
Abstract
DNA is constantly attacked by different damaging agents; therefore, it requires frequent repair. On the one hand, the base excision repair (BER) system is responsible for the repair of the most frequent DNA lesions. On the other hand, the formation of poly(ADP-ribose) is one of the main DNA damage response reactions that is catalysed by members of the PARP family. PARP1, which belongs to the PARP family and performs approximately 90% of PAR synthesis in cells, could be considered a main regulator of the BER process. Most of the experimental data concerning BER investigation have been obtained using naked DNA. However, in the context of the eukaryotic cell, DNA is compacted in the nucleus, and the lowest compaction level is represented by the nucleosome. Thus, the organization of DNA into the nucleosome impacts the DNA-protein interactions that are involved in BER processes. Poly(ADP-ribosyl)ation (PARylation) is thought to regulate the initiation of the BER process at the chromatin level. In this review, we focus on the mechanisms involved in BER in the nucleosomal context and the potential effect of PARylation, which is catalysed by DNA-dependent PARP1, PARP2 and PARP3 proteins, on this process.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Nucleosome core particle
- NCP
- Base excision repair
- DNA damge response
- Poly(ADP-ribosyl)ation
- PARP1
- PARP2
- PARP3
4.1 Base Excision Repair
DNA, as a carrier of genetic information, is constantly being repaired to cleanse from damage. Much damage occurs due to the actions of exogenous and endogenous reagents every day (Gates 2009; Lindahl 1993; Swenberg et al. 2011; Hoeijmakers 2001). Maintaining the integrity of genetic information in eukaryotic cells is largely based on the functioning of DNA repair systems (Sancar et al. 2004; Chatterjee and Walker 2017). There are several repair systems, including nucleotide excision repair, base excision repair (BER), mismatch repair, non-homologous DNA end joining, homologous recombination and direct DNA repair. To date, the main biochemical steps utilized by these processes have been fairly well studied. However, the interests of researchers are now focused on the clarification of the details of DNA repair in the context of chromatin. Regardless of the type of DNA damage that occurs, the initiation of the repair process requires chromatin decompaction. In this regard, an important role is played by the binding of poly(ADP-ribose)polymerase1 (PARP1) to the nucleosome or its catalytic activation via poly(ADP-ribosyl)ation (PARylation) of the participating proteins. The study of chromatin preservation is a difficult task because of the size of the corresponding nucleoprotein and the complex arrangement of this structure. For this reason, in addition to a more complicated model of the polynucleosome structures, the mononucleosome is used as a simpler model.
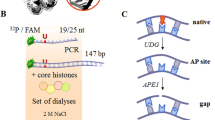
In eukaryotic cells, the BER system is responsible for repairing lesions that do not lead to significant distortion of the DNA double helix structure. In particular, methylated or oxidized bases and apurinic/apyrimidinic (AP) sites are corrected by this repair system (Kim and Wilson 2012; Krokan and Bjørås 2013; Khodyreva and Lavrik 2011). The BER system includes the following basic stages: DNA damage recognition, excision of the damaged base, incision of the sugar-phosphate backbone, incorporation of dNMP, and ligation (Fig. 4.1). The initiation stage involves activity of a specific DNA glycosylase, the identity of which depends on the type of damage that occurred. The enzyme recognizes the damaged DNA nucleobase and hydrolyses the N-glycosidic bond, leading to the formation of an AP site in the DNA. In addition, the appearance of an AP site in the DNA structure could be a consequence of spontaneous hydrolysis of the N-glycosidic bond. Bifunctional DNA glycosylases can cleave DNA to generate 3′-phospho alpha,beta-unsaturated aldehyde (PUA) and 5′-phosphate (P) ends or 3′-P and 5′-P ends depending on the reaction mechanism (β- or β/δ-elimination). AP sites are predominantly cleaved via hydrolytic mechanisms, resulting in a single-strand break containing 5′-deoxyribose phosphate (dRP) and 3′-hydroxyl (OH) groups. In mammalian cells, this reaction is mostly catalysed by AP endonuclease 1 (APE1). Moreover, APE1 cleaves 3′-PUA, producing 3′-OH. Thus, APE1 is the main enzyme that creates single-strand breaks with 3′-OH groups during the synthetic stage of the BER process. In the next stage, the single nucleotide gap is filled by DNA polymerase beta (Polβ) activity, and the 5′-dRP group is removed via its dRP-lyase activity, resulting in a 5′-P end. For dNMP incorporation, Polβ requires 3′-OH. If a 3′-P end occurs, it is normally converted to 3′-OH via the phosphatase activity of polynucleotide kinase phosphatase (PNKP). If the 5′-dRP group cannot be removed due to chemical reasons, DNA synthesis is prolonged by strand displacement. In this case, Polβ initiates synthesis that could be extended by Polβ itself or DNA polymerases δ/ε (Pol δ/ε). The flap of the DNA is removed by flap endonuclease 1 (FEN 1). Finally, the single-strand break is ligated by the DNA ligase IIIα/XRCC1 complex or DNA ligase I. A detailed review of the main BER stages has been presented in several articles (Kim and Wilson 2012; Krokan and Bjørås 2013; Khodyreva and Lavrik 2011; Abbotts and Wilson III 2017).

Base excision repair system. The damaged DNA base is recognized by specific DNA glycosylases that hydrolyse the N-glycosidic bond, leading to the formation of an AP site (for monofunctional DNA glycosylases) or, for bifunctional DNA glycosylases, either 3′- phospho alpha,beta-unsaturated aldehyde (PUA) and 5′-phosphate (P) ends or 3′-P and 5′-P ends, depending on the reaction mechanism. The AP site is cleaved by APE1, resulting in gapped DNA with 5′-deoxyribose phosphate (dRP) and 3′-hydroxyl (OH) groups. 3′-P is converted to 3′-OH via the phosphatase activity of polynucleotide kinase phosphatase (PNKP). 3′-PUA is converted to 3′-OH by APE1 via its phosphodiesterase activity. In the next stage, the single nucleotide gap is filled by the activity of DNA polymerase β (Polβ). The 5′-dRP group is removed via the dRP-lyase activity of Polβ, resulting in 5′-P. If for some reason the 5′-dRP group cannot be removed, Pol β is replaced by DNA polymerases δ/ε (Pol δ/ε), which prolong strand displacement synthesis, and the DNA flap is removed by flap endonuclease 1 (FEN1). Finally, the single-strand break is ligated by the DNA ligase IIIα/XRCC1 complex or DNA ligase I
This multistep system requires precise regulation via multiple protein-nucleic acid and protein–protein interactions. The most common model involves the consistent operation of repair enzymes coordinated by scaffold and regulatory proteins (Moor et al. 2015; Moor and Lavrik 2018). XRCC1 performs scaffold functions during BER, while both PARP1 and PARP2 are regulatory components in BER. Indeed, XRCC1 interacts with Polβ as well as with PARP1, DNA ligase IIIa and the other components of this system. The involvement of PARP1 and PARP2, as well as poly(ADP-ribosyl)ation, contributes to BER (Khodyreva and Lavrik 2011; Moor et al. 2015; Moor and Lavrik 2018; Kutuzov et al. 2013, 2015). Therefore, BER is implemented by mutual enzyme cooperation that underlies the mechanisms used in each step. Additionally, accessory proteins regulate functional activity and DNA-protein interactions. The processes involved in the operation of this repair complex at the chromatin level are unclear.
4.2 Nucleosome Structure
In eukaryotic cells, DNA exists in a compacted state in the nucleus. The lowest compaction level is nucleosomal. In these structures, DNA is wrapped around a histone core of approximately 1.7 turns that consisting of eight histones – two dimers, H2A-H2B and H3-H4. The structure constructed from the histone core and the 147 nucleotides of DNA is known as the nucleosome core particle (NCP). The structure and function of nucleosomes are reviewed in detail in (McGinty and Tan 2015).
The reconstitution of DNA into nucleosome particles requires a specific nucleotide sequence that defines the positioning of the histone core on the DNA molecule. The first demonstration of the predictable positioning of nucleosomes was provided by using the 5S rDNA sequence (Simpson and Stafford 1983). Later, using the SELEX technique, Widom’s group constructed model DNAs that allow nucleosome formation (Lowary and Widom 1998). Currently, the most commonly used sequences with defined nucleosome positioning are the 5S rDNA and Widom 601 sequences. NCP is a dynamic nucleoprotein complex. Depending on the DNA sequence, the free energy for binding of the nucleosome core and DNA molecule can be different. Based on the stability of this interaction, the motility of the complex also differs, and it can control access to the DNA. The 601 sequence is designed to form a strong complex with a nucleosome core. In comparison to NCPs formed based on the naturally occurring 5S rDNA sequence, NCPs formed based on the 601 sequence exhibit decreased dynamics and display higher stability in the presence of restriction enzymes (Polach and Widom 1999).
The organization of DNA into nucleosome particles impacts DNA-protein interactions. It should be noted that under DNA wrapping, the accessibility of nucleotide bases for DNA-protein interactions varies greatly depending on the DNA sequence. One of the consequences is that the base rotational orientation is related to the nucleosomal core and is defined as “in” or “out” depending upon whether the nucleobase is facing in or out in relation to the histone octamer. Another consequence is that the orientation of the coupled bases is related to the dyad axes. As a result, deviations in enzymatic functioning in the presence of naked DNA and DNA consisting of nucleosomes could be observed (Balliano and Hayes 2015).
4.3 Base Excision Repair in the Nucleosome Context
Most of the experimental data concerning the investigation of BER protein activities have been obtained when using naked DNA. A number of studies have investigated the efficiency of individual stages during the BER process using NCP. In the first step, the damaged nucleobase in the DNA is recognized and eliminated by a specific DNA glycosylase. The activity of several DNA glycosylases has been studied (Olmon and Delaney 2017; Beard et al. 2003; Czaja et al. 2014; Hinz et al. 2010; Prasad et al. 2007; Maher et al. 2019). Even though the enzyme activity varied depending on the position and the base rotational orientation, the general effect was the same. The structural architecture of NCP suppresses the functioning of DNA glycosylases. An extensive study of the hydrolytic activity of several DNA glycosylases belonging to different structural superfamilies was performed on nucleosomal DNA (Olmon and Delaney 2017). E. coli uracil DNA glycosylase (UDG) acts on U, E. coli formamidopyrimidine DNA glycosylase (Fpg) and human 8-oxoguanine glycosylase 1 (hOGG1) act on 8-oxoG, human alkyladenine DNA glycosylase (hAGG) acts on ethenoadenine, and E. coli Endonuclease III (EndoIII) acts on 5-hydroxyU. Based on this list, only UDG and hAGG displayed activity towards lesions located in the dyad position, and a slight influence of the damage orientation relative to the nucleosome core was observed. The activity of the other DNA glycosylases was completely inhibited for lesions positioned on the dyad axis, regardless of rotational position. The efficiency of UDG and hAGG can probably be explained by the structural features of these enzymes. Their binding to NCP is subject to relatively slight steric obstruction from the histone octamer core compared to other DNA glycosylases.
After removing the damaged nucleobase, the next stage of the BER process is AP site cleavage. The ability of APE1 to cleave AP sites in the context of NCP has been demonstrated in several publications (Hinz et al. 2010; Hinz 2014; Rodriguez and Smerdon 2013). Initially, it was shown that the activity of APE1 is almost entirely independent of the AP site orientation (Hinz et al. 2010; Rodriguez and Smerdon 2013). Later, the data were clarified, and it was revealed that the activity of APE1 is strongly dependent on the rotational orientation of the AP site precursor (Hinz 2014). For outward-oriented damage, the efficiency of AP site cleavage was much higher than that for inward-oriented damages. When a damaged base is removed by a bifunctional DNA glycosylase, a 3′-PUA residue can appear. APE1 was shown to be able to remove the 3′-PUA after cleavage of AP site in the nucleosome (Maher et al. 2019; Odell et al. 2011). APE1 digested the inward-oriented substrate twice as slowly as the outward-oriented substrate (Odell et al. 2011).
These first two steps of BER produce a single nucleotide gap, which is usually filled by DNA polymerase β. It was shown that in the nucleosomal context, DNA synthesis is presumably carried out through single-nucleotide gap filling (Meas and Smerdon 2016). In several studies, a significant reduction in pol β polymerase activity on NCPs was demonstrated (Rodriguez and Smerdon 2013; Odell et al. 2011; Nilsen et al. 2002; Beard et al. 2003; Balliano et al. 2017). The results demonstrated the dependence of Polβ activity on the position of the damage. The location of the damage near the dyad leads to a decrease in Polβ activity compared to that in a location near the edge (Nilsen et al. 2002; Beard et al. 2003). It should be noted that the magnitude of the effect is significantly dependent on the stability of the NCP model. The authors of (Beard et al. 2003) used DNA that formed more stable NCP and observed a total suppression of Polβ activity. The orientation of the damage relative to the nucleosome core also contributes to Polβ activity. In general, the inward-oriented position is filled by Polβ less efficiently (Rodriguez and Smerdon 2013; Odell et al. 2011). Reduced strand displacement activity of Polβ is also observed on NCPs compared with that on naked DNA (Balliano et al. 2017).
In the case of strand displacement DNA synthesis, FEN1 follows the DNA polymerase and normally removes the flap DNA strand. Its activity on an NCP also depends on the DNA sequence. Experiments with 5S rDNA demonstrated that FEN1 had equal activity on NCPs and on naked DNA (Huggins et al. 2002). Alternatively, the data obtained by using 601 DNA for NCP reconstitution demonstrated the inability of FEN1 to process the flap structure (Jagannathan et al. 2011). These controversial results could be explained by differences in the stability of NCPs formed using different DNA sequences. It is possible that due to the high affinity of histone octamers for the 601 DNA sequence, the formation of the correct DNA-protein complex between DNA and FEN1 that will facilitate the enzymatic activity of FEN1 is prevented.
The final stage of BER, which produces an intact DNA strand, is ligation of the nick formed in the previous stage. Several research groups have also studied the functioning of DNA ligases within NCPs. Overall, their data showed that a reduction in the activity of DNA ligase was observed when using NCPs compared to that observed when using naked DNA (Odell et al. 2011; Chafin et al. 2000). The strongest suppression was detected when using the NCPs based on 601 DNA (Chafin et al. 2000). This finding is in accordance with the hypothesis concerning the restriction of the conformational mobility of DNA in 601 NCPs compared to that observed in more “relaxed” NCPs based on 5S rDNA.
In summary, we can conclude that the enzymatic activity of the main BER proteins is reduced due to NCP compaction. The outcome depends on the type of DNA used for NCP reconstitution and on the rotational orientation of the damage relative to the nucleosome core. The most significant effect is due to the type of DNA used in the NCP. From this point of view, 601 DNA exhibits stronger binding to the histone octamer core and suppression of the activity of BER enzymes than those observed in NCPs based on the 5S rDNA sequence (Polach and Widom 1999). Regarding the orientation of the nucleobase, inward-facing damage is usually more resistant to the action of BER enzymes.
4.4 PARP1, PARP2, PARP3 and the Nucleosome
One of the main reactions involved in the DNA damage response is poly(ADP-ribosyl)ation (Khodyreva and Lavrik 2016). The interaction of PARP1/2/3 with different types of DNA damage has been extensively studied by many research groups (Kutuzov et al. 2013; D’Amours et al. 1999; Amé et al. 1999; Langelier et al. 2014; Pion et al. 2005; Potaman et al. 2005; Jorgensen et al. 2009). The DNA substrates included in previous studies contained different types of breaks or DNA ends. It should be noted that the affinity of PARP1/2/3 proteins for different DNA structures does not correlate with their activation efficacy. For example, the most efficient activation of PARP1 can be achieved using nick-containing DNA duplexes, but the protein does not display a high affinity for such DNA. At the same time, PARP1 displays the highest affinity for blunt-ended DNA but is only weakly activated by this DNA structure (Pion et al. 2005; D’Silva et al. 1999). It was shown that PARPs are preferably activated by DNA breaks, whereas the activation of PARP1 and PARP2 by DNA structures containing AP sites, hairpins and junction points is very low (Kutuzov et al. 2013; Potaman et al. 2005; Jorgensen et al. 2009; Khodyreva et al. 2010).
Thus, the BER system could be regulated through PARylation-derived signals. PARylation is catalysed by members of the poly(ADP-ribose)polymerase family. This family consists of 17 proteins that are encoded by different genes (Amé et al. 2004; Hottiger et al. 2010). Despite the fact that membership in the PARP family is defined by the existence of a conservative motif in a catalytic domain, not all PARP proteins possess catalytic activity (Amé et al. 2004; Hottiger et al. 2010; Vyas et al. 2014). Moreover, only three proteins are activated in response to DNA damage: PARP1, PARP2 and PARP3 (Langelier et al. 2012; Ame et al. 1999; Grundy et al. 2016). Usually, PARPs utilize protein acceptors, but recent data suggest that PARP1/2/3 are able to modify DNA (Talhaoui et al. 2016; Zarkovic et al. 2018; Munnur and Ahel 2017; Belousova et al. 2018a). PARP enzymes transfer the ADP-ribose moiety to acceptors and then covalently attach it, resulting in the formation of mono(ADP-ribosyl)ated (MARylated) or poly(ADP-ribosyl)ated (PARylated) protein/DNA. The substrate used during MAR- or PARylation is NAD+. The formation of ADP-ribose polymers is a reversible process (Crawford et al. 2018). Moreover, PAR has been revealed to be highly toxic for cells (Andrabi et al. 2006). During PAR catabolism, the main enzyme degrading this polymer in eukaryotic cells is poly(ADP-ribose)glycohydrolase (PARG) (Lin et al. 1997). Therefore, PAR formation constitutes a temporary intracellular signal.
In this review, we address three enzymes, PARP1, PARP2, and PARP3, that are members of the PARP family. The rate of PAR formation catalysed by PARP1 is the highest. Approximately 90% of PAR in cells is synthesised by PARP1 (Ame et al. 1999). It was previously shown that PARP1 can synthesize linear and branched PAR polymers (Hassa et al. 2006). However, according to recent data, PARP1 predominantly generates a linear PAR polymer, while PARP2 produces branched polymer (Chen et al. 2018). PARP3 is able to transfer only mono(ADP-ribose) (MAR) moieties (Vyas et al. 2014). Authors of (Chen et al. 2018) hypothesized that PARP1 and PARP2 can work together in tandem, in which PARP1 is responsible for the rate of PAR formation while PARP2 is required for branching of the polymer. Moreover, PARP3 may serve as an initiator of PARylation by transferring the first ADP-ribose moiety, which is subsequently elongated by PARP1/PARP2. The data regarding the feasibility of this mechanism were obtained for DNA poly(ADP-ribosyl)ation (Belousova et al. 2018b). The length and branching type of PAR molecules could lead to variation in the functioning of the acceptor molecule; however, the unique correlations that occur between the PAR type and the observed molecular behaviour are still unclear.
There are numerous data regarding the participation of PARP1 in BER regulation (Khodyreva and Lavrik 2016). PARP1 recognizes DNA repair intermediates generated during different BER stages, such as AP sites, nicked and gapped DNAs, and flaps in DNA duplexes (Khodyreva et al. 2010; Lavrik et al. 2001; Sukhanova et al. 2010, 2004, 2015). The affinity of PARP1 for these structures is not correlated with the efficiency of catalytic activation and varies widely (Langelier et al. 2014; Pion et al. 2005). PARP2 also interacts with the main BER DNA intermediates (Kutuzov et al. 2013; Langelier et al. 2014; Pion et al. 2005). Generally, PARP2 displays much lower affinity and specificity by DNA binding than PARP1. PARylation catalysed by PARP2 is also less efficient than that catalysed by PARP1 (Kutuzov et al. 2013; Langelier et al. 2014).
There is little information concerning the participation of PARP3 in different cellular processes. Most of the data describe the involvement of PARP3 in double-strand break repair (Rulten et al. 2011; Beck et al. 2014). Recent data have demonstrated probable involvement of PARP3 in single-strand break repair (Grundy et al. 2016). In particular, PARP3 was shown to accelerate the repair of γ-ray-induced SSBs in chicken DT40 cells. PARP3 like PARP1 is defined as a nick-sensor. It is noteworthy that PARP3 displays high specificity in recognizing nick-flanking DNA ends. In contrast to PARP1, PARP3 activity was stimulated only if the 5′- and 3′-ends contained canonical 5′-P and 3′-OH moieties (Potaman et al. 2005; Khodyreva et al. 2010; Crawford et al. 2018). There are no direct data indicating PARP3 participation in BER, but a number of enzymes that carry out SSBR also participate in BER (Chatterjee and Walker 2017). Moreover, a single-strand break is a DNA intermediate that appears during BER. Therefore, PARP3 on BER process is expected.
It was suggested that PARP1 is recruited to BER complexes to nicked DNA or DNA containing AP sites during the early stages of the process, where it aids in coordinating subsequent stages via both protein-protein interactions and PARylation (Fig. 4.1) (Khodyreva and Lavrik 2016). For example, autoPARylation of PARP1 leads to the recruitment of another BER scaffold protein, XRCC1, that recognizes PAR molecules (Masson et al. 1998; Hanzlikova et al. 2017). The role of PARP2 is less well characterized but is considered to partially overlap with that of PARP1. For example, PARP2, similar to PARP1, is essential for XRCC1 and PNKP recruitment to oxidative single-strand breaks (Hanzlikova et al. 2017). Most likely, these enzymes can act together as a heterodimer (Schreiber et al. 2002). PARP3 has also been shown to act as a heterodimer with PARP1 (Loseva et al. 2010). Moreover, during an investigation of the PARP1 and PARP3 interaction, Helleday’s group observed that the PARylation of both proteins was performed by PARP1 in a DNA-independent manner. This intriguing finding could be related an additional regulatory mechanism that influences PARP1 activity.
One of the greatest challenges in maintaining genome integrity in response to DNA damage is the existence of the chromatin structure and its rearrangements that allow access to repair proteins. PARylation is one of the mechanisms that produces such changes in chromatin architecture (Fig. 4.2) (Martinez-Zamudio and Ha 2012). Thus, functionally active proteins that are necessary for DNA damage repair could be recruited via the PAR recognition mechanism (Pleschke et al. 2000). At the same time, PARylation of chromatin-associated components could alter the chromatin structure itself. PARP1 can catalyse autoPARylation. Depending on the length of the polymer, it can perform different functions. PAR can serve as an intracellular signal of DNA damage recruiting repair factors (Liu et al. 2017). Alternatively, the electrostatic repulsion between long polymers can lead to the dissociation of target proteins from complexes with chromatin (Kurgina et al. 2018).

Suggested role of PARP1 in nucleosome metabolism. Poly(ADP-ribose)polymerase 1 (PARP1) binding with nucleosomes leads to local relaxation of nucleosome structure, which may provide additional access to proteins during their interaction with DNA. Two types of modification could occur during the interaction of PARP1 with the nucleosome in the presence of the nicotinamide adenine dinucleotide (NAD+) molecule. During autoPARylation (autopoly(ADP-ribosyl)ation), PARP1 dissociates from its complex with the nucleosome, resulting in nucleosome compaction, which may be more favourable for the screening of DNA damage and initiation of repair processes. Alternatively, if under binding of PARP1 to an NCP the main acceptor is the histone core, it can lead to nucleosome destabilization and subsequent escape of DNA from the NCP structure. NCP destabilization was also observed during transcription regulation
Histones have been shown to be acceptors of PAR (Ueda et al. 1975). PARylation of chromatin is predominantly associated with relaxation of the chromatin structure (Fig. 4.2) (Poirier et al. 1982; Ciccarone et al. 2017). Core histone PARylation can destabilize the nucleosome (Huletsky et al. 1989; Realini and Althaus 1992). While the ability of PARP1 to PARylate histones H1, H2A, H2B, H3 and H4 has been shown (Messner et al. 2010), the main target histone of PARP1 is H1. PARP2 has not been detected in PARylation of core histones (Messner et al. 2010); the main target of PARP3 is H2B (Grundy et al. 2016).
According to (Kim et al. 2004), binding of PARP1 to chromatin leads to additional chromatin compaction. However, recent data obtained by V. M. Studitsky’s group demonstrated the local relaxation of nucleosome structure upon PARP1 binding (Sultanov et al. 2017). Such an increase in NCP motility probably provides alternative access for repair proteins for their interaction with DNA. Moreover, autoPARylation of PARP1 promotes the reversing into solid NCP structure and leads to nucleosomal compaction.
Two types of modification could occur during the interaction of PARP1 with NCP in the presence of the NAD+-molecule (Fig. 4.2). First, PARP1 could perform autoPARylation. Second, PARP1 could catalyse PARylation of the histone core. It is likely that the synthesis of PAR attached to different acceptors is carried out under various conditions and thus results in different effects. Therefore, one scenario could be that during autoPARylation, PARP1 dissociates from the complex with NPC, resulting in NCP compaction. In such a case, the initial nucleosomal decompaction upon PARP1 binding is short-lived and is not critical to the maintenance of the NCP structure. It is conceivable that such a scenario is preferable for screening DNA damage and initiating repair processes. Based on the literature data, the fact that the DNA is tightly wrapped is affected to a large extent by the functional activity of the majority of the BER proteins. However, even the negligible difference in the degree of wrapping observed between 601 and 5S rDNA allows to rise up the repair activity in the case of 5S DNA.
Alternatively, if under binding of PARP1 to an NCP the main acceptor is the histone core, it can lead to nucleosome destabilization and subsequent escape of DNA from the NCP structure. NCP destabilization was also observed during transcription regulation (Martinez-Zamudio and Ha 2012). For example, Martinez-Zamudio and Hyo Chol Ha have shown that the enzymatic activity of PARP1 is stimulated via the TLR4-dependent intracellular signalling pathway and that subsequent PARylation of the core histone proteins H3/H2B promoted the accessibility of the promoter regions to transcription factors involved in the NF-κB-dependent inflammatory response.
Another outstanding issue is the functional significance of PARP1, PARP2 and PARP3. The rate of PAR synthesis is the highest for PARP1. PARP2 was found to be able to produce branching during PAR synthesis. PARP3 is exclusively a mono(ADP-ribosyl)transferase. It is remarkable that these DNA-dependent PARPs are able to act pairwise, for instance, PARP1 and PARP2 or PARP1 and PARP3 (Chen et al. 2018; Loseva et al. 2010). Moreover, each of these proteins is able to interact with the other BER proteins. An additional point is the suggestion that PAR functions as an organizer of cellular architecture. Consistent with this theory, local PAR synthesis may lead to the formation of a compartment in which PAR itself can orchestrate to outcompete interactions during the repair process (Leung 2014; Altmeyer et al. 2015). The formation of PAR compartments stabilized by Mg2+ ions and destroyed by the activity of PARG was recently detected by using the light-scattering technique (Kurgina et al. 2018; Vasil’eva et al. 2019). Therefore, it can be suggested that PARP1, PARP2 and PARP3 function together within the context of the BER process to form an expanding system of PAR signals. The precise role of these signal needs to be investigated.
References
Abbotts R, Wilson DM III (2017) Coordination of DNA single strand break repair HHS public access. Radic Biol Med 107:228–244
Altmeyer M, Neelsen KJ, Teloni F et al (2015) Liquid demixing of intrinsically disordered proteins is seeded by poly(ADP-ribose). Nat Commun 6:8088
Amé JC, Rolli V, Schreiber V et al (1999) PARP-2, a novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. J Biol Chem 274:17860–17868
Amé JC, Spenlehauer C, De Murcia G (2004) The PARP superfamily. BioEssays 26:882–893
Andrabi SA, Kim NS, Yu S-W, National Academy of Sciences et al (2006) Poly(ADP-ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci U S A 103:18308–18313
Balliano AJ, Hayes JJ (2015) Base excision repair in chromatin: insights from reconstituted systems. DNA Repair 36:77–85
Balliano A, Hao F, Njeri C et al (2017) HMGB1 stimulates activity of polymerase β on nucleosome substrates. Biochemistry 56:647–656
Beard BC, Wilson SH, Smerdon MJ (2003) Suppressed catalytic activity of base excision repair enzymes on rotationally positioned uracil in nucleosomes. Proc Natl Acad Sci U S A 100:7465–7470
Beck C, Boehler C, Guirouilh Barbat J et al (2014) PARP3 affects the relative contribution of homologous recombination and nonhomologous end-joining pathways. Nucleic Acids Res 42:5616–5632
Belousova EA, Ishchenko АA, Lavrik OI (2018a) DNA is a new target of Parp3. Sci Rep 8:4176
Belousova EA, Ishchenko АA, Lavrik OI et al (2018b) DNA is a new target of Parp3. Sci Rep 8:1–12
Chafin DR, Vitolo JM, Henricksen LA et al (2000) Human DNA ligase I efficiently seals nicks in nucleosomes. EMBO J 19:5492–5501
Chatterjee N, Walker GC (2017) Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen 58:235–263
Chen Q, Kassab MA, Dantzer F, Springer US et al (2018) PARP2 mediates branched poly ADP-ribosylation in response to DNA damage. Nat Commun 9:3233
Ciccarone F, Zampieri M, Caiafa P (2017) PARP1 orchestrates epigenetic events setting up chromatin domains. Semin Cell Dev Biol 63:123–134
Crawford K, Bonfiglio JJ, Mikoč A et al (2018) Specificity of reversible ADP-ribosylation and regulation of cellular processes. Crit Rev Biochem Mol Biol 53:64–82
Czaja W, Mao P, Smerdon MJ (2014) Chromatin remodelling complex RSC promotes base excision repair in chromatin of Saccharomyces cerevisiae. DNA Repair (Amst) 16:35–43
D’Amours D, Desnoyers S, D’Silva I et al (1999) Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J 342(Pt 2):249–268
D’Silva I, Pelletier JD, Lagueux J et al (1999) Relative affinities of poly(ADP-ribose) polymerase and DNA-dependent protein kinase for DNA strand interruptions. Biochim Biophys Acta 1430:119–126
Gates KS (2009) An overview of chemical processes that damage cellular DNA: spontaneous hydrolysis, alkylation, and reactions with radicals. Chem Res Toxicol 22:1747–1760
Grundy GJ, Polo LM, Zeng Z et al (2016) PARP3 is a sensor of nicked nucleosomes and monoribosylates histone H2BGlu2. Nat Commun 7:12404
Hanzlikova H, Gittens W, Krejcikova K et al (2017) Overlapping roles for PARP1 and PARP2 in the recruitment of endogenous XRCC1 and PNKP into oxidized chromatin. Nucleic Acids Res 45:2546–2557
Hassa PO, Haenni SS, Elser M, American Society for Microbiology (ASM) et al (2006) Nuclear ADP-ribosylation reactions in mammalian cells: where are we today and where are we going? Microbiol Mol Biol Rev 70:789–829
Hinz JM (2014) Impact of abasic site orientation within nucleosomes on human APE1 endonuclease activity. Mutat Res Fundam Mol Mech Mutagen 766–767:19–24
Hinz JM, Rodriguez Y, Smerdon MJ (2010) Rotational dynamics of DNA on the nucleosome surface markedly impact accessibility to a DNA repair enzyme. Proc Natl Acad Sci 107:4646–4651
Hoeijmakers JHJ (2001) Genome maintenance mechanisms for preventing cancer. Nature 411:366–374
Hottiger MO, Hassa PO, Lüscher B et al (2010) Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem Sci 35:208–219
Huggins CF, Chafin DR, Aoyagi S et al (2002) Flap endonuclease 1 efficiently cleaves base excision repair and DNA replication intermediates assembled into nucleosomes. Mol Cell 10:1201–1211
Huletsky A, de Murcia G, Muller S et al (1989) The effect of poly(ADP-ribosyl)ation on native and H1-depleted chromatin. A role of poly(ADP-ribosyl)ation on core nucleosome structure. J Biol Chem 264:8878–8886
Jagannathan I, Pepenella S, Hayes JJ (2011) Activity of FEN1 endonuclease on nucleosome substrates is dependent upon DNA sequence but not flap orientation. J Biol Chem 286:17521–17529
Jorgensen TJ, Chen K, Chasovskikh S et al (2009) Binding kinetics and activity of human poly(ADP-ribose) polymerase-1 on oligo-deoxyribonucleotide substrates. J Mol Recognit 22:446–452
Khodyreva SN, Lavrik OI (2011) Affinity modification in a proteomic study of DNA repair ensembles. Bioorg Khim 37:91–107
Khodyreva SN, Lavrik OI (2016) Poly(ADP-Ribose) polymerase 1 as a key regulator of DNA repair. Mol Biol (Mosk) 50:655–673
Khodyreva SN, Prasad R, Ilina ES et al (2010) Apurinic/apyrimidinic (AP) site recognition by the 5’-dRP/AP lyase in poly(ADP-ribose) polymerase-1 (PARP-1). Proc Natl Acad Sci 107:22090–22095
Kim Y-J, Wilson DM (2012) Overview of base excision repair biochemistry. Curr Mol Pharmacol 5:3–13
Kim MY, Mauro S, Gévry N et al (2004) NAD+-dependent modulation of chromatin structure and transcription by nucleosome binding properties of PARP-1. Cell 119:803–814
Krokan HE, Bjørås M (2013) Base excision repair. Cold Spring Harb Perspect Biol 5:a012583
Kurgina TA, Anarbaev RO, Sukhanova MV et al (2018) A rapid fluorescent method for the real-time measurement of poly(ADP-ribose) polymerase 1 activity. Anal Biochem 545:91–97
Kutuzov MM, Khodyreva SN, Amé J-C et al (2013) Interaction of PARP-2 with DNA structures mimicking DNA repair intermediates and consequences on activity of base excision repair proteins. Biochimie 95:1208–1215
Kutuzov MMM, Khodyreva SNSN, Ilina ESES et al (2015) Interaction of PARP-2 with AP site containing DNA. Biochimie 112:10–19
Langelier M-F, Planck JL, Roy S et al (2012) Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1. Science 336:728–732
Langelier M-F, Riccio AA, Pascal JM (2014) PARP-2 and PARP-3 are selectively activated by 5′ phosphorylated DNA breaks through an allosteric regulatory mechanism shared with PARP-1. Nucleic Acids Res 42:7762–7775
Lavrik OI, Prasad R, Sobol RW et al (2001) Photoaffinity labeling of mouse fibroblast enzymes by a base excision repair intermediate. Evidence for the role of poly(ADP-ribose) polymerase-1 in DNA repair. J Biol Chem 276:25541–25548
Leung AKL (2014) Poly(ADP-ribose): an organizer of cellular architecture. J Cell Biol 205:613–619
Lin W, Amé JC, Aboul-Ela N et al (1997) Isolation and characterization of the cDNA encoding bovine poly(ADP-ribose) glycohydrolase. J Biol Chem 272:11895–11901
Lindahl T Nature Publishing Group (1993) Instability and decay of the primary structure of DNA. Nature 362:709–715
Liu L, Kong M, Gassman NR et al (2017) PARP1 changes from three-dimensional DNA damage searching to one-dimensional diffusion after auto-PARylation or in the presence of APE1. Nucleic Acids Res 45:12834–12847
Loseva O, Jemth A-S, Bryant HE et al (2010) PARP-3 is a mono-ADP-ribosylase that activates PARP-1 in the absence of DNA. J Biol Chem 285:8054–8060
Lowary PT, Widom J (1998) New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J Mol Biol 276:19–42
Maher RL, Wallace SS, Pederson DS (2019) The lyase activity of bifunctional DNA glycosylases and the 3′-diesterase activity of APE1 contribute to the repair of oxidized bases in nucleosomes. Nucleic Acids Res 47:2922–2931
Martinez-Zamudio R, Ha HC (2012) Histone ADP-ribosylation facilitates gene transcription by directly remodeling nucleosomes. Mol Cell Biol 32:2490–2502
Masson M, Niedergang C, Schreiber V et al (1998) XRCC1 is specifically associated with poly(ADP-ribose) polymerase and negatively regulates its activity following DNA damage. Mol Cell Biol 18:3563–3571
McGinty RK, Tan S (2015) Nucleosome structure and function. Chem Rev 115:2255–2273
Meas R, Smerdon MJ (2016) Nucleosomes determine their own patch size in base excision repair. Sci Rep 6:27122
Messner S, Altmeyer M, Zhao H et al (2010) PARP1 ADP-ribosylates lysine residues of the core histone tails. Nucleic Acids Res 38:6350–6362
Moor NA, Lavrik OI (2018) Protein-protein interactions in DNA base excision repair. Biochemistry (Mosc) 83:411–422
Moor NA, Vasil’eva IA, Anarbaev RO et al (2015) Quantitative characterization of protein-protein complexes involved in base excision DNA repair. Nucleic Acids Res 43:6009–6022
Munnur D, Ahel I (2017) Reversible mono-ADP-ribosylation of DNA breaks. FEBS J 284:4002–4016
Nilsen H, Lindahl T, Verreault A (2002) DNA base excision repair of uracil residues in reconstituted nucleosome core particles. EMBO J 21:5943–5952
Odell ID, Barbour J-E, Murphy DL et al (2011) Nucleosome disruption by DNA ligase III-XRCC1 promotes efficient base excision repair. Mol Cell Biol 31:4623–4632
Olmon ED, Delaney S (2017) Differential ability of five DNA glycosylases to recognize and repair damage on nucleosomal DNA. ACS Chem Biol 12:692–701
Pion E, Ullmann GM, Amé J-C et al (2005) DNA-induced dimerization of poly(ADP-ribose) polymerase-1 triggers its activation. Biochemistry 44:14670–14681
Pleschke JM, Kleczkowska HE, Strohm M et al (2000) Poly(ADP-ribose) binds to specific domains in DNA damage checkpoint proteins. J Biol Chem 275:40974–40980
Poirier GG, de Murcia G, Jongstra-Bilen J et al (1982) Poly(ADP-ribosyl)ation of polynucleosomes causes relaxation of chromatin structure. Proc Natl Acad Sci U S A 79:3423–3427
Polach KJ, Widom J (1999) Restriction enzymes as probes of nucleosome stability and dynamics. Methods Enzymol 304:278–298
Potaman VN, Shlyakhtenko LS, Oussatcheva EA et al (2005) Specific binding of poly(ADP-ribose) polymerase-1 to cruciform hairpins. J Mol Biol 348:609–615
Prasad A, Wallace SS, Pederson DS (2007) Initiation of base excision repair of oxidative lesions in nucleosomes by the human, bifunctional DNA glycosylase NTH1. Mol Cell Biol 27:8442–8453
Realini CA, Althaus FR (1992) Histone shuttling by poly(ADP-ribosylation). J Biol Chem 267:18858–18865
Rodriguez Y, Smerdon MJ (2013) The structural location of DNA lesions in nucleosome core particles determines accessibility by base excision repair enzymes. J Biol Chem 288:13863–13875
Rulten SL, Fisher AEO, Robert I et al (2011) PARP-3 and APLF function together to accelerate nonhomologous end-joining. Mol Cell 41:33–45
Sancar A, Lindsey-Boltz LA, Unsal-Kaçmaz K et al (2004) Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 73:39–85
Schreiber V, Amé J-C, Dollé P et al (2002) Poly(ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision DNA repair in association with PARP-1 and XRCC1. J Biol Chem 277:23028–23036
Simpson RT, Stafford DW (1983) Structural features of a phased nucleosome core particle. Proc Natl Acad Sci U S A 80:51–55
Sukhanova MV, Khodyreva SN, Lavrik OI (2004) Poly(ADP-ribose) polymerase-1 inhibits strand-displacement synthesis of DNA catalyzed by DNA polymerase beta. Biochemistry (Mosc) 69:558–568
Sukhanova M, Khodyreva S, Lavrik O (2010) Poly(ADP-ribose) polymerase 1 regulates activity of DNA polymerase beta in long patch base excision repair. Mutat Res 685:80–89
Sukhanova MV, Abrakhi S, Joshi V et al (2015) Single molecule detection of PARP1 and PARP2 interaction with DNA strand breaks and their poly(ADP-ribosyl)ation using high-resolution AFM imaging. Nucleic Acids Res 44(6):e60
Sultanov DC, Gerasimova NS, Kudryashova KS et al (2017) Unfolding of core nucleosomes by PARP-1 revealed by spFRET microscopy. AIMS Genet 4:21–31
Swenberg JA, Lu K, Moeller BC et al (2011) Endogenous versus exogenous DNA adducts: their role in carcinogenesis, epidemiology, and risk assessment. Toxicol Sci 120:S130–S145
Talhaoui I, Lebedeva NA, Zarkovic G et al (2016) Poly(ADP-ribose) polymerases covalently modify strand break termini in DNA fragments in vitro. Nucleic Acids Res 44(19):9279–9295
Ueda K, Omachi A, Kawaichi M et al (1975) Natural occurrence of poly(ADP-ribosyl) histones in rat liver. Proc Natl Acad Sci U S A 72:205–209
Vasil’eva IA, Anarbaev RO, Moor NA et al (2019) Dynamic light scattering study of base excision DNA repair proteins and their complexes. Biochim Biophys Acta, Proteins Proteomics 1867:297–305
Vyas S, Matic I, Uchima L et al (2014) Family-wide analysis of poly(ADP-ribose) polymerase activity. Nat Commun 5:4426
Zarkovic G, Belousova EA, Talhaoui I et al (2018) Characterization of DNA ADP-ribosyltransferase activities of PARP2 and PARP3: new insights into DNA ADP-ribosylation. Nucleic Acids Res 46:2417–2431
Acknowledgements
This work was supported by RSF project № 17-74-20075.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Kutuzov, M.M., Belousova, E.A., Ilina, E.S., Lavrik, O.I. (2020). Impact of PARP1, PARP2 & PARP3 on the Base Excision Repair of Nucleosomal DNA. In: Zharkov, D. (eds) Mechanisms of Genome Protection and Repair. Advances in Experimental Medicine and Biology, vol 1241. Springer, Cham. https://doi.org/10.1007/978-3-030-41283-8_4
Download citation
DOI: https://doi.org/10.1007/978-3-030-41283-8_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-41282-1
Online ISBN: 978-3-030-41283-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)