., Jüppner H. 2020. Preferential maternal transmission of STX16-GNAS mutations responsible for autosomal dominant pseudohypoparathyroidism type Ib (PHP1B): another example of transmission ratio distortion. J. Bone Miner. Res. 36(4), 696–703. https://doi.org/10.1002/jbmr.4221
Rezwan F.I., Poole R.L., Prescott T., Walker J.M., Karen Temple I., Mackay D.J. 2015. Very small deletions within the NESP55 gene in pseudohypoparathyroidism type 1b. Eur. J. Hum. Genet. 23, 494–499.
Takatani R., Molinaro A., Grigelioniene G., Tafaj O., Watanabe T., Reyes M., Sharma A., Singhal V., Raymond F.L., Linglart A., Jüppner H. 2016. Analysis of multiple families with single individuals affected by pseudohypoparathyroidism type Ib (PHP1B) reveals only one novel maternally inherited GNAS deletion: Only one novel maternally inherited GNAS deletion among multiple PHP1B patients. J. Bone Miner. Res. 31, 796–805.
Swieringa F., Solari F.A., Pagel O., Beck F., Huang J., Feijge M.A.H., Jurk K., Körver-Keularts I.M.L.W., Mattheij N.J.A., Faber J., Pohlenz J., Russo A., Stumpel C.T.R.M., Schrander D.E., Zieger B., et al. 2020. Impaired iloprost-induced platelet inhibition and phosphoproteome changes in patients with confirmed pseudohypoparathyroidism type Ia, linked to genetic mutations in GNAS. Sci. Rep. 10, 11389.
Jüppner H. 2021. Molecular definition of pseudohypoparathyroidism variants. J. Clin. Endocrinol. Metab. 106 (6), 1541–1552. https://doi.org/10.1210/clinem/dgab060
Bastepe M. 2018. GNAS mutations and heterotopic ossification. Bone. 109, 80–85.
Turan S., Bastepe M. 2018. GNAS complex locus. In: Encyclopedia of Signaling Molecules. Ed. Choi S. Cham: Springer, 2173–2185. doi.org/https://doi.org/10.1007/978-3-319-67199-4_101631
Colson C., Decamp M., Gruchy N., Coudray N., Ballandonne C., Bracquemart C., Molin A., Mittre H., Takatani R., Jüppner H., Kottler M-L., Richard N. 2019. High frequency of paternal iso or heterodisomy at chromosome 20 associated with sporadic pseudohypoparathyroidism 1B. Bone. 123, 145–152.
Mulchandani S., Bhoj E.J., Luo M., Powell-Ha-milton N., Jenny K., Gripp K.W., Elbracht M., Eggermann T., Turner C.L.S., Temple I.K., Mackay D.J.G., Dubbs H., Stevenson D.A., Slattery L., Zackai E.H., et al. 2016. Maternal uniparental disomy of chromosome 20: a novel imprinting disorder of growth failure. Genet. Med. 18, 309–315.
Eggermann T., Oehl-Jaschkowitz B., Dicks S., Thomas W., Kanber D., Albrecht B., Begemann M., Kurth I., Beygo J., Buiting K. 2017. The maternal uniparental disomy of chromosome 6 (upd(6)mat) “phenotype”: Result of placental trisomy 6 mosaicism? Mol. Genet. Genom. Med. 5, 668–677.
Mackay D.J.G., Boonen S.E., Clayton-Smith J., Goodship J., Hahnemann J.M.D., Kant S.G., Njølstad P.R., Robin N.H., Robinson D.O., Siebert R., Shield J.P.H., White H.E., Temple I.K. 2006. A maternal hypomethylation syndrome presenting as transient neonatal diabetes mellitus. Hum. Genet. 120, 262–269.
Boonen S.E., Pörksen S., Mackay D.J., Oestergaard E., Olsen B., Brondum-Nielsen K., Temple I.K., Hahne-mann J.M. 2008. Clinical characterisation of the multiple maternal hypomethylation syndrome in siblings. Eur. J. Hum. Genet. 16, 453–461.
Mackay D.J.G., Callaway J.L.A., Marks S.M., White H.E., Acerini C.L., Boonen S.E., Dayanikli P., Firth H.V., Goodship J.A., Haemers A.P., Hahnemann J.M.D., Kordonouri O., Masoud A.F., Oestergaard E., Storr J., et al. 2008. Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in ZFP57. Nat. Genet. 40, 949–951.
Quenneville S., Verde G., Corsinotti A., Kapopoulou A., Jakobsson J., Offner S., Baglivo I., Pedone P.V., Grimaldi G., Riccio A., Trono D. 2011. In embryonic stem cells, ZFP57/KAP1 recognize a methylated hexanucleotide to affect chromatin and DNA methylation of imprinting control regions. Mol. Cell. 44, 361–372.
Ecco G., Imbeault M., Trono D. 2017. KRAB zinc finger proteins. Development. 144, 2719–2729.
Farhadova S., Gomez-Velazquez M., Feil R. 2019. Stability and lability of parental methylation imprints in development and disease. Genes (Basel). 10 (12), 999. https://doi.org/10.3390/genes10120999
Baglivo I., Esposito S., De Cesare L., Sparago A., Anvar Z., Riso V., Cammisa M., Fattorusso R., Grimaldi G., Riccio A., Pedone P.V. 2013. Genetic and epigenetic mutations affect the DNA binding capability of human ZFP57 in transient neonatal diabetes type 1. FEBS Lett. 587, 1474–1481.
Monteagudo-Sánchez A., Hernandez Mora J.R., Simon C., Burton A., Tenorio J., Lapunzina P., Clark S., Esteller M., Kelsey G., López-Siguero J.P., de Nanclares G.P., Torres-Padilla M-E., Monk D. 2020. The role of ZFP57 and additional KRAB-zinc finger proteins in the maintenance of human imprinted methylation and multi-locus imprinting disturbances. Nucleic Acids Res. 48, 11394–11407.
Takahashi N., Coluccio A., Thorball C.W., Planet E., Shi H., Offner S., Turelli P., Imbeault M., Ferguson-Smith A.C., Trono D. 2019. ZNF445 is a primary regulator of genomic imprinting. Genes Dev. 33, 49–54.
Kim J.D., Kim H., Ekram M.B., Yu S., Faulk C., Kim J. 2011. Rex1/Zfp42 as an epigenetic regulator for genomic imprinting. Hum. Mol. Genet. 20, 1353–1362.
Monk D., Sanchez-Delgado M., Fisher R. 2017. NLRPs, the subcortical maternal complex and genomic imprinting. Reproduction. 154, R161–170.
Begemann M., Rezwan F.I., Beygo J., Docherty L.E., Kolarova J., Schroeder C., Buiting K., Chokkalingam K., Degenhardt F., Wakeling E.L., Kleinle S., González Fassrainer D., Oehl-Jaschkowitz B., Turner C.L.S., Patalan M., et al. 2018. Maternal variants in NLRP and other maternal effect proteins are associated with multilocus imprinting disturbance in offspring. J. Med. Genet. 55, 497–504.
Eggermann T., Kadgien G., Begemann M., Elbracht M. 2020. Biallelic PADI6 variants cause multilocus imprinting disturbances and miscarriages in the same family. Eur. J. Hum. Genet. 29 (4), 575–580. https://doi.org/10.1038/s41431-020-00762-0
Mackay D.J.G., Eggermann T., Buiting K., Garin I., Netchine I., Linglart A., de Nanclares G.P. 2015. Multilocus methylation defects in imprinting disorders. Biomol. Concepts. 6, 47–57.
Nakka P., Pattillo Smith S., O’Donnell-Luria A.H., McManus K.F., Mountain J.L., Ramachandran S., Sathirapongsasuti J.F., Agee M., Auton A., Bell R.K., Bryc K., Elson S.L., Fontanillas P., Furlotte N.A., Hicks B., et al. 2019. Characterization of prevalence and health consequences of uniparental disomy in four million individuals from the general population. Am. J. Hum. Genet. 105, 921–932.
Yauy K., de Leeuw N., Yntema H.G., Pfundt R., Gilissen C. 2020. Accurate detection of clinically relevant uniparental disomy from exome sequencing data. Genet. Med. 22, 803–808.
Scuffins J., Keller-Ramey J., Dyer L., Douglas G., Torene R., Gainullin V., Juusola J., Meck J., Retterer K. 2021. Uniparental disomy in a population of 32 067 clinical exome trios. Genet Med. 23 (6), 1101–1107. https://doi.org/10.1038/s41436-020-01092-8
Gigante S., Gouil Q., Lucattini A., Keniry A., Beck T., Tinning M., Gordon L., Woodruff C., Speed T.P., Blewitt M.E., Ritchie M.E. 2019. Using long-read sequencing to detect imprinted DNA methylation. Nucleic Acids Res. 47, e46–e46.
Klobučar T., Kreibich E., Krueger F., Arez M., Pólvora-Brandão D., von Meyenn F., da Rocha S.T., Eckersley-Maslin M. 2020. IMPLICON: An ultra-deep sequencing method to uncover DNA methylation at imprinted regions. Nucleic Acids Res. 48, e92–e92.
Santoni F.A., Stamoulis G., Garieri M., Falconnet E., Ribaux P., Borel C., Antonarakis S.E. 2017. Detection of imprinted genes by single-cell allele-specific gene expression. Am. J. Hum. Genet. 100, 444–453.
ACKNOWLEDGMENTS
The authors are grateful to I.V. Bure for assistance in preparation of this paper.
Funding
This study was supported by the Russian Foundation for Basic Research, project no. 20-14-50138.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Сonflict of interest. The authors declare that they have no conflict of interest.
Statement on the welfare of humans or animals. This article does not contain any studies involving animals or human participants performed by any of the authors.
Additional information
Translated by M. Novikova
Abbreviations: lncRNA, Long Non-Coding RNA; UPD, Uniparental Disomy; AS-IC, Angelman Syndrome Imprinting Center; BP, Break Point; DMR, Differentially Methylated Regions; GOM, Gain of Methylation; IC, Imprinting Centre; LOM, Loss of Methylation; MLID, Multi-Locus Imprinting Disturbances; PWS-IC, Prader-Willi Syndrome Imprinting Center; snoRNA, small nucleolar RNA; miRNA, microRNA.
Rights and permissions
About this article

Cite this article
Zaletaev, D.V., Nemtsova, M.V. & Strelnikov, V.V. Epigenetic Regulation Disturbances on Gene Expression in Imprinting Diseases. Mol Biol 56, 1–28 (2022). https://doi.org/10.1134/S0026893321050149
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0026893321050149