
Abstract
The hydrates Ln(Htba)3 ∙ 3H2O (Ln = Yb (I), Er (II), Ho (III); Н2tba = 2-thiobarbituric acid), Ln(Htba)3 ∙ 2H2O and Ln(Htba)3 ∙ 8H2O were crystallized from aqueous solutions. According to single-crystal X-ray diffraction analysis data, the structure of monoclinic crystals of isostructural complexes I–III was [Ln2(H2O)6(μ2-Htba-О,O')4(Htba-О)2]n. The formation of isostructural Ln(Htba) ∙ 2H2O (Ln = La, Ce, Eu, Yb, Lu), Ln(Htba)3 ∙ 8H2O (Ln = Eu, Tb, Ho, Yb) and Y(Htba)3 ∙ nH2O (n = 2, 8) was confirmed by the comparison of X-ray diffraction patterns, and their composition was determined by elemental and thermal analyses. The stability of crystal hydrates under heating in an air atmosphere and in contact with their saturated solutions was studied.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
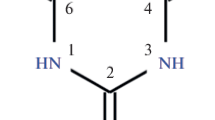
Thiobarbituric acid (H2tba) as a polyfunctional N,N',O,O',S-donating ligand with various binding centers (Fig. 1) forms coordination polymers of diverse structures with metal ions [1, 2]. Noteworthy are neutral homogeneous complexes (NHCs) with ligands of only one sort and water molecules, as they do not contain counterions, which frequently fill the potentially useful channels or cavities in the crystal lattice. Taking into account the various coordination modes of Htba– ions and H2O molecules (Table S1) [3] in the crystallization of NHCs from an aqueous medium, it is possible to obtain several compounds with a different number of coordinated water molecules. We have synthesized hydrates of HNCs with thiobarbituric, barbituric (H2ba), and 1,3-diethyl-2-thiobarbituric (HDetba) acids and s-, p-, and d-metal ions (Table S1) [2, 3]. It is of interest to study the structural and hydrate variety of compounds of this class for the practically important rare-earth elements (REEs) [4–7]. The information about the synthesis, structure, and properties of REE thiobarbiturate complexes, which may have luminescent properties and serve as precursors for the synthesis of oxysulfates [8] and oxides [9], is limited by five isostructural compounds Ln(Htba)3 ∙ 3H2O [9]. In the present work, the hydrates Ln(Htba)3 ∙ 3H2O, Ln(Htba)3 ∙ 2H2O, Ln(Htba)3 ∙ 8H2O and Y(Htba)3 ∙ nH2O (n = 2, 8) were synthesized. They were studied by powder X-ray diffraction and thermal analysis. The structures of Ln(Htba)3 ∙ 3H2O (Ln = Yb (I), Er (II), Ho (III)) were characterized by X-ray diffraction. Only one crystal structure was characterized for each NHC [10].

Graphical formula for a 2-thiobarbituric acid (H2tba) molecule.
EXPERIMENTAL
LuCl3 · 6H2O, YbCl3 · 6H2O, Er2O3, Ho2O3, YCl3 · 6H2O, Eu(CH3COO)3 · 4H2O, TbCl3 ∙ 6H2O, GdCl3 ∙ 6H2O, CeCl3 · 7H2O, La(NO3)3 · 6H2O, H2tba, HCl, and NaOH (all of chemically pure grade) were used for synthesis without additional purification. The oxides Er2O3 and Ho2O3 were dissolved in concentrated HCl and then carefully evaporated to dry chloride salts.
Synthesis of Ln(Htba)3∙ 3H2O(Ln = Yb (I), Er (II), Ho (III)). Hydrates of lanthanide salts (0.11 mmol) were dissolved in water (10 mL) to add solid H2tba (0.05 g, 0.35 mmol), thereupon the mixture was neutralized with a 1 M NaOH solution to pH of 4. The formed fine-crystalline precipitates composed, according to X-ray diffraction data, predominantly of corresponding octahydrates were dissolved under heating in a great excess of distilled water (200–300 mL), and the solutions allowed to vaporize for several weeks at room temperature to the formation of crystals suitable for X-ray diffraction analysis. The yield of complexes I–III increased during the evaporation of water to attain 20–30% at a water volume of 100–150 mL. The elemental analysis results for complexes I–III are given in Table 1.
A key factor influencing the composition of the synthesized isostructural hydrates M(Htba)3 ∙ 2H2O and M(Htba)3 ∙ 8H2O (M = Ln, Y) is the temperature. As a rule, the interaction between a REE salt and a NaOH neutralized H2tba solution (рН 4–5) leads to the formation of colorless octahydrates M(Htba)3 ∙ 8H2O at room temperature and pale yellow dihydrates M(Htba)3 ∙ 2H2O at 90°C. The methods of their synthesis are described below in the general form.
Synthesis of M(Htba)3∙ 2H2O (M = La, Ce, Eu, Yb, Lu, Y). To H2tba (0.20 g, 1.4 mmol) in water (5 mL), NaOH (0.056 g, 1.4 mmol) was added, thereupon the mixture was allowed to stand at 90°C for 5 min to the complete dissolution of thiobarbituric acid. To the hot yellow-orange solution, an aqueous solution (1 mL) containing a M(III) salt (0.46 mmol) was added under stirring. The initially formed white bulky precipitate (M(Htba)3 ∙ 8H2O) became more compact and yellow in color. It was filtered out, washed with acetone (2 mL), and dried in the air to a constant weight. The yield of these compounds was 40–50%. The results of their elemental analysis agree with the proposed composition (Table 1).
Synthesis of M(Htba)3∙ 8H2O (M = Eu, Tb, Ho, Yb, Y). The methods for the syntehsis of these isostructural compounds differ from the previous technique in that their synthesis was performed at room temperature, and a small excess of NaOH (1.6 mmol) was used for the faster dissolution of 2-thiobarbituric acid. The yield of these compounds was 50–60%. Their composition was confirmed by the chemical analysis data (Table 1).
X-ray diffraction analysis. Yellow crystals oc complex I (Ln = Yb) of 0.3 × 0.3 × 0.2 mm in size, complex II (Ln = Er) of 0.35 × 0.35 × 0.3 mm in size, and complex III (Ln = Ho) of 0.3 × 0.2 × 0.2 mm in size were studied at 296 K. Reflection intensities were measured on a Bruker AXS SMART APEX II single-crystal diffractometer (MoKα radiation) with a CCD detector. The experimental absorption corrections were applied with the SADABS software [11] by multiscanning. The structural model was established by direct methods and refined with the SHELXTL software suite [12]. Electron density difference syntheses were used to determine the hydrogen atom positions, which were further idealized and refined as bonded with the basic atoms. X-ray diffraction experiment parameters and structure refinement results are given in Table 2. The structures of complexes I–III were deposited with the Cambridge Structure Database (nos. 1971780–1971782; deposit@ccdc.cam.ac.uk or http://www.ccdc.cam.ac.uk/data_request/cif).
The powder X-ray diffraction patterns of these complexes were recorded at room temperature on a Bruker D8 Advance diffractometer with a Vantec linear detector and CuKα radiation. The phase purity of powders of complexes I–III was confirmed with the use of unit cell parameters from the single-crystal experiment, and the profile of refining the unit cell parameters was fitted by the Le Bail method using the TOPAS 4.2 software [13]. Refinement was stable and gave good difference X-ray diffraction patterns (Fig. 2).

Experimental, theoretical, and difference X-ray diffraction patterns for crystals of (a) Y(Htba)3 ∙ 8H2O, (b) Ho(Htba)3 ∙ 3H2O, and (c) Yb(Htba)3 ∙ 2H2O.
The thermal analysis of the complexes was performed on a TA Instruments SDT-Q600 analyzer in an air flow (50 mL/min) within a range of 22–850°C at a heating rate of 10 K/min. The composition of gaseous products was determined on a Thermo Scientific Nicolet380 IR spectrometer integrated with a thermal analyzer.
RESULTS AND DISCUSSION
The formation of three groups of hydrates M(Htba)3 ∙ 8H2O, M(Htba)3 ∙ 3H2O and M(Htba)3 ∙ 2H2O agrees with the results of chemical and thermal analyses. An isostructural character of the hydrates with the same chemical composition is confirmed by the coincidence of their powder X-ray diffraction patterns and calculated crystallographic parameters (Tables 2–4). For comparison Fig. 2 shows experimental X-ray diffraction patterns, which appreciably differ from each other, for three hydrates of REE thiobarbiturates with different compositions.
Crystallization from aqueous solution at room temperature primarily results in white bulky M(Htba)3 ∙ 8H2O hydrates, which are then converted into M(Htba)3 ∙ 3H2O for 1–3 months at room temperature. At 90°C, M(Htba)3 ∙ 8H2O is usually converted into more compact yellow fine-crystalline M(Htba)3 ∙ 2H2O precipitates. Such transformations agree with the empirical rule according to which the compounds containing a smaller number of water molecules crystallize at a higher temperature. All our attempts to separate these complexes in the form of crystals suitable for single-crystal X-ray diffraction analysis were unsuccessful. When brough in contact with the mother solution for 1–3 months at room temperature, M(Htba)3 ∙ 2H2O crystals were partially converted into the hydrates M(Htba)3 ∙ 3H2O, which were more thermodynamically stable in an aqueous solution.
The independent part of a cell in isostructural Ln(Htba)3 ∙ 3H2O (Ln = Yb, Er, Ho) contains two Ln3+ and three Htba– ions and three water molecules in shared positions. The coordination of Htba– ions to Ln3+ ions is implemented only through O atoms. One of the Ln3+ ion is bonded with six Htba– ions (two terminal and four bridging ones) and two water molecules, and the other is linked to four bridging Htba– ions and four water molecules (Fig. 3). The Ln(1)O8 and Ln(2)O8 polyhedra are square antiprisms linked to each other by means of bridging Нtba– ions with the formation of an infinite layer in the plane perpendicular to direction a + c (Fig. S1). Taking into account the different coordination environments of two independent Ln3+ ions and the fact that the structure contains both μ2-bridging and terminal Htba– ligands, the structure of the considered complexes can be described best of all by the formula [Ln2(H2O)6(Htba-О,O')4(Htba-О)2]n (Fig. 3) or merely Ln2(H2O)6(Htba)6. They are isostructural to the earlier characterized compounds Eu2(Htba)6(H2O)6 [8], Sm2(Htba)6(H2O)6, and Ln2(Htba)6(H2O)6 (Ln = Tb, Gd, Nd) [9]. The Ln–O bonds (Table S2) in complexes I–III (2.263(3)–2.406(3) Å) have typical lengths [10] and naturally decrease from complex I to complex III. The structure contains three independent, one terminal (C), and two bridging (An ad B) Htba– ions. Their corresponding geometric parameters are in satisfactory agreement with the literature data [11–19] and almost coincide with each other for complexes I–III, e.g., the lengths of the bonds С–О (1.250(8)–1.274(8) Å), C(4)–C(5) and C(5)–C(6) (1.382(10)–1.402(6) Å), and C–S (1.664(4)–1.697(7) Å).

Structure of [Ln2(H2O)6(HTBA-О,O')4(HTBA-О)2]n.
Structural analysis showed that complexes I–III have twelve hydrogen bonds N–H∙∙∙O, N–H∙∙∙S, O–H∙∙∙O, and O–H∙∙∙S (Table S3) with participation of all the Htba– ions and water molecules. These hydrogen bonds form a three-dimensional framework, in which the supramolecular \({\text{R}}_{2}^{2}\left( 8 \right),\) S(6), \({\text{R}}_{2}^{2}\left( {28} \right)\), and \({\text{R}}_{4}^{4}\left( {26} \right)\) motifs can be distinguished [20] similarly to the other isostructural Ln(III) thiobarbiturates [8, 9]. The π–π interactions [21] between Нtba– ions (Table S4) of head-to-tail type additionally stabilize the structure of the complexes.
At the beginning of thermal decomposition, all the synthesized hydrates are subjected to complete dehydration confirmed by the results of the IR spectroscopic analysis of gaseous products. We have not managed to identify the crystalline phases formed upon the complete dehydration of the synthesized compounds using X-ray diffraction. The compositions of intermediate and final thermolysis products were not studied in this work. The dehydration of Y(Htba)3 ∙ 8H2O occurs in two stages; the first begins at ~80°C, and the second is started at ~100°C (Fig. 4a). They are accompanied by the appearance of endotherms at 85 and 129°C, respectively. The weight loss (Δmexp) at ~200°C is close to its theoretically calculated value (Δmcalcd) under the assumption of complete dehydration (Δmexp = 20.9%, Δmcalcd = 21.7%, –8Н2О). At T > 370°C, the sample sustains oxidative decomposition, which corresponds to two exotherms at 422 and 598°C on the DSC curve. According to the results of IR spectroscopic analysis of gaseous products, water is the only gaseous product below 370°C, and СО2, CS2, and NН3 were detected among the thermolysis products within a range of 380–800°C.

TG and DSC curves for the oxidative dehydration of (a) Y(Htba)3 ∙ 8H2O, (b) Ho(Htba)3 ∙ 3H2O, and (c) Yb(Htba)3 ∙ 2H2O.
As follows from Fig. 4b, the dehydration of Ho(Htba)3 ∙ 3H2O (III) begins at 180°C and ends at 270–280°C. The experimental weight loss is close to the calculated value under the assumption of the removal of all Н2О molecules (Δmexp = 7.97%, Δmcalcd = 8.33%, –3Н2О). Dehydration is accompanied by the appearance of an endotherm at 234°C. At T > 320°C, the sample sustains oxidative decomposition, which corresponds to the exotherm at 570°C on the DSC curve. Below 320°C, the only gaseous product is water, and CS2, NН3, SO2, and СО2 were revealed in the thermolysis products within a range of 380–800°C.
The dehydration of Yb(Htba)3 ∙ 2H2O begins at 230°C and almost terminates at 300°C (Fig. 4c). It corresponds to the endotherm at 237°C. The weight loss at 250°C exceeds the theoretically calculated value under the assumption of the complete dehydration of this compound (Δmexp = 6.48%, Δmcalcd = 5.64%, ‒2Н2О). The observed difference seems to be due to a hygroscopic of this compound. At T > 370°C, the organic ligand is subject to oxidation, which corresponds to the strong exotherm at 451°C in the DSC curve. At T < 370°, the only gaseous product is water, and CS2, NН3, SO2, and СО2 were revealed among the thermolysis products within a range of 380–800°C.
The removal of all the water molecules from Y(Htba)3 ∙ 8H2O occurring as soon as at T < 150°C allows us to classify them as crystallization molecules. In contrast to them, the water molecules in Ho(Htba)3 ∙ 3H2O and Yb(Htba)3 ∙ 2H2O are most likely to be coordinated, as their dehydration begins at much higher temperatures in comparison with Y(Htba)3 ∙ 8H2O. As “hard” acids, Ln3+ ions are not prone to form any bonds with the ligands through a sulfur atom, as indirectly confirmed by the crystallization of their rare complexes with S-coordinated ligands only from non-polar solvents [4]. For this reason, when considering the possible structure of the hydrates Ln(Htba)3 ∙ 2H2O and Ln(Htba)3 ∙ 8H2O, it is possible to take into account only the Ln–OHtba and Ln–Ow bonds. Since CN(Ln(III)) ≥ 8 in common practice, and Нtba– ions do not form any O,O′-coordinated chelates [1, 2, 10], Yb(Htba)3 ∙ 2H2O and the hydrates isostructural to it are coordination polymers. Taking into account a relatively low complete dehydration temperature typical for uncoordinated water molecules, it is also possible to presume that Y(Htba)3 ∙ 8H2O and the complexes isostructural to it have a polymeric structure. Therefore, taking into account the earlier observed highest thermodynamic stability of M(Htba)3 ∙ 3H2O crystals in a solution, the mutual transformations of polymeric Ln(III) hydrates in their saturated aqueous solutions can be described by the scheme

CONCLUSIONS
Three types of hydrates of thiobarbiturate REE complexes (Htba)3 ∙ nH2O (n = 2, 3, 8) with different crystal structures and thermal stabilities in the processes of dehydration have been crystallized from aqueous solution under different conditions. The oxidative decomposition of coordinated Htba– ligands is started at a higher temperature than for free H2tba, which melts with decomposition as soon as ~250°C [22].
As already mentioned, hydrates of different composition and structure were synthesized earlier for neutral homogeneous complexes formed by H2tba and HDetba with s, p, and d metals (Table S1) [3]. The existence of several hydrates along with the possibility of formation of bond isomers by polyfunctional ligands enriches the coordination chemistry of the metal complexes with barbituric acids [2]. The separation of dihydrates, trihydrates, and octahydrates of REE thiobarbiturates is an example of the structural and hydrate variety of compounds of this class.
REFERENCES
K. T. Mahmudov, M. N. Kopylovich, A. M. Maharramov, et al., Coord. Chem. Rev. 265, 1 (2014). https://doi.org/10.1016/j.ccr.2014.01.002
N. N. Golovnev and M. S. Molokeev, 2-Thiobarbituric Acid and Its Metal Complexes: Synthesis, Structure, and Properties (Sib. Feder. Univ., Krasnoyarsk, 2014) [in Russian].
N. N. Golovnev, M. S. Molokeev, and M. K. Lesnikov, Zh. Sibirsk. Fed. Univ., Khim., No. 10, 401 (2017). https://doi.org/10.17516/1998-2836-0036
S. Cotton, Lanthanide and Actinide Chemistry (Wiley, Rutland (UK), 2006).
M. C. Heffern, L. M. Matosziuk, and T. J. Meade, Chem. Rev. 114, 4496 (2014). https://doi.org/10.1021/cr400477t
Y. Yang, Q. Zhao, W. Feng, and F. Li, Chem. Rev. 113, 192 (2013). https://doi.org/10.1021/cr2004103
K. Binnemans, Chem. Rev. 109, 4283 (2009). https://doi.org/10.1021/cr8003983
N. N. Golovnev and M. S. Molokeev, Russ. J. Coord. Chem. 40, 648 (2014). https://doi.org/10.1134/S1070328414090036
N. N. Golovnev, M. S. Molokeev, and I. V. Sterkhova, Zh. Neorg. Khim. 64, 965 (2019). https://doi.org/10.1134/S0044457X19090137
Cambridge Structural Database, Version 5.37 (Univ. of Cambridge, Cambridge (UK), 2015).
G. M. Sheldrick, SADABS, Version 2.01 (Bruker, Madison, WI, 2004).
G. M. Sheldrick, SHELXTL, Version 6.10 (Bruker, Madison, WI, 2004).
Bruker AXS TOPAS V4: General Profile and Structure Analysis Software for Powder Diffraction Data. User’s Manual (Bruker, Karlsruhe, 2008).
V. I. Balas, I. I. Verginadis, G. D. Geromichalos, et al., Eur. J. Med. Chem. 46, 2835 (2011). https://doi.org/10.5517/ccv3tc3
M. Kubicki, A. Owczarzak, V. I. Balas, and S. K. Hadjikakou, J. Coord. Chem. 65, 1107 (2012). https://doi.org/10.1080/00958972.2012.660148
Y. Gong, Z. Hao, J. Li, et al., Dalton Trans. 42, 6489 (2013). https://doi.org/10.1039/C3DT32380C
W. M. Hutzler, E. Egert, and M. Bolte, Acta Crystallogr., Sect. C 72, 705 (2016). https://doi.org/10.1107/S205322961601336X
S. Gomathi, J. S. Nirmalram, and P. T. Muthiah, Acta Crystallogr., Sect. B 71, 144 (2015). https://doi.org/10.1107/S2052520615001729
C. Wang, M.-S. Zhou, L.-J. Yang, et al., Jiegou Huaxue 36, 1210 (2017). https://doi.org/10.14102/j.cnki.0254-5861.2011-1494
J. W. Steed and J. L. Atwood Supramolecular Chemistry 1st Ed. (CRC Press, 2004; Akademkniga, Moscow, 2007).
PLATON: A Multipurpose Crystallographic Tool (Utrecht Univ., Utrecht, The Netherlands, 2008).
M. V. Roux, R. Notario, M. Segura, and J. S. Chickos, J. Chem. Eng. Data 57, 249 (2011). https://doi.org/10.1021/je200420u
ACKNOWLEDGMENTS
X-ray diffraction data were measured on the equipment of the Krasnoyarsk Regional Shared Facilities Center of the Federal Research Center “Krasnoyarsk Scientific Center” of the Siberian Branch of the Russian Academy of Sciences.
Funding
This study was financially supported by the Russian Foundation for Basic Research within scientific project no. 19-52-80003.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflict of interests.
Additional information
Translated by E. Glushachenkova
Supplementary material
Rights and permissions
About this article

Cite this article
Golovnev, N.N., Molokeev, M.S., Lesnikov, M.K. et al. Hydrates of Lanthanide(III) 2-Thiobarbiturates: Synthesis, Structure, and Thermal Decomposition. Russ. J. Inorg. Chem. 65, 999–1005 (2020). https://doi.org/10.1134/S0036023620070098
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0036023620070098