
Abstract
The crystal structure and the phonon spectrum of a La2Zr2O7 crystal have been investigated in terms of the MO LCAO approach using DFT hybrid functionals that take into account the contribution of the nonlocal exchange within the Hartree–Fock formalism. The frequencies, symmetry species, and intensities of IR and Raman active fundamental vibrations are determined. Elastic constants are calculated. The calculations have been carried out using the new version of the CRYSTAL program—CRYSTAL17—designed to model periodic structures within the MO LCAO approach.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
The interest in studies of rare-earth zirconates R2Zr2O7 (R is a rare-earth ion) is due to the variety of their properties and applications [1–5]. One representative of this series, La2Zr2O7, has been experimentally studied by the X-ray diffraction analysis, Raman spectroscopy, and IR spectroscopy methods [6–12]. The structure and the lattice dynamics of the La2Zr2O7 rare-earth zirconate were modeled more than dozen years ago by the molecular dynamics method [13], and, later, within the ab initio approach in the basis of plane waves [14, 15]. Recently, using the same approach, other rare-earth zirconates with a pyrochlore structure were also investigated [16]. The band structure and elastic constants were calculated in [14–16], but the phonon spectra were not studied. In this case, there is no information in the literature on the study of rare-earth zirconates, including La2Zr2O7, in terms of the MO LCAO approach. It seems relevant to perform such an investigation.
In this work, using the MO LCAO approach with DFT hybrid functionals, we will study the structure and the lattice dynamics of one of the representatives of this series—La2Zr2O7 rare-earth zirconate with the pyrochlore structure (\(Fd\overline 3 m\)).
CALCULATION METHODS
Our ab initio calculations were carried out within the density functional theory (DFT) using hybrid functionals that could take into account both local and nonlocal (in the Hartree–Fock (HF) formalism) exchange. Calculations were performed using the PBE0 [17], B3PW [18], and HSE06 [19, 20] functionals, which have been widely used in recent times [21], and also using the PBESOL0 functional incremented in the CRYSTAL17 program [22, 23]. The use of hybrid functionals that could take into account both local and nonlocal HF exchange makes it possible to describe well compounds with ionic–covalent bonds, their band structure, infrared (IR) and Raman spectra, and elastic properties [24–26]. Recently, a comparative analysis of the B3LYP, PBE0, and other functionals has been carried out with respect to CCSD calculations (altogether, 128 functionals of different levels were tested) [27]. It was shown that the PBE0 functional is characterized by a small error for functionals of its level upon reproduction of the electron density and other characteristics with respect to CCSD calculations [27]. In our previous works, using the PBE0 hybrid functional, the structure, the dynamics of the crystal lattice, and the elastic properties of rare-earth titanates R2Ti2O7 (R is a rare-earth ion) with the structure of pyrochlore were successfully described [28, 29].
Crystalline La2Zr2O7 was investigated for a highly symmetric structure of pyrochlore (\(Fd\overline 3 m\), Z = 2). Ions occupy the following positions: Zr—16c (0, 0, 0); La—16d (1/2, 1/2, 1/2); O1—48f (x, 1/8, 1/8); O2—8b (3/8, 3/8, 3/8) [6]. Oxygen is contained in all the structural units of this compound, being occupying two symmetrically nonequivalent positions. Therefore, the reproduction of the structure and the properties of the crystal will significantly depend on the basis set of oxygen. In this work, we used the TZVP basis set of the type of [30], which is available on website of the CRYSTAL program [31]. The basis set of zirconium [32] is also available on website of CRYSTAL. This basis set was used by the authors of the CRYSTAL program to calculate the structure and IR spectra of z-irconium complexes in which oxygen was a ligand of zirconium [32]. To describe the inner shells of lanthanum, the ECP46MWB quasirelativistic pseudopotential was used (ECP stands for the effective core potential; 46 is the number of inner electrons replaced by the pseudopotential; WB—quasi-relativistic [33, 34]). To describe the outer shells, 5s25p6, involved in the formation of a chemical bond, the TZVP valence basis set with diffuse and polarization orbitals was used [33, 35, 36]. Both the pseudopotential and the valence basis set (“ECP46MWB-II”) are available on the Stuttgart website [37]. Gaussian primitives with an exponent of smaller than 0.1 a.u.–2 were eliminated from the valence basis set, since these calculations are periodic. The last diffuse orbital of the type of 4f with an exponent of 0.17033 was also eliminated. In calculations, the crystal structure was initially optimized. Then, for the crystal structure corresponding to a minimum of energy, the phonon spectrum (at the Γ point) was calculated or the elastic constants were determined. The calculation error of the self-consistent field was set to be 10–10 a.u. (TOLDEE = 10), the parameters that determine the calculation accuracy of integrals, “TOLINTEG,” were equal to 8, 8, 8, 8, and 16. The parameters “SHRINK,” which determine the frequency of the Monkhorst–Pack grid in the reciprocal space, were equal to 8. Details of the calculation algorithm are available in [38].
DISCUSSION OF RESULTS
The results of calculations of the crystal lattice are given in Table 1. The hybrid functionals reproduce quite well the crystal structure of lanthanum zirconate. The HSE06 functional, which calculates the HF nonlocal exchange using the screening of the Coulomb potential, yields quite acceptable results. It seems that the separation of the exchange contribution to HSE06 into the short-range (SR) and long-range (LR, which is neglected) contributions, has been done successfully. The HSE06 functional is based on the PBE0 functional; correspondingly, the share of the HF exchange in it is the same—25% [19]. Using this approximation, i.e., screening, reduces considerably the computation time; for example, the calculation of the crystal structure with the HSE06 functional requires a fivefold shorter amount of time than the calculation with the PBE0 functional. Such a decrease in the cost of computer resources deteriorates the results insignificantly—the discrepancy between the calculated and experimental values of the lattice constant increases by only 0.004 Å in comparison with the PBE0 calculation (Table 1). The PBESOL0 functional differs from the PBE0 functional in that it uses PBESOL instead of the PBE local exchange-correlation functional [39]. The calculation with this functional underestimates the lattice constant, the discrepancy with experiment is comparable with that of the PBE0 and HSE06 calculations (Table 1). The B3PW functional gives the maximum discrepancy with experiment (Table 1), which may be caused by a lower fraction of HF the exchange. We note that, upon modeling of rare-earth titanates R2Ti2O7, the PBE0 functional reproduced the lattice structure much better than B3LYP [28]. In general, the calculation of the crystal structure of La2Zr2O7 reproduces well the interionic distances and the lattice constant.
The lanthanum zirconate under investigation, La2Zr2O7, has the structure of pyrochlore, and its ph-onon modes at the Γ point are as follows: Γ = A1g + Eg + 2F1g + 4F2g + 3A2u + 3Eu + 8F1u + 4F2u. Among these modes, the F1u mode is translational, the 4F2u, 3Eu, 3A2u, and 2F1g modes are “silent,” which are IR and Raman inactive. The A1g + Eg + 4F2g modes are Raman active (Raman modes), and seven F1u modes are IR active.
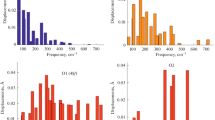
The results of our calculations of phonon modes at the Γ point are given in Table 2. The frequencies and symmetry species of phonon modes were determined from the ab initio calculation. From the analysis of displacement vectors obtained by the ab initio calculation, the degrees of participation of ions in each mode were determined (the “Ions participants” column). Calculations predict that vibrations of structural units are strongly mixed. It is interesting that, in this case, modes in which only oxygen ions participate can be distinguished. For example, in the F1u IR active mode with a frequency of 246 cm–1 (the wavenumber was determined in accordance with the PBE0 calculation), only oxygen ions participate. Moreover, O1 ions occupying position 48f, which is characterized by displacement x, predominantly participate in this mode. We note that the most intense Raman mode—F2g (308 cm–1)—involves the participation of O1 and O2 ions, with the participation of O1 ions being predominant. In the Eg Raman mode with the frequency at 325 cm–1, only O1 ions participate. In accordance with our calculations, this is the second intense mode in the Raman spectrum (Table 2; Fig. 1). In the A1g and F2g Raman modes, which are located at frequencies of 506 and 758 cm–1, respectively, also only O1 ions participate. In the F2g modes (413 and 536 cm–1), only oxygen ions are involved, with the involvement of O2 ions in positions 8b (3/8, 3/8, 3/8) being predominant.

Raman spectrum of La2Zr2O7. The calculation was made for the excitation radiation at a wavelength of 488 nm and T = 298 K (PBE0 functional).
In general, we can note that only oxygen ions participate in Raman-active modes. Moreover, in the A1g and Eg modes, only oxygen O1 ions that are located at the 48f position are involved. Thus, the behavior of these modes can carry information on displacement x of oxygen in this position under the influence of external actions on the crystal.
In IR active modes (F1u), all ions participate: La, Zr, O1, and O2, but with different degrees of participation in each particular mode. (Ions, the degree of participation of which is insignificant, are not shown in the column “Ions participants.”) In the most intense IR mode (F1u, 329 cm–1), zirconium and both oxygen ions participate, with the degree of participation of O1 being maximum. The second intense IR mode (F1u, 203 cm–1) involves lanthanum, zirconium and, substantially, O1. In the third intense IR mode (F1u, 499 cm–1) oxygen ions participate, with the degree of participation of O1 ions being also significant. The predominant participation of lanthanum manifests itself in the low-lying F2u mode (55 cm–1); however, this mode is inactive both in IR and in Raman spectra. In the low-lying F1u mode (102 cm–1), all ions are involved, but, mainly, zirconium. Lanthanum and zirconium participate in IR and Raman modes with frequencies up to 200 cm–1, in addition, two modes with higher frequencies can be distinguished: zirconium is involved in the F1u mode with a frequency of 329 cm–1, while lanthanum is involved in the F1u mode with a frequency of 384 cm–1 (Table 2). A significant participation of lanthanum and zirconium in “silent” modes can be noted, which are inactive either in IR absorption or in Raman scattering.
The results of modeling of the Raman and IR spectra are shown in Figs. 1 and 2. In [2, 6], in the measured Raman spectrum, the most intense peak was observed at about 300 cm–1, which agrees well with our calculation results. The presence of two peaks at about 350 and 500 cm–1 (Fig. 2) is in good agreement with the results of measurements of the transmission coefficient of the IR spectrum of La2Zr2O7 in [12].

Results of modeling of the IR spectrum of La2Zr2O7. (calculations with the PBE0 functional). All IR modes belong to the F1u symmetry species.
The conclusions that were drawn in experimental work [11] that the ZrO6 structural group participates in the IR mode with a frequency of 508 cm–1 are consistent with the predictions of the calculations on a strong displacement (participation) of O1 ions in this mode, which are zirconium ligands. The conclusions of [11] that the La–O and ZrO6 structural units participate in the Raman modes with frequencies of 299 and 506 cm–1 (stretching modes) are consistent with the predictions of the calculations about a strong displacement of oxygen O1 in these modes and about the participation of oxygen O2 (Table 2). The calculation results are also consistent with experiment [12], where intense IR modes at 366 and 508 cm–1 were revealed, for which the calculation predicts a strong participation of oxygen O1, i.e., a change in the Zr–O bond length.
The results of calculations of the intensity of Raman modes are given in Tables 3 and 4. Calculations with the B3PW and PBE0 functionals give close results.
The results of calculations of elastic constants, bulk modulus, shear modulus, etc., are given in Tables 5 and 6. Calculations with the B3PW and PBE0 functionals give close results, which are in good agreement with the calculations of elastic constants in the basis set of plane waves [14]. The available experimental data, e.g., for the Young modulus of La2Zr2O7, differ significantly, varying from 141 GPa [40] to 280 GPa [6].
Our calculations of the bulk and shear moduli in the Voigt, Reuss, and Hill approximations (Table 6) give close results, which allows us to infer that the isotropy of chemical bonds and elastic properties of La2Zr2O7 is quite high, which is also seen from Fig. 3 (the dependence of the Young modulus on the direction in the crystal was constructed using the ELATE program [41]).

Dependences of the Young modulus (GPa) on the crystal direction.
The stability conditions of the crystal lattice to hydrostatic compression for cubic crystals [42]:
are implemented (calculation at P = 2GPa with the PBE0 functional).
CONCLUSIONS
In this work, from unified ab initio calculations, we determined the frequencies and symmetry species of the fundamental vibrations of La2Zr2O7 lanthanum zirconate with the pyrochlore structure (\(Fd\overline 3 m\)), which may be useful in interpretation of its measured IR and Raman spectra. From the analysis of displacement vectors obtained from the ab initio calculations, the degree of participation of ions in each mode was determined. Modes with absolute or predominant participation of oxygen ions in position 48f, which is characterized by department x, were distinguished. It was shown that, within the MO LCAO approach with hybrid functionals that take into account the nonlocal exchange in the Hartree–Fock formalism, it becomes possible to describe the structure and lattice dynamics of lanthanum zirconate.
REFERENCES
M. C. Hatnean, C. Decorse, M. R. Lees, O. A. Petrenko, and G. Balakrishnan, Crystals 6, 79 (2016). https://doi.org/10.3390/cryst6070079
V. V. Popov, A. P. Menushenkov, B. R. Gaynanov, Ya. V. Zubavichus, R. D. Svetogorov, A. A. Yastrebtsev, A. A. Pisarev, L. A. Arzhatkina, and K. V. Ponkratov, J. Phys.: Conf. Ser. 941, 012079 (2017). https://doi.org/10.1088/1742-6596/941/1/012079
L. Kong, I. Karatchevtseva, D. J. Gregg, M. G. Blackford, R. Holmes, and G. Triani, J. Am. Ceram. Soc. 96, 935 (2013). https://doi.org/10.1111/jace.12060
D. R. Modeshia and R. I. Walton, Chem. Soc. Rev. 49, 4303 (2010). https://doi.org/10.1039/B904702F
A. Chen, J. R. Smith, K. L. Duncan, R. T. DeHoff, K. S. Jones, and E. D. Wachsman, J. Electrochem. Soc. B 157, 1624 (2010). https://doi.org/10.1149/1.3484092
K. Shimamura, T. Arima, K. Idemitsu, and Y. Inagaki, Int. J. Thermophys. 28, 1074 (2007). https://doi.org/10.1007/s10765-007-0232-9
B. Paul, K. Singh, T. Jaroń, A. Roy, and A. Chowdhury, J. Alloys Compd. 686, 130 (2016). https://doi.org/10.1016/j.jallcom.2016.05.347
M. Subramanian, G. Aravamudan, and G. Subba Rao, Progr. Solid State Chem. 15, 55 (1983). dx.doi.org/https://doi.org/10.1016/0079-6786(83)90001-8
L. N. Komissarova, N. V. Gundobin, F. M. Spiridonov, and K. I. Petrov, Zh. Neorg. Khim. 20, 582 (1975).
X. Cheng, Z. Qi, T. Li, G. Zhang, C. Li, H. Zhou, Y. Wang, and M. Yin, Phys. Status Solidi B 249, 854 (2011). https://doi.org/10.1002/pssb.201147313
Y. Tong, Y. Wang, Z. Yu, X. Wang, X. Yang, and L. Lu, Mater. Lett. 62, 889 (2008). https://doi.org/10.1016/j.matlet.2007.07.005
D. Chen and R. Xu, Mater. Res. Bull. 33, 409 (1998). https://doi.org/10.1016/S0025-5408(97)00242-0
A. Chartier, C. Meis, J.-P. Crocombette, L. R. Corrales, and W. J. Weber, Phys. Rev. B 67, 174102 (2003). https://doi.org/10.1103/PhysRevB.67.174102
X. Guo and J. Zhang, Mater. Today: Proc. 15, 25 (2014). https://doi.org/10.1016/j.matpr.2014.09.006
J. Feng, B. Xiao, C. L. Wan, Z. X. Qu, Z. C. Huang, J. C. Chen, R. Zhou, and W. Pan, Acta Mater. 59, 1742 (2011). https://doi.org/10.1016/j.actamat.2010.11.041
S. Zhang, H. B. Zhang, F. A. Zhao, M. Jiang, H. Y. Xiao, Z. J. Liu, and X. T. Zu, Sci. Rep. 7, 6399 (2017). https://doi.org/10.1038/s41598-017-06725-8
J. P. Perdew, M. Ernzerhof, and K. Burke, J. Chem. Phys. 105, 9982 (1996). https://doi.org/10.1063/1.472933
A. D. Becke, J. Chem. Phys. 98, 5648 (1993). https://doi.org/10.1063/1.464913
J. Heyd, G. E. Scuseria, and M. Ernzerhof, J. Chem. Phys. 118, 8207 (2003). https://doi.org/10.1063/1.1564060
G. J. Benjamin, M. H. Thomas, and E. S. Gustavo, Phys. Chem. Chem. Phys. 11, 443 (2009). https://doi.org/10.1039/b812838c
K. Burke, J. Chem. Phys. 136, 150901 (2012). https://doi.org/10.1063/1.4704546
R. Dovesi, V. R. Saunders, C. Roetti, R. Orlando, C. M. Zicovich-Wilson, F. Pascale, B. Civalleri, K. Doll, N. M. Harrison, I. J. Bush, Ph. D’Arco, M. Llunel, M. Causa, Y. Noel, L. Maschio, A. Erba, M. Rerat, and S. Casassa, CRYSTAL17 User’s Manual. http://www.crystal.unito.it/index.php.
R. Dovesi, A. Erba, R. Orlando, Zicovich, C. M. Wilson, B. Civalleri, L. Maschio, M. Rérat, S. Casassa, J. Baima, S. Salustro, and B. Kirtman, Comp. Molec. Sci. 8, e1360 (2018). dx.doi.org/https://doi.org/10.1002/wcms.1360
R. A. Evarestov, A. V. Bandura, and V. E. Aleksandrov, Phys. Solid State 47, 2248 (2005).
D. V. Korabel’nikov and Yu. N. Zhuravlev, Phys. Solid State 58, 1166 (2016).
M. L. Pierre, R. Orlando, L. Maschio, K. Doll, P. Ugliengo, and R. Dovesi, J. Comp. Chem. 32, 1775 (2011). https://doi.org/10.1002/jcc.21750
M. G. Medvedev, I. S. Bushmarinov, J. Sun, J. P. Perdew, and K. A. Lyssenko, Science (Washington, DC, U. S.) 355, 49 (2017). https://doi.org/10.1126/science.aah5975
V. A. Chernyshev, V. P. Petrov, and A. E. Nikiforov, Phys. Solid State 57, 996 (2015).
V. A. Chernyshev, V. P. Petrov, A. E. Nikiforov, P. A. Agzamova, and N. M. Avram, Opt. Mater. 72, 565 (2017). https://doi.org/10.1016/j.optmat.2017.06.062
M. F. Peintinger, D. V. Oliveira, and T. Bredow, J. Comp. Chem. 34, 451 (2012). https://doi.org/10.1002/jcc.23153
http://www.crystal.unito.it/index.php.
L. Valenzano, B. Civalleri, S. Chavan, S. Bordiga, M. Nilsen, S. Jakobsen, K. P. Lillerud, and C. Lamberti, Chem. Mater. 23, 1700 (2011). https://doi.org/10.1021/cm1022882
M. Dolg, H. Stoll, A. Savin, and H. Preuss, Theor. Chim. Acta 75, 173 (1989). https://doi.org/10.1007/BF00528565
M. Dolg, H. Stoll, and H. Preuss, Theor. Chim. Acta 85, 441 (1993). https://doi.org/10.1007/BF01112983
J. Yang and M. Dolg, Theor. Chem. Acc. 113, 212 (2005). https://doi.org/10.1007/s00214-005-0629-0
A. Weigand, X. Cao, J. Yang, and M. Dolg, Theor. Chem. Acc. 126, 117 (2009). https://doi.org/10.1007/s00214-009-0584-2
Energy-Consistent Pseudopotentials of the Stuttgart. http://www.tc.uni-koeln.de/PP/clickpse.en.html
V. A. Chernyshev, A. E. Nikiforov, V. P. Petrov, A. V. Serdtsev, M. A. Kashchenko, and S. A. Klimin, Phys. Solid State 58, 1642 (2016).
J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou, and K. Burke, Phys. Rev. Lett. 100, 136406 (2008). https://doi.org/10.1103/PhysRevLett.100.136406
G. di Girolamo, F. Marra, M. Schioppa, C. Blasi, G. Pulci, and T. Valente, Surf. Coat. Technol. 268, 298 (2015). https://doi.org/10.1016/j.surfcoat.2014.07.067
http://progs.coudert.name/elate.
Yu. Kh. Vekilov and O. M. Krasilnikov, Phys. Usp. 52, 831 (2009). https://doi.org/10.3367/UFNr.0179.200908f.0883
Funding
This work was financially supported by the Ministry of Education and Science of the Russian Federation, project no. 3.9534.2017/8.9.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The author declares that he has no conflict of interest.
Additional information
Translated by V. Rogovoi
Rights and permissions
About this article

Cite this article
Chernyshev, V.A. Phonon Spectrum of La2Zr2O7: ab initio Calculation. Opt. Spectrosc. 127, 825–831 (2019). https://doi.org/10.1134/S0030400X19110067
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0030400X19110067