
Abstract
Base excision repair (BER) is aimed at repair of damaged bases, which are the largest group of DNA lesions. The main steps of BER are recognition and removal of the aberrant base, cutting of the DNA sugar-phosphate backbone, gap processing (including dNMP insertion), and DNA ligation. The precise function of BER depends on the regulation of each step by regulatory/accessory proteins, the most important of which is poly(ADP-ribose) (PAR) polymerase 1 (PARP1). PARP1 plays an important role in DNA repair, maintenance of genome integrity, and regulation of mRNA stability and decay. PARP1 can therefore affect BER both at the level of BER proteins and at the level of their mRNAs. There is no systematic data on how the PARP1 content affects the activities of key BER proteins and the levels of their mRNAs in human cells. Whole-cell extracts and RNA preparations obtained from the parental HEK293T cell line and its derivative HEK293T/P1-KD cell line with reduced PARP1 expression (shPARP1-expressing cells, a PARP1 knockdown) were used to assess the levels of mRNAs coding for BER proteins: PARP1, PARP2, uracil DNA glycosylase (UNG2), AP endonuclease 1 (APE1), DNA polymerase β (POLβ), DNA ligase III (LIG3), and XRCC1. Catalytic activities of the enzymes were evaluated in parallel. No significant effect of the PARP1 content was observed for the mRNA levels of UNG2, APE1, POLβ, LIG3, and XRCC1. The amount of the PARP2 mRNA proved to be reduced two times in HEK293T/P1-KD cells. Activities of these enzymes in whole-cell extracts did not differ significantly between HEK293T and HEK293T/P1-KD cells. No significant change was observed in the efficiencies of the reactions catalyzed by UNG2, APE1, POLβ, and LIG3 in conditions of PAR synthesis. A DNA PARylation pattern did not dramatically change in a HEK293T/P1-KD cell extract with a reduced PARP1 content as compared with an extract of the parental HEK293T cell line.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Base excision repair (BER) is aimed at repair of damaged bases, which are the largest group of DNA lesions. Single-strand breaks and spontaneous apurinic/apyrimidinic (AP) sites can also be repaired via the BER mechanism [1–5]. BER proceeds through the following main steps: an aberrant base is recognized and excised, the sugar-phosphate backbone is cleaved, the gap is processed (including dNMP insertion), and DNA is ligated. Two pathways are known for BER: a short-patch BER (SP-BER) pathway replaces only one nucleotide, while long-patch BER (LP-BER) is responsible for replacing 2–13 nucleotides [4, 5].
The same event starts both SP-BER and LP-BER; i.e., a damaged nitrogenous base is recognized by a proper DNA glycosylase, which hydrolyze the N-glycosidic bond between deoxyribose and an incorrect base. DNA glycosylases that recognize uracil and alkylated bases are monofunctional enzymes; i.e., they excise the base without affecting the sugar-phosphate backbone. Then AP endonuclease 1 (APE1) cleaves the sugar-phosphate backbone 5' of the AP site to produce 3'-OH and 5'-dRP. Oxidized bases are excised mostly by bifunctional DNA glycosylases, which additionally possess lyase activity. These enzymes excise the base and cleave the sugar-phosphate backbone via a β- or β,δ-elimination mechanism. The former yields a break flanked with 5'-phosphate and 3'-unsaturated aldehyde; the latter mechanism produces a single-nucleotide gap with 5'-phosphate and 3'-phosphate. These blocked 3' ends should be processed prior to DNA synthesis. The APE1 role is reduced to removing the 3'-unsaturated aldehyde in the case of β-elimination, and polynucleotide kinase mostly removes the 3'‑phosphate produced as a result of β,δ-elimination. In SP-BER, DNA polymerase β (POLβ) inserts a single dNMP and removes the 5'-dRP residue. Then the break is ligated by DNA ligase III (LIG3), which acts in complex with its cofactor X-ray repair cross-complementing protein 1 (XRCC1) [1–3]. LP-BER is triggered when POLβ is incapable of removing the 5'‑dRP group. POLβ adds the first nucleotide and dissociates from DNA. Then POLδ or POLε carries out strand displacement synthesis to produce a flap structure (a DNA duplex in which one of the strands has a single-stranded overhang). Flap endonuclease 1 (FEN1) removes the single-stranded overhang, and DNA ligase I (LIG1) ligates the break ends [1–5].
The precise function of BER depends on the regulation of each step by regulatory/accessory proteins, the most important of which is poly(ADP-ribose) (PAR) polymerase 1 (PARP1) [6–8]. PARP1 belongs to a protein family whose three members (PARP1, PARP2, and PARP3) bind to DNA lesions and catalyze the (ADP-ribosyl)lation reaction utilizing NAD+ as a substrate [6–10]. PARPs add single ADP-ribose residues to their targets or synthesize PAR on a target. The targets include PARPs themselves (autoPARylation) and several other proteins or DNA (PARylation) [6–10]. PARP1 and PARP2 have been shown to play important roles in DNA repair and maintenance of genome integrity [6–10].
The interactions of PARP1 with key proteins and DNA intermediates at various BER steps have been studied using various methods [9–13]. In particular, PARP1 and PARP2 have been found to specifically interact with the AP site, which is an early BER intermediate, before a break is introduced [13, 14]. PARylation has been studied in terms of its effect on activities of POLβ and other BER enzymes in SP and LP pathways [12, 15, 16].
Recently, PARP1 has been identified as a factor in regulating the mRNA stability and decay [17]. PARP1 can therefore affect BER both at the level of proteins involved in the process and at the level of expression of their mRNAs.
It should be noted that discrepant data on PARP1 functions have accumulated because PARP1 can act simultaneously as both catalytic and acceptor protein. The role of PARP1 in regulating BER has been studied using various experimental models, including purified proteins, cell extracts, cells, and organisms. However, a systematic study has not been performed to understand how various PARP1 amounts affect the activities of key BER proteins and the levels of their mRNAs in human cells.
We used whole-cell extracts and RNA preparations from the HEK293T parental cell line and its derivative HEK293T/P1-KD with lower PARP1 expression (shPARP1-expressing cells, a PARP1 knockdown) to assess the levels of mRNAs coding for several BER proteins, including PARP1, PARP2, uracil-DNA glycosylase (UNG2), APE1, POLβ, LIG3, and XRCC1. Catalytic activities of the enzymes were assayed in parallel in these experiments. The PARP1 amount did not significantly affect the UNG2, APE1, POLβ, LIG3, and XRCC1 mRNA levels. At the same time, the PARP2 mRNA level proved to be reduced two times in HEK293T/P1-KD cells. Activities of the above enzymes in whole-cell extracts did not significantly differ between HEK293T and HEK293T/P1-KD cells. No significant change was observed in the efficiencies of the reactions catalyzed by UNG2, APE1, POLβ, and LIG3 in conditions of PAR synthesis. The pattern of DNA PARylation, which has recently been discovered [18–23], did not dramatically change in a HEK293T/P1-KD cell extract with a reduced PARP1 amount as compared with an extract of the parental HEK293T cell line.
EXPERIMENTAL
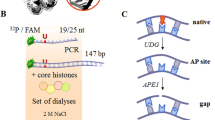
Materials. Oligonucleotides, including PCR primers, were synthesized in the Laboratory of Biomedicinal Chemistry (Institute of Chemical Biology and Fundamental Medicine). We used [γ-32P]ATP and [α‑32P]dATP (>3000 Ci/mol) (Laboratory of Biotechnology, Institute of Chemical Biology and Fundamental Medicine), Escherichia coli uracil-DNA glycosylase (UDG), T4 polynucleotide kinase (Biosan, Russia), horseradish peroxidase-conjugated secondary antibodies (Laboratory of Biotechnology, Institute of Chemical Biology and Fundamental Medicine), and BioMaster RT–PCR SYBR Blue (×2) (Biolabmix, Russia).
Plasmids carrying human APE1 and mouse POLβ cDNAs were kindly provided by Dr. S.H. Wilson (NIEHS, NIH, United States). Recombinant POLβ and APE1 were purified as in [24, 25]. Vectors coding for PARP1 and PARP2 were kindly provided by Dr. V. Schreiber (ESBS, Ilkirch, France). PARP1 and PARP2 were isolated according to a published protocol [26]. A plasmid carrying a PARP3 cDNA was kindly provided by Dr. G. Zarkovich (Gustave Roussy, Université Paris-Saclay, France). PARP3 was purified as in [21].
The cell lines HEK293T and HEK293T/P1-KD (with a lower PARP1 content) were kindly provided by researchers of the Laboratory of Nucleoproteins headed by O.A. Dontsova (Moscow State University, Moscow). Whole-cell extracts were prepared as in [27]. RNA was isolated using an Invitrogen kit (Thermo Fisher Scientific, United States) according to the manufacturer’s protocol [28]. The purity was checked by the absorption ratio at 260 and 230 nm (A260/230), which varies in a range of 1.8−2.2 in pure preparations. RNA preparations were digested with DNase prior to use in reverse transcription–quantitative polymerase chain reaction (RT–qPCR). Protein concentrations were measured by the Bradford assay [29], using bovine serum albumin (BSA) as a standard.
Oligonucleotide 5'-end labeling with [32P] and production of substrate DNAs. DNA-1, DNA-2, and DNA-3 (Table 1) were 5'-end labeled with [32P] using T4 polynucleotide kinase and [γ-32P]ATP, and the labeled oligonucleotides were isolated as in [30].
The 32P-labeled oligonucleotides were used to obtain DNA duplexes; their structures and designations are summarized in Table 1. The oligonucleotides were combined in an equimolar proportion, and hybridization was performed by heating the mixture at 95°C for 5 min and cooling slowly at room temperature.
Quantitative analysis of autoradiography results. Gel was exposed with a radiosensitive screen to visualize the radioactive products. The radioactive products were visualized using a Typhoon FLA 9000 scanner (GE Healthcare, United States). Band intensities were evaluated using Quantity One software (Bio-Rad, United States).
RT–qPCR was used to estimate the relative gene expression levels in HEK293T and HEK293T/P1-KD cells and was carried out using BioMaster RT–PCR SYBR Blue (×2). The reaction mixture (20 µL) contained 10 ng of total RNA, 0.5 µM primers, and a mixture of reverse transcriptase (75 units/ µL) and DNA polymerase (4 units/ µL) in an RT–qPCR buffer (2 µL of the mixture per 25 µL of RT–PCR SYBR Blue (×2)). The reaction was run on a LigthCycler96 thermal cycler (Roche, Switzerland) and included reverse transcription at 45°C for 1800 s; preincubation at 95°C for 300 s; and 40 cycles of denaturation at 95°C for 10 s, primer annealing at 60°C for 10 s, and elongation at 72°C for 10 s. The signal was detected at 84°C for 5 s. Experiments were performed in triplicate at each point; the GAPDH, B2M, and TUBβ housekeeping genes were used as calibrators. Primer sequences are summarized in Table 2. All primers were designed using Primer-BLAST (NCBI, United States); the amplification efficiency was determined for each primer pair.
The amplification efficiencies of the primer pairs, expression levels normalized to the calibrator levels (ΔΔCt), and standard errors were calculated using LigthCycler96 software (Roche). Results were represented as means and standard deviations. Relative levels of gene expression were compared between the HEK293T and HEK293T/P1-KD cell lines by calculating the p‑value and using the Mann–Whitney test; results were considered significant at p ≤ 0.05 ((*) p ≤ 0.05). Calculations were performed using the PRISM program (GraphPad Software, United States).
Comparisons of PARP1 amounts in cell extracts by Western blotting. Western blotting was carried out according to a published protocol [31]. Briefly, 2.5 µg of total protein of a cell extract or 0.05 µg of individual PARP1 was resolved electrophoretically according to Laemmli [32]. Proteins were transferred onto a nitrocellulose membrane using a Trans-Blot Turbo kit as recommended by the manufacturer. The nitrocellulose membrane was incubated consecutively with primary antibodies (anti-PARP1 rabbit antibodies diluted 1 : 1000), horseradish peroxidase-conjugated anti-rabbit Ig secondary antibodies, and the commercial Super Signal West Pico PLUS substrate (Thermo Scientific). Chemiluminescence was detected using an Amersham Imager 600 instrument (GE Healthcare, United States).
Generation of DNA structures containing an AP site or a cleaved AP site with 5'-dRP. The structures were produced from uracil-containing DNAs with the use of UDG immediately before an experiment. U-DNA was used at a ratio of 1 µM U-DNA : 0.1 units/µL UDG. The reaction was carried out in a TE buffer (10 mM Tris-HCl, pH 7.8, 1 mM EDTA) at 37°C for 30 min.
Estimation of the PARylation level in cell extracts. The reaction mixture (10 µL) contained 0.6 A260/mL activated DNA, 20 µM radiolabeled [32P]NAD+ ([32P]NAD+ : NAD+= 1 : 9), 0.5 mg/mL extract or 30 nM PARP1, 5 mM MgCl2, and buffer components (50 mM Tris-HCl, pH 8.0, 40 mM NaCl, 0.1 mg/mL BSA, 1 mM DTT). The mixture was incubated at 37°C for 1 min. An aliquot (4 µL) was taken and put onto a paper target soaked with trichloroacetic acid (TCA) to stop the reaction. Targets were washed three times with 5% TCA and two times with ethanol, dried, and autoradiographed.
UDG assays of cell extracts. The reaction mixture (10 µL) contained 0.1 µM labeled [32P]U-DNA (DNA-1 or DNA-4, Table 1), 0.05 mg/mL extract or 0.1 units/µL UDG, 10 mM EDTA, 400 µM NAD+ (where specified), and buffer components (50 mM Tris-HCl pH 8.0, 40 mM NaCl, 5% glycerol, 0.1% Nonidet P-40, 1 mM DTT). The reaction was carried out at 37°C. Aliquots (2 µL) were taken after 2, 5, and 10 min of incubation; combined with an equal volume of 100 mM NaOH; and incubated at 60°C for 10 min. The reaction products were resolved by PAGE in 20% gel in the presence of 7 M urea and 10% formamide [33].
AP site processing by cell extract proteins. The reaction mixture (10 µL) contained 0.1 µM [32P]-labeled AP-DNA duplex (DNA-4 pretreated with UDG, Table 1), 0.01 mg/mL extract or 1 nM APE1, 5 mM MgCl2, 400 µM NAD+ (where specified), and buffer components (50 mM Tris-HCl, pH 8.0, 50 mM NaCl, 1 mM DTT, 5% glycerol, 0.1% Nonidet P-40, 0.1 mg/mL BSA). The reaction was carried out at 37°C. Aliquots (2 µL) were taken after 2, 4, and 8 min of incubation; combined with an equal volume of a mixture of methoxyamine (100 mM) and EDTA (50 mM); and incubated at 0°C for 30 min. The reaction products were resolved by PAGE in 20% gel in the presence of 7 M urea and 10% formamide; gels were autoradiographed [33]. The efficiency of AP site cleavage by the lyase mechanism was assayed similarly, except that the reaction mixture contained 20 mM EDTA in place of magnesium ions and was incubated for 10, 15, and 30 min.
DNA polymerase activity assays of cell extracts. The reaction mixture contained 0.1 µM [32P]-labeled DNA duplex (DNA-5 pretreated with UDG, Table 1), 0.05 mg/mL extract or 50 nM POLβ, 5 mM MgCl2, 0.1 µM each dNTP, 400 µM NAD+ (where specified), and buffer components (50 mM Tris-HCl, pH 8.0, 50 mM NaCl, 1 mM DTT, 5% glycerol, 0.1% Nonidet P-40, 0.1 mg/mL BSA). The mixture was incubated at 37°C. Aliquots (2 µL) were collected after 5, 10, and 15 min of incubation and combined with 1 µL of 25 mM EDTA to stop the reaction. The reaction products were resolved by PAGE in 20% gel in the presence of 7 M urea and 10% formamide; gels were autoradiographed [33].
DNA ligase activity assays of cell extracts. The reaction mixture contained 0.1 µM [32P]-labeled DNA duplex (DNA-6, Table 1), 0.05 mg/mL extract or 0.1 units/µL T4 DNA ligase, 5 mM MgCl2, 1 mM ATP, 400 µM NAD+ (where specified), and buffer components (50 mM Tris-HCl, pH 8.0, 50 mM NaCl, 1 mM DTT, 5% glycerol, 0.1% Nonidet P-40, 0.1 mg/mL BSA). The mixture was incubated at 37°C. Aliquots (2 µL) were taken after 5, 15, and 30 min of incubation and combined with 1 µL of 25 mM EDTA to stop the reaction. The reaction products were resolved by PAGE in 20% gel in the presence of 7 M urea and 10% formamide; gels were autoradiographed [33].
DNA PARylation. The standard reaction mixture (10 µL) contained 50 mM Tris-HCl (pH 8.0), 50 mM NaCl, 1 mM DTT, 0.1 mg/mL BSA, 2 mM spermine, 0.15 mM NAD+, 10 mM EDTA, 20 nM [5'-32P]-labeled substrate DNA (DNA-7, Table 1), and 200 nM PARP3. The reaction mixture was incubated at 37°C for 15 min, supplemented with a cell extract (to 1.25 mg/mL by protein) and/or PARP1 (200 nM) or PARP2 (200 nM), and incubated again at 37°C for 15 min. The reaction was terminated by adding an equal volume of formamide. The reaction products were resolved by PAGE in 20% gel in the presence of 7 M urea and 10% formamide; gels were autoradiographed [33].
Synthesis of [32P]NAD+. Radiolabeled NAD+ was synthesized from [α-32P]ATP as described in [19]. The reaction mixture contained 1 mM ATP, 10 MBq of [α-32P]ATP, 20 mM MgCl2, 2 mM β-nicotinamide mononucleotide, and 5 mg/mL nicotinamide-nucleotide adenylyltransferase in 25 mM Tris-HCl (pH 7.5). The mixture was incubated at 37°C for 60 min and then heated at 90°C for 3 min to stop the reaction. Denatured protein was removed by centrifugation, and the supernatant was used as a source of NAD+ without purification.
RESULTS AND DISCUSSION
PARP1 is a high-copy-number protein and performs pleiotropic functions in the cell. The role of PARP1 in various DNA repair mechanisms has been studied with various experimental models, which each have their advantages and drawbacks. At a particular stage, in vitro reconstruction of a repair system from whole-cell extracts and DNA intermediates that contain DNA lesions characteristic of a particular repair step provides a convenient and productive means to analyze activities of DNA repair enzymes, including BER. Cells with a lower PARP1 level open new opportunities to study how PARP1 influences the BER efficiency both at the level of proteins or their activities and at the level of their mRNAs. We used the HEK293T/P1-KD cell line, in which the PARP1 level was decreased by introducing a construct (the LeGO Cer lentivirus vector) that inhibits PARP1 expression via the RNA interference mechanism.
First, the PARP1 levels were assessed in HEK293T and HEK293T/P1-KD cells. RT–qPCR showed that the PARP1 mRNA level in the HEK293T/P1-KD cell line is reduced almost two times as compared with the parental HEK293T cell line (Fig. 1a).

Results of (a) RT–qPCR and (b) Western blot analyses of the PARP1 levels in the HEK293T and HEK293T/P1-KD cell lines and (c) PAR synthesis in whole-cell extracts. Relative mRNA levels were measured in three independent experiments and represented as means and standard deviations. The PAR amount synthesized at 37°C within 1 min is shown with a column chart. The PAR amount synthesized by endogenous PARPs of an extract was normalized to the PAR amount synthesized using 30 nM recombinant PARP1 in each experiment. Results are shown as mean ± standard deviation as obtained in three independent experiments.
The results of Western blotting (Fig. 1b) agreed with the RT–qPCR results and showed a substantial, but incomplete decrease in PARP1 content. It should be noted that a moderate decrease in PARP1 in HEK293T/P1-KD cells has been noted by the researchers who provided the cell line [34].
The efficiency of PAR synthesis catalyzed by endogenous PARPs of whole-cell extracts was estimated to further characterize the cell lines. Radiolabeled [32P]NAD+ and activated DNA as a cofactor were used for the purpose (Fig. 1c). Based on the kinetics of PAR synthesis by endogenous PARPs of extracts, a time point of 1 min was chosen in the linear part of the kinetic curve. To ensure adequate comparisons, the PAR amount synthesized in extracts was normalized to the PAR amount synthesized using 30 nM recombinant PARP1. The PAR synthesis levels correlated well with the PARP1 mRNA levels and the PARP1 amounts estimated by Western blotting. A comparison with the PAR levels synthesized in extracts is correct because PARP1 synthesizes more than 90% of PAR in the cell according to published data [6–10]. Thus, a decrease in the PAR amount synthesized in cell extracts corresponded to the extent of inhibition of PARP1 expression.
Comparisons of Relative Levels of PARP2, UNG, APE1, POLβ, LIG3, and XRCC1 mRNAs between HEK293T and HEK293T/P1-KD Cells
Total RNAs isolated from HEK293T and HEK293T/P1-KD cells were used to estimate how the change in PARP1 content affects the levels of mRNAs coding for SP-BER proteins. Figure 2 shows the relative levels estimated for several key SP-BER proteins, including PARP2, UNG, APE1, POLβ, LIG3, and XRCC1, as estimated by RT–qPCR. The PARP2 mRNA level in PARP1 knockdown cells was reduced approximately two times as compared with cells of the parental line. Expression of UNG2, APE1, POLβ, and LIG3 decreased to a lower extent in knockdown cells, while XRCC1 expression was moderately increased.

Comparison of the PARP2, UNG2, APE1, POLβ, LIG3, and XRCC1 mRNA levels between HEK293T and HE-K293T/P1-KD cells. The mRNA levels were measured in three independent experiments. The p-value was calculated using the Mann–Whitney test. Differences were considered significant at *p ≤ 0.05.
Efficiencies of BER Steps in Extracts of HEK293T and HEK293T/P1-KD Cells
Functional tests with [32P]-labeled DNA structures mimicking the intermediates of respective BER steps and whole-cell extracts were used to study the effect of PARP1 on activities of SP-BER proteins at various repair steps, including uracil excision, AP site processing, DNA synthesis, and end ligation. Additional tests were carried out in the presence of NAD+ to estimate the possible effect of PAR synthesis on the BER steps.
Uracil excision from DNA. In a general case, BER starts with excision of a damaged nitrogenous base by DNA glycosylases. Uracil is one of the base lesions repaired via BER. Uracil is occasionally added in place of thymine during DNA synthesis or arises as a result of spontaneous and enzymatic cytosine deamination. It is thought that 70–200 spontaneous cytosine deamination events occur in a single cell daily [35].
Three types of uracil-DNA glycosylases are found in higher eukaryotic cells [36]: single strand-selective monofunctional uracil glycosylase (SMUG), mitochondrial UNG1, and nuclear UNG2. UNG2 and SMUG1 initiate BER in the majority of DNA contexts [36].
To detect uracil excision from DNA, we used alkaline treatment, which cleaves the sugar-phosphate backbone at AP sites, which result from uracil excision. The uracil excision efficiency was compared between double- and single-stranded DNAs (DNA-1 and DNA-4, Table 1).
No significant difference in uracil excision efficiency was observed between single- and double-stranded substrates in extracts of the two cell lines (Fig. 3). In addition, NAD+ did not significantly affect the uracil excision efficiency.

Efficiency of uracil excision from DNA. The reaction mixture (10 µL) contained 0.1 µM [32P]-labeled uracil-containing DNA, a whole-cell extract (final concentration 0.05 mg/mL) or UDG, and buffer components.
It should be noted that a DNA substrate mimicking the intermediate of a certain BER step served as a cofactor necessary for activating PARP1 when we studied how PAR synthesis affects the activities of endogenous enzymes in cell extracts. Preliminary experiments with recombinant PARP1 were performed to compare the cofactor characteristics of activated DNA conventionally used in PARP1 activity assays and BER DNA substrates used at 0.1 µM. This concentration of BER DNA substrates was used to measure the activities of BER enzymes in extracts. The PAR synthesis rate in the presence of BER DNA substrates was approximately half as high as in a standard mixture with activated DNA (0.6 A260/mL). Uracil excision from a strand yields an AP site, which is processed then.
AP site processing. Hydrolysis or cleavage via a β‑or β,δ-elimination mechanism mediates the breakage of the sugar-phosphate backbone at the AP site. Hydrolysis makes the major contribution to AP site processing in mammalian cells, producing a break with 3'-hydroxyl and 5'-deoxyribose phosphate groups [37]. APE1 hydrolyzes 95% of AP sites in mammalian cells [38].
Discrepant data have been reported on how PARP1 and APE1 affect each other during BER. Both stimulatory and inhibitory effects have been observed upon cooperation of the two enzymes [12, 13, 39–41]. The efficiency of AP site cleavage in our experiments is shown in Fig. 4. The extent of AP site hydrolysis in a HEK293T/P1-KD cell extract was somewhat lower than in a cell extract of the parental line. Thus, PARP1 exerted a weak stimulatory effect in the conditions used. NAD+ as a PARylation substrate did not considerably affect the process.

Efficiency of AP site hydrolysis. The reaction mixture (10 µL) contained 0.1 µM [32P]-labeled AP-DNA, a whole-cell extract (final concentration 0.01 mg/mL) or 1 nM АРE1, 5 mM MgCl2, 400 µM NAD+ (where specified), and buffer components. The results are shown as mean ± standard deviation as obtained in three independent experiments.
To estimate the contribution of Mg2+-independent AP site cleavage, EDTA was added to the reaction mixture to inhibit the binding of metal ions with APE1. In these conditions, AP sites can be cleaved via a lyase mechanism by bifunctional DNA glycosylases and other enzymes that possess AP lyase activity [1, 2, 6, 13, 14]. In particular, PARP1 and PARP2 possess lyase activity according to our data [6, 13, 14]. A hydrolytic mechanism that does not require magnesium ions for catalysis is also possible for enzymatic cleavage of AP sites. Tyrosyl-DNA phosphodiesterase is one of the enzymes that utilize this mechanism [42]. The contribution of magnesium-independent AP site processing was low in our conditions, amounting to 4–6%. These variants of AP site cleavage are capable of providing for APE1-independent AP site processing, but are far less efficient and may help to sustain cell viability when APE1 is lost.
DNA synthesis in the gap resulting from AP site processing is the next step of BER. POLβ makes the greatest contribution to filling in the gap in a SP-BER substrate [43].
Experiments with recombinant proteins and several cell extracts in our lab have shown that PARP1 can decrease DNA-polymerase activity of POLβ, especially in strand displacement synthesis [40, 41, 44]. Automodification decreases the inhibitory effect of PARP1. Dana on DNA synthesis are shown in Fig. 5. The primer extension efficiency in a HEK293T/P1-KD cell extract with a lower PARP1 content was somewhat higher than in an extract of the parental cell line. This was observed for both addition of a single nucleotide in the case of SP-BER and addition of several nucleotides in the case of LP-BER. Primer extension by a higher number of nucleotides was disregarded in our quantitative analysis. In conditions of PAR synthesis (in the presence of NAD+), the DNA synthesis efficiency was slightly higher in both of the extracts. The results can be explained in full by competition for substrate binding between POLβ and PARP1 and, possibly, PARP2. PARP1 and PARP2 have high affinities for DNAs with gaps [44–46]. Automodified PARP1 shows lower affinity for DNA as compared with unmodified PARP1 because of electrostatic repulsion between DNA and PAR. This decreases the enzyme–DNA complexation and consequently leads to less efficient primer extension.

Efficiency of DNA synthesis. The reaction mixture (10 µL) contained 0.05 mg/mL extract protein or 50 nM POLβ, 5 mM MgCl2, 0.1 mM each dNTP, 0.1 µM [32P]-labeled DNA, 400 µM NAD+ (where specified), and buffer components. The results are shown as mean ± standard deviation as obtained in three independent experiments.
DNA ligation. Restoration of the integrity of the sugar-phosphate backbone is the last step of SP-BER and is carried out by ATP-dependent LIG3. LIG3 forms a stable complex with the XRCC1 architectural protein, and the complex acts in BER [46]. XRCC1 lacks catalytic activity, but forms binary and ternary complexes with several BER enzymes (proteins) to ensure their concerted action, which is due, to a great extent, to the XRCC1 capabilities of binding with BER proteins, undergoing PARylation, and binding with PAR [46, 47].
A DNA substrate with a break in one of the strands (DNA-6, Table 1) was used to assay DNA ligase activity in extracts of the two cell lines. Figure 6 shows the efficiency of nick ligation in the extracts within 5, 15, and 30 min. The incubation duration was increased as compared with the other BER reactions because the ligation efficiency is lower, and partly hydrolyzed oligonucleotides were detected in the samples as a result. A significant difference in ligation efficiency was not observed between the extracts with a normal or modified PARP1 content. PAR synthesis did not affect the ligation efficiency.

Efficiency of DNA ligation. The reaction mixture (10 µL) contained 0.1 µM [32P]-labeled DNA duplex, 0.05 mg/mL extract protein or 0.1 units/µL T4 DNA ligase, 5 mM MgCl2, 1 mM ATP, 400 µM NAD+ (where specified), and buffer components. The results are shown as mean ± standard deviation as obtained in three independent experiments.
DNA PARylation is another process recently found to involve PARPs [18–23]. PARylation of DNA ends by PARP1 and PARP2 activities and mono(ADP-ribosyl)ation by PARP3 have been demonstrated in our and other studies [18–23]. Moreover, 5′-mono(ADP-ribosyl)ated DNAs are more efficient substrates of PAR chain extension by PARP1 and PARP2 as compared with unmodified DNAs. DNA PARylation by endogenous PARPs has been observed in several mammalian cell extracts with different PARP1 contents. The DNA PARylation pattern has been found to depend on the PARP1 content [18, 19, 21–23]. DNA PARylation is possible to consider as a mechanism that protects the double-stranded ends from degradation by cell nucleases. Given the specific requirements of PARPs to the DNA structure in the DNA (ADP-ribosyl)ation reaction, the protective effect is possible to occur when double-stranded ends and breaks or gaps are within one or two helix turns to ensure the time interval necessary for single-strand break repair [20, 21, 23]. The difference in PARP1 and, probably, PARP2 contents between extracts of the two cell lines made it possible to expect a difference in DNA modification pattern. DNA PARylation in extracts was studied with DNA-7, which contained a single-strand gap (Fig. 7). DNA-7 is a model BER substrate and has been used in our previous experiments [21, 23]. DNA PARylation by endogenous PARPs took place in both of the extracts (Fig. 7; 11, 12), but its low yield complicated the analysis. Much the same results have been obtained with structurally similar DNA duplexes and mammalian cell extracts, and the yield of PARylation products has been found to substantially increase when DNA is preliminarily modified with a single ADP-ribose residue [19]. PARP3 is the most convenient to use to introduce this modification (Fig. 7, 1) [18]. Subsequent incubation with extract protein alone or in combination with recombinant PARP3 and/or recombinant PARP1 and PARP2 (Fig. 7, 4–8) yielded PARylated DNA, which contained products of step-by-step primer extension with ADP-ribose residues and high-molecular-weight products, which were not resolved by electrophoresis (shown with a bracket on the left).

DNA PARylation. The standard reaction mixture (10 µL) contained 50 mM Tris-HCl, pH 8.0, 50 mM NaCl, 1 mM DТТ, 0.1 mg/mL BSA, 2 mM spermine, 0.15 mM NAD+, 10 mM EDТА, 20 nM [5'-32P]-labeled DNA substrate (DNA-7, Table 1), and cell extract protein (1.25 mg/mL). Mixtures were incubated at 37°C for 15 min (11 and 12). In some cases, mixtures were supplemented with 200 nM PARP3, incubated at 37°C for 15 min, combined with cell extract protein (to 1.25 mg/mL) and/or PARP1 (200 nM) and PARP2 (200 nM), and further incubated at 37°C for 15 min.
Primer PARylation in a HEK293T/P1-KD cell extract was less efficient than in a HEK293T extract; the product yields were 71 and 84%, respectively (Fig. 7; 6, 7). Recombinant PARP1 and PARP2 produced different oligonucleotide PARylation patterns. Products of step-by-step primer extension with ADP-ribose residues and a minor amount of high-molecular-weight products were observed in the case of PARP1, whereas PARP2 produced mostly high-molecular-weight products, which remained unresolved upon electrophoresis, while primer molecules with relatively few ADP-ribose residues were almost undetectable (Fig. 7; 8, 9). When both PARP1 and PARP2 were present in the reaction mixture, the primer PARylation pattern was characteristic of PARP2, indicating that PARP2 efficiently functions in the process. When PARP1 and PARP2 were added to cell extracts, the product distribution pattern changed insignificantly as compared with the patterns obtained using the extracts alone (Fig. 7; compare lanes 2 + 3 and 4 + 5 with 6 + 7). However, note that the amount of the unmodified mono(ADP-ribosyl)ated primer decreased when PARP2 was added to the extracts (from 16 to 5% in the case of HEK293T and from 29 to 6% in the case of HEK293T/P1-KD). PARP1 and PARP2 added to the extracts each shifted the product distribution to their characteristic patterns. The effect was more distinct in the case of PARP2. However, the effect of PARP2 added to the extracts was far lower than in a system reconstructed from purified proteins, where the product distribution pattern obtained with equimolar amounts of PARP1 and PARP2 was virtually undistinguishable from the pattern obtained with PARP2 alone. This was most likely explained by competition with other cell DNA-binding proteins. In total, a lower PARP1 content in the extract did not principally change the DNA PARylation pattern. The results clearly demonstrate that the three PARPs activated upon interaction with DNA cooperate in DNA PARylation.
To summarize, the PARP1 content in extracts did not significantly affect the activities of the main SP-BER enzymes and DNA PARylation. The mRNA levels of the BER enzymes were also not affected significantly by the PARP1 content. It seems that relatively low effects of PARP1 can contribute to the fine tuning of the activities of BER enzymes in the absence of severe cell stress. Our finding that a weak effect is exerted by PARP1 on activity of the BER system agree with the current views that the throughput capacity of the BER system is redundant and that extra amounts of non-demanded proteins are processed, for example, by the ubiquitin-dependent proteolysis system [48–50].
REFERENCES
Hegde M.L., Hazra T.K., Mitra S. 2008. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 18, 27–47.
Almeida K.H., Sobol R.W. 2007. A unified view of base excision repair: lesion-dependent protein complexes regulated by post-translational modification. DNA Repair. 6, 695–711.
Schärer O.D. 2003. Chemistry and biology of DNA repair. Angew. Chem. Int. Ed. Engl. 42, 2946–2974.
Frosina G., Fortini P., Rossi O., Carrozzino F., Raspaglio G., Cox L.S., Lane D.P., Abbondandolo A., Dogliotti E. 1996. Two pathways for base excision repair in mammalian cells. J. Biol. Chem. 271, 9573–9578.
Klungland A., Lindahl T. 1997. Second pathway for completion of human DNA base excision-repair: reconstitution with purified proteins and requirement for DNase IV (FEN1). EMBO J. 16, 3341–3348.
Khodyreva S.N., Lavrik O.I. 2016. Poly(ADP-ribose) polymerase 1 as a key regulator of DNA repair. Mol. Biol. (Moscow). 50, 580–595.
Langelier M.F., Eisemann T., Riccio A.A., Pascal J.M. 2018. PARP family enzymes: regulation and catalysis of the poly(ADP-ribose) posttranslational modification. Curr. Opin. Struct. Biol. 53, 187−198.
Alemasova E.E., Lavrik O.I. 2019. Poly(ADP-ribosyl)ation by PARP1: reaction mechanism and regulatory proteins. Nucleic Acids Res. 47, 3811–3827.
Hanzlikova H., Gittens W., Krejcikova K., Zeng Z., Caldecott K.W. 2017. Overlapping roles for PARP1 and PARP2 in the recruitment of endogenous XRCC1 and PNKP into oxidized chromatin. Nucleic Acids Res. 45, 2546–2557.
De Vos M., Schreiber V., Dantzer F. 2012. The diverse roles and clinical relevance of PARPs in DNA damage repair: current state of the art. Biochem. Pharmacol. 84, 137–146.
Mortusewicz O., Amé J.-C., Schreiber V., Leonhardt H. 2007. Feedback-regulated poly(ADP-ribosyl)ation by PARP-1 is required for rapid response to DNA damage in living cells. Nucleic Acids Res. 35, 7665–7675.
Sukhanova M.V., Khodyreva S.N., Lebedeva N.A., Prasad R., Wilson SH., Lavrik O.I. 2005. Human base excision repair enzymes apurinic/apyrimidinic endonuclease1 (APE1), DNA polymerase beta and poly(ADP-ribose) polymerase 1: interplay between strand-displacement DNA synthesis and proofreading exonuclease activity. Nucleic Acids Res. 33, 1222–1229.
Khodyreva S.N., Prasad R., Ilina E.S., Sukhanova M.V., Kutuzov M.M., Liu Y., Hou E.W., Wilson S.H., Lavrik O.I. 2010. Apurinic/apyrimidinic (AP) site recognition by the 5'-dRP/AP lyase in poly(ADP-ribose) polymerase-1 (PARP-1). Proc. Natl. Acad. Sci. U. S. A. 107, 22090–22095.
Kutuzov M.M., Khodyreva S.N., Ilina E.S., Sukhanova M.V., Amé J.C., Lavrik O.I. 2015. Interaction of PARP-2 with AP site containing DNA. Biochimie. 112, 10–19.
Sukhanova M.V., Hamon L., Kutuzov M.M., Joshi V., Abrakhi S., Dobra I., Curmi P.A., Pastre D., Lavrik O.I. 2019. A single-molecule atomic force microscopy study of PARP1 and PARP2 recognition of base excision repair DNA intermediates. J. Mol. Biol. 431, 2655–2673.
Kurgina T.A., Anarbaev R.O., Sukhanova M.V., Lavrik O.I. 2018). A rapid fluorescent method for the real-time measurement of poly(ADP-ribose) polymerase 1 activity. Anal. Biochem. 545, 91–97.
Matveeva E.A., Mathbout L.F., Fondufe-Mittendorf Y.N. 2019. PARP1 is a versatile factor in the regulation of mRNA stability and decay. Sci. Rep. 91, 1–12.
Belousova E.A., Ishchenko A.A., Lavrik O.I. 2018. DNA is a new target of PARP3. Sci. Rep. 8, 4176.
Kosova A.A., Kutuzov M.M., Evdokimov A.N., Ilina E.S., Belousova E.A., Romanenko S.A., Trifonov V.A., Khodyreva S.N., Lavrik O.I. 2019. Poly(ADP-ribosyl)ation and DNA repair synthesis in the extracts of naked mole rat, mouse, and human cells. Aging (Albany NY). 11, 2852–2873.
Belousova E.A., Kutuzov M.M., Ivankina P.A., Ischenko A.A., Lavrik O.I. 2018. A new DNA break repair pathway involving PARP3 and base excision repair proteins. Dokl. Biochem. Biophys. 482, 233–237.
Talhaoui I., Lebedeva N.A., Zarkovic G., Saint-Pierre C., Kutuzov M.M., Sukhanova M.V., Matkarimov B.T., Gasparutto D., Saparbaev M.K., Lavrik O.I., Ishchenko A.A. 2016. Poly(ADP-ribose) polymerases covalently modify strand break termini in DNA fragments in vitro. Nucleic Acids Res. 44, 9279−9295.
Munnur D., Ahel I. 2017. Reversible mono-ADP-ribosylation of DNA breaks. FEBS J. 284, 4002–4016.
Zarkovic G., Belousova E.A., Talhaoui I., Saint-Pierre C., Kutuzov M.M., Matkarimov B.T., Biard D., Gasparutto D., Lavrik O.I., Ishchenko A.A. 2018. Characterization of DNA ADP-ribosyltransferase activities of PARP2 and PARP3: new insights into DNA ADP-ribosylation. Nucleic Acids Res. 46, 2417−2431.
Lebedeva N.A., Khodyreva S.N., Favre A., Lavrik O.I. 2003. AP endonuclease 1 has no biologically significant 3'–5'-exonuclease activity. Biochem. Biophys. Res. Commun. 300, 182–187.
Drachkova I.A., Petruseva I.O., Safronov I.V., Zakharenko A.L., Shishkin G.V., Lavrik O.I., Khodyreva S.N. 2001. Reagents for modification of protein–nucleic acid complexes: II. Site-specific photomodification of mammalian DNA polymerase complexes with primers extended by dCTP exo-N-substituted arylazido derivatives, Russ. J. Bioorg. Chem. 27, 173–179.
Amé J.C., Kalisch T., Dantzer F., Schreiber V. 2011. Purification of recombinant poly(ADP-ribose) polymerases. Methods Mol. Biol. 780, 135–152.
Biade S., Sobol R.W., Wilson S.H., Matsumoto Y. 1998. Impairment of proliferating cell nuclear antigen-dependent apurinic/apyrimidinic site repair on linear DNA. J. Biol. Chem. 273, 898–902.
Rio D.C., Ares M. Jr.., Hannon G.J., Nilsen T.W. 2010. Purification of RNA using TRIzol (TRI Reagent). Cold Spring Harb. Protoc. pdb.prot.5439.
Bradford M.A. 1976. Rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal. Biochem. 72, 248–254.
Ilina E.S., Lavrik O.I., Khodyreva S.N. 2008. Ku antigen interacts with abasic sites. Biochim. Biophys. Acta. 1784, 1777–1785.
Ilina E.S., Khodyreva S.N., Lavrik O.I. 2018. Unusual interaction of human apurinic/apyrimidinic endonuclease 1 (APE1) with abasic sites via the Schiff-base-dependent mechanism. Biochimie. 150, 88–99.
Laemmli U.K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 277, 680–685.
Sambrook J., Fritsch E.F., Maniatis T. 1989. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Lab. Press, 2nd ed.
Savelyev N.V., Shepelev N.M., Lavrik O.I., Rubtsova M.P., Dontsova O.A. 2021. PARP1 regulates the biogenesis and activity of telomerase complex through modification of H/ACA-proteins. Front. Cell Dev. Biol. 9, 621134.
Doseth B., Visnes T., Wallenius A., Ericsson I., Sarno A., Pettersen HS., Flatberg A., Catterall T., Slupphaug G., Krokan H.E., Kavli B. 2011. Uracil-DNA glycosylase in base excision repair and adaptive immunity: Species differences between man and mouse. J. Biol. Chem. 286, 16669–16680.
Doseth B., Ekre C., Slupphaug G., Krokan H.E., Kavli B. 2012. Strikingly different properties of uracil-DNA glycosylases UNG2 and SMUG1 may explain divergent roles in processing of genomic uracil. DNA Repair (Amst.). 11, 587−593.
Mol C.D., Hosfield D.J., Tainer J.A. 2000. Abasic site recognition by two apurinic/apyrimidinic endonuclease families in DNA base excision repair: the 3' ends justify the means. Mutat. Res. 460, 211–229.
Wilson D.M., Barsky D. 2001. The major human abasic endonuclease: formation, consequences and repair of abasic lesions in DNA. Mutat. Res. 485, 283–307.
Prasad R., Dyrkheeva N., Williams J., Wilson S.H. 2015. Mammalian base excision repair: functional partnership between PARP-1 and APE1 in AP-site repair. PLoS One. 10, e0124269.
Sukhanova M.V., Khodyreva S.N., Lavrik O.I. 2004. Poly(ADP-ribose) polymerase-1 inhibits strand-displacement synthesis of DNA catalyzed by DNA polymerase beta, Biochemistry (Moscow). 69, 558−568.
Sukhanova M., Khodyreva S., Lavrik O. 2010. P-oly(ADP-ribose) polymerase 1 regulates activity of DNA polymerase beta in long patch base excision repair. Mutat. Res., Fundam. Mol. Mech. Mutagen. 685 (1−2), 80−89.
Lebedeva N.A., Rechkunova N.I., El-Khamisy S.F., Lavrik O.I. 2012. Tyrosyl-DNA phosphodiesterase 1 initiates repair of apurinic/apyrimidinic sites. Biochimie. 94, 1749−1753.
Srivastava D.K., Berg B.J., Prasad R., Molina J.T., Beard W.A., Tomkinson A.E., Wilson S.H. 1998. Mammalian abasic site base excision repair. Identification of the reaction sequence and rate-determining steps. J. Biol. Chem. 273, 21203–21209.
Kutuzov M.M., Khodyreva S.N., Amé J.C., Ilina E.S., Sukhanova M.V., Schreiber V., Lavrik O.I. 2013. Interaction of PARP-2 with DNA structures mimicking DNA repair intermediates and consequences on activity of base excision repair proteins. Biochimie. 95, 1208−1215.
Kutuzov M.M., Belousova E.A., Kurgina T.A., Ukraintsev A.A., Vasil’eva I.A., Khodyreva S.N., Lavrik O.I. 2021. The contribution of PARP1, PARP2 and poly(ADP-ribosyl)ation to base excision repair in the nucleosomal context. Sci. Rep. 11, 4849.
Caldecott K.W. 2019. XRCC1 protein; form and function. DNA Repair (Amst.). 81, 102664.
Moor N.A., Lavrik O.I. 2018. Protein-protein interactions in DNA base excision repair. Biochemistry (Moscow). 83, 411–422.
Parsons J.L., Dianov G.L. 2013. Co-ordination of base excision repair and genome stability. DNA Repair (Amst.). 12, 326−333.
Edmonds M.J., Parsons J.L. 2014. Regulation of base excision repair proteins by ubiquitylation. Exp. Cell. Res. 329, 132−138.
Fang Q., Inanc B., Schamus S., Wang X.H., Wei L., Brown A.R., Svilar D., Sugrue K.F., Goellner E.M., Zeng. X, Yates N.A., Lan L., Vens C., Sobol R.W. 2014. HSP90 regulates DNA repair via the interaction between XRCC1 and DNA polymerase β. Nat. Commun., 5, 5513.
Funding
This work was supported by the Russian Science Foundation (project no. 19-14-00204, a major part of the work) and the Russian Foundation for Basic Research (project no. 20-04-00674 A, DNA PARylation experiments).
Author information
Authors and Affiliations
Contributions
E. S. Ilina and A. S. Kochetkova contributed equally to this study.
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of interest. This article does not contain any studies involving animals or human subjects performed by any of the authors.
Additional information
Translated by T. Tkacheva
Abbreviations: AP, apurinic/apyrimidinic (site); APE1, apurinic/apyrimidinic endonuclease 1; BER, base excision repair; dRP, deoxyribose phosphate; LIG3, DNA ligase III; PARP1, PARP2, and PARP3, poly(ADP-ribose) polymerases 1, 2, and 3; PAR, poly(ADP-ribose); PARylation, poly(ADP-ribosyl)ation; POLβ, DNA polymerase β; UNG, uracil-DNA glycosylase; UDG, Escherichia coli uracil-DNA glycosylase; XRCC1, X-ray repair cross-complementing protein 1.
Rights and permissions
About this article

Cite this article
Ilina, E.S., Kochetkova, A.S., Belousova, E.A. et al. Influence of the Poly(ADP-Ribose) Polymerase 1 Level on the Status of Base Excision Repair in Human Cells. Mol Biol 57, 272–283 (2023). https://doi.org/10.1134/S0026893323020097
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0026893323020097