
Currently, more than 37 million individuals worldwide are infected with the human immunodeficiency virus (HIV). Antiretroviral therapy may control the viral infection but is incapable of eradicating it. It is important to understand how cells respond to HIV-1 infection and what cellular factors are involved in this process to develop novel classes of antiviral drugs. This review summarizes the current understanding of the HIV restriction mechanism. We discuss the ambiguous role of HIV restriction factors in viral infection and counteraction mediated by HIV-1 accessory proteins.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The human immunodeficiency virus (HIV) is a member of the family Retroviridae, genus Lentivirus, with a long incubation period. HIV includes the following species: HIV-1, the most widespread and pathogenic, and HIV-2, differing from HIV-1 in structure and pathogenic effect. There are four groups of HIV-1: M (Major), N (Non-M, Non-O), O (Outlier), and P (Putative). It is currently believed that HIV-1 group M originated from the chimpanzee immunodeficiency virus (SIV) at the beginning of the XX century shown in [1]. More than 70 million people are infected with HIV-1 group M; for 30 million people, this disease has already led to fatal outcomes. HIV-1 from other groups (N, O, P) also originated from SIV (chimpanzee in group N and gorilla in groups O and P) but occurs in humans much less frequently. HIV-1 group O was found in approximately 100 thousand people in Cameroon and in neighboring countries in [2]; group N and P viruses have been found only in a few individuals in [3–5].
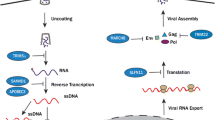
After decoding the nucleotide sequence of HIV-1, it became clear that its genome was much more complex compared to those of many animal retroviruses. The latter genomes include the gag, pol, and env genes encoding only structural proteins (e.g., the murine leukemia virus (MuLV) genome). Open reading frames uncharacteristic of other members of the family have been found in the 3'-region of the HIV-1 genome. Here, regulatory proteins Tat and Rev and accessory proteins Vif, Vpr, Vpu, and Nef are encoded (in HIV-2, they also include Vpx). Tat and Rev are necessary for HIV-1 replication in host cells: they activate transcription of genes from proviral DNA and provide viral RNA transport from nucleus to cytoplasm. Accessory proteins are needed for replication only in some types of cells, though they are commonly believed to counteract the cellular mechanisms of antiviral protection. It took years to find out the targets of HIV accessory proteins: restriction factors (Fig. 1). Although there is a considerable body of literature on restriction factors (e.g., reviews in [6–8]), its amount continuously increases, while the role of some of these factors is not quite clear or revised and, therefore, new data have to be periodically generalized.

The counteraction of accessory viral proteins to cellular restriction factors.
The best-known restriction factors with established mechanisms of action are IFITM1–3 in [9], APOBEC3 in [10–12], TRIM5α in [13–15], Mx2 in [16–18], Schlaffen11 in [19], SAMHD1 in [20–22], and BST-2 (also known as Tetherin) in [23, 24].
Below we will thoroughly consider the cellular proteins involved in HIV restriction, in the order they influence different stages of the HIV replication cycle: from early to late.
IFITM
The penetration of HIV-1 into a cell causes activation of pattern-recognizing receptors (PRR), including Toll-like receptor 7 (TLR7), and the production of the type I interferons (IFN) in [25]. Numerous genes of restriction factors of different viruses have been found in recent years, their expression being dependent on type I IFN production. Some of them (MxB, BST-2, APOBEC3) were thoroughly studied in the context of specific viral infections, in particular, retroviral infection. Among such IFN-stimulated genes (ISGs), the interferon-induced transmembrane gene family (IFITM) with broad-spectrum antiviral activity was identified.
The IFITM protein family was described 30 years ago, with the simultaneously revealed induction of their expression via type I and II IFN in [26]. The antiviral function of IFITM1, IFITM2 and IFITM3 proteins was found in 2009, as a result of screening small interfering RNA (siRNA) in [9]. These proteins performed the function of restriction factors at the early stages of cell infection with influenza A virus, subtype H1N1, as well as flaviviruses: the Dengue and West Nile fever viruses in [9]. The antiviral effect of IFITM proteins against HIV-1 was shown in 2011: the IFITM1, IFITM2, and IFITM3 knockdown resulted in enhanced infection in [27].
IFITM1 is localized on the cell plasma membrane, while IFITM2 and IFITM3 are localized in late endosomes and lysosomes in [28, 29]. The proteins contain two transmembrane domains and an extremely conservative intracellular domain, which interestingly mirrors the structure of another restriction factor, BST-2, acting at the stage of particle release from infected cells in [24] (see below). The principle of restriction of virus penetration into a cell by IFITM proteins is associated with two aspects: the regulation of cell membrane cholesterol content and the inhibition of virus–cell fusion in [30–32]. In addition, IFITM proteins can be incorporated into viral particles, which leads to a decrease in their infectivity in [33]. Cell membrane fusion is impaired in this virions. In human SupT1 T cells with a Tet-On-regulated expression of different IFITM, all three proteins inhibit the infection with cell-free HIV-1 in [27]. However, IFITM3 demonstrates low efficiency in HIV-1 restriction during cell-to-cell transmission in [33].
The study of IFITM-mediated HIV-1 restriction depending on viral tropism (the virus uses CCR5 or CXCR4 as coreceptors to penetrate into a cell) has shown that the X4-tropic virus effectively undergoes restriction via the IFITM2 and IFITM3 proteins in endosomes and lysosomes, whereas the R5-tropic virus undergoes restriction via IFITM1 expressed on the cell plasma membrane in [34]. This selectivity is determined when the HIV-1 virion penetrates into a cell by fusing with the endosomal membrane, while CXCR4 is used as a coreceptor, and with the plasma membrane for CCR5-tropic viruses in [34].
The IFITM-mediated restriction goes on until integration of the provirus into the cell genome and, therefore, HIV-1 with IFITM restriction can be controlled in one of the following ways: modification of the site for particle penetration into a cell or the presence of proteins counteracting the restriction factor in virions. The accumulation of mutations in the Env protein in due course allows the virus to avoid restriction in [35].
Thus, IFITM proteins are integral to the process of viral and cell membrane fusion at the stage of virion incorporation into a host cell, as well as during cell-to-cell transmission. This process involves the envelope protein of HIV-1; however, its exact mechanism is still unclear.
TRIM
The proteins of the TRIM family, E3-ligases, participate in cell cycle regulation, autophagy, and innate immune responses. TRIM proteins are involved in the NF-κB and type I IFN signaling pathways and indirectly influence HIV infection. However, several proteins of this family directly inhibit viral infection at different stages of virus replication, inter alia, providing proteasomal degradation of viral proteins in [36–41].
The TRIM family was characterized, for the first time as early as in 2001, as a group of proteins with a conservative three-component (TRIpartite Motif) N-terminal domain RBCC (Really Interesting New Gene (RING) E3 ligase domain, B boxes and Сoiled-Сoil domains) in [42]. The RBCC domain provides the E3-ligase function of the protein, as well as oligomerization, which is necessary for its functional activity in [43, 44].
The C-terminal domain, on the contrary, is variable and responsible for the interaction between TRIM and partner proteins. TRIM proteins are divided into 11 subfamilies by type of C-terminal domain. The PRY/SPRY (SPla and the RYanodine Receptor) motif occurs at the C-terminus of more than 30 TRIM proteins and is associated with the antiviral activity of TRIM5, TRIM22, TRIM11, and TRIM15 proteins of the respective subfamilies in [40, 45, 46].
The TRIM5α protein of the TRIM family is considered the key molecule providing resistance of Old World monkeys (African green monkeys, rhesus macaque, etc.) to the HIV-1 infection in [13]; in New World monkeys (e.g., night monkeys), this function is performed via TRIM5 fused with cyclophilin A (TRIMCyp) in [14, 47].
Several models describing the mechanism of HIV-1 restriction via the TRIM5α protein have been proposed; each is based on interaction between the SPRY domain and the viral capsid, which leads to the impairment of virus disassembly (early “uncoating”). However, the exact mechanism of TRIM5α interference in unpacking of the virion has not yet been elucidated. For binding with the capsid, TRIM5α forms antiparallel dimers and trimers due to coiled-coil domains. TRIM5α forms a hexagonal lattice surrounding the capsid in [48–51]. Interestingly, the formation of hexagonal latice is also true for TRIM-Cyp, suggesting the existence of a common mechanism of HIV restriction via different TRIM proteins in [52].
One more consequence of the formation of hexagonal structures via TRIM5α molecule on the viral capsid is the triggering of the antiviral response of the cell in [53] (Fig. 2). Dimerization of the RING domains of TRIM5α on the capsid surface results in enhanced E3-ligase activity of TRIM5α and activated synthesis of polyubiquitin chains linked to the side chain of Lys63. These ubiquitin chains activate the TAK1 kinase complex through autophosphorylation which, in turn, leads to the translocation of transcription factors AP-1 and NF-κB to the nucleus, followed by the expression of type-I IFN in [51].

The role of TRIM proteins in HIV replication. Ub, ubiquitin; K48-, K63-polyUb, ubiquitin chains linked via Lys48 and Lys63, respectively; P, phosphorylation.
One more successful antiviral strategy is implemented via TRIM proteins from addressing viral components to proteasomal degradation. However, it is still an open question whether TRIM5α performs this function with respect to HIV-1 capsid. At the moment, no ubiquitination sites have been found at the HIV-1 capsid protein in [54], but the ability of TRIM5α to promote self-ubiquitination is known, followed by degradation in the proteasome where it probably carries viral components in [55] (Fig. 2). In addition, virus nucleocapsid components, including integrase, undergo effective proteasomal degradation (not without the involvement of TRIM5α) in [56].
Interestingly, the human homologue of TRIM5α has no marked antiviral activity. However, the only amino acid substitution in PRY-SPRY domains of the human TRIM5α is able to appreciably rehabilitate it as a HIV-1 restriction factor in [57]. Another protein of the family, TRIM22, being a human paralog of TRIM5α, participates in the inhibition of HIV-1 replication via type I IFN in [58, 59]. In addition, TRIM22, irrespective of the inherent ligase function, inhibits the Tat- and NF-κB-independent transcription of viral genes from the LTR-promoter of HIV-1 through direct interaction with the Sp1 factor in [60, 61]. These observations are especially noteworthy for clinicians: the possibility of controlling viral genome transcription in the latent period is under discussion in [62].
It is yet unclear how exactly HIV-1 encounters TRIM-mediated restriction. The level of TRIM11 expression in the cell is regulated via accessory viral protein Vpr at low intracellular concentrations in [63]; however, this regulatory mechanism is still far from being understood.
SAMHD1
The role of SAMHD1 (sterile alpha motif and histidine-aspartate domain containing protein 1) in HIV restriction was independently determined via two research teams in 2011. The protein was identified using mass spectrometry as a partner of accessory protein Vpx of the HIV-2 virus (in HIV-1, there is no Vpx) in [20, 64]. SAMHD1 is expressed at a high level in myeloid cells and resident CD4+ T cells and is a deoxynucleoside triphosphatase responsible for the regulation of dNTP pool and inhibition of HIV reverse transcription in [20, 22, 64, 65].
The histidine/aspartate (HD) catalytic domain of SAMHD1 protein hydrolyzes dNTP with the formation of deoxyribonucleoside and triphosphate, thereby removing structural blocks for the synthesis of viral cDNA in [22]. The enzyme activity is determined through tetramerization and allosterically controlled by the GTP and all four dNTPs in [66–70].
SAMHD1 also has a metal-dependent 3′→5′ exonuclease activity against single-stranded DNA and RNA, suggesting the existence of one more HIV restriction mechanism peculiar to this enzyme: viral RNA binding and degradation in [71]. dNTP triphosphate hydrolase and 3′→5′ exonuclease activities of SAMHD1 are performed via the HD domain. In the SAMHD1 point mutants with D137N or Q548A substitutions, Ryoo et al. in [72] have demonstrated the ability of SAMHD1 to inhibit HIV-1 infection without dNTP triphosphate hydrolase activity and by maintaining 3′→5′ exonuclease activity. However, the inhibitory effect of SAMHD1 was not observed in the reverse situation. Thereby, the authors concluded that it is the 3′→5′ exonuclease activity of SAMHD1 that majorly contributes to HIV-1 restriction in [72]. Although these results were not confirmed later in [73, 74], Ryoo et al. explained this with the differences in experimental setup in [75]. The significance of the 3′→5′ exonuclease activity of SAMHD1 for HIV restriction is an open question to date.
The accessory HIV-2 protein Vpx binds to the SAMHD1 C-terminus and adapter molecule DCAF1 and initiates the formation of E3-ubiquitin ligase complex, followed by proteasomal degradation of SAMHD1 in [20, 64, 76, 77]. The mechanism of this HIV-1 resistance to SAMHD1 restriction has not yet been completely studied. Vpr has carried the same motif as Vpx, which is necessary for binding with the DCAF1 adapter and arresting the cell cycle in the G2 phase in [77]. In addition, Kyei et al. in [78] have recently shown that HIV-1 can use cell cycle regulator cyclin L2 to neutralize SAMHD1 in macrophages. The importance of SAMHD1 in HIV-1 restriction has been shown via macrophages isolated from patients with the Aicardi–Goutieres syndrome caused by mutations in SAMHD1. The macrophages of such patients are characterized by much higher sensitivity to HIV-1 infection compared to the macrophages of healthy people in [21].
APOBEC3 (CEM15)
APOBEC3 (apolipoprotein Bediting complex 3) is a family of cytidine deaminases comprising seven proteins in primates: APOBEC3A (A3A), A3C, A3H with one catalytic domain and A3B, A3D, A3F, A3G with two domains in [79, 80]. APOBEC3 proteins, especially A3G, are characterized by the high level of expression in many types of cells including CD4+ Т cells, dendritic cells, and macrophages in [81]. In addition, the expression of APOBEC3 is stimulated via type I IFN in [81]. A3G was originally given precedence over other proteins of the family, but later it became clear that A3H, A3F, A3D and A3B were also active against HIV, while the viral protein Vif counteracted each in [82].
The antiviral role of APOBEC3 proteins is evident once they have entered viral particles, i.e., only in the next cycle of virus replication in [83, 84]. Restriction occurs via deaminase-induced C→U transformation in the (–)-strand of HIV DNA. Uracil is recognized by polymerase as thymine, which leads to the G→A mutation in the synthesis of DNA (+)-strand in [83]. Hypermutation during the reverse transcription of viral RNA into DNA has two important consequences: first, aberrant sequences are recognized via the cells and eliminated; second, the mutated DNA of the provirus is integrated in the cell genome, however, due to the great number of premature stop codons (formed, e.g., after TGG→TAG substitution) and other mutations, infectious viral particles are not produced in [10, 83]. APOBEC3-dependent mutagenesis occurs at a higher frequency at the 3'-end of viral sequence. In contrast to viral DNA (–)-strand synthesized from a single site (primary binding site) in the 5'‑LTR region, the synthesis of DNA (+)-strand is initiated from two polypurine tracts: the central (cPPT) and that at the 3'-end (3'-PPT). The presence of two sites for initiation of the synthesis of DNA (+)-strand and simultaneous RNA cleavage creates the prerequisites for the nonequilibrium and longer single-stranded state of DNA (–)-strand (which is a substrate for APOBEC3) in the 3'-region of the HIV genome in [85, 86].
The proteins of the APOBEC3 family inhibit lentiviral infection via the alternative mechanism not associated with deaminase activity. A3-deaminases impair reverse transcription by preventing (–)-strand synthesis and tRNA binding with the viral mRNA in [10, 87]. However, this hypothesis was not confirmed in some works in [88, 89] as they showed that APOBEC3 proteins were incapable of HIV restriction without deaminase catalytic domain.
HIV uses accessory protein Vif, which induces degradation of deaminases prior to their incorporation into viral particles, as a tool for counteracting APOBEC3-mediated restriction. Vif binds to APOBEC3 molecule in the infected cell, followed by the involvement of the E3-ubiquitin ligase complex comprising elongin B, elongin C, and RBX1, with the subsequent degradation of APOBEC3 in [84, 90, 91]. However, Vif cannot completely eliminate APOBEC3 in virus-producing cells, as has been demonstrated by the presence of numerous mutations in proviruses of the cells of patients with acute and chronic HIV infection, as well as during the vertical transmission of HIV to newborn infants in [82, 92]. Probably, the induction by type I IFN shifts the APOBEC3–Vif balance towards restriction factors, so they have time to enter viral particles before binding with Vif.
The virus seems to gain additional benefits in partial containment of APOBEC3-mediated restriction. It may happen that A3 deaminases do not completely suppress virus replication but provide a high level of mutagenesis and, therefore, a higher risk of appearance of escape-mutants resistant to antiretroviral drugs. The low efficiency of antiretroviral therapy in some patients is already associated with defective Vif in [93]. Thus, APOBEC3 can be considered an example of a restriction factor the virus turns to its own advantage.
MxB
MxA and MxB (Mx1 and Mx2 in mice) are proteins of the GTPase family; the expression of both proteins is induced by type-I IFN. MxA is known to have a broad-spectrum antiviral activity against DNA- and RNA-containing viruses but not retroviruses in [94, 95]. Recently, three independent research teams have shown that MxB is involved in HIV-1 restriction: without affecting the reverse transcription of viral RNA, it destabilizes the preintegration complex and thereby reduces the integration of proviral DNA into the cell genome in [16–18, 96].
In contrast to MxA, MxB has an extended N-terminal domain necessary for HIV-1 restriction in addition to the GTPase domain in [16, 96, 97]. This domain carries a nuclear localization signal (NLS) and can bind to the viral capsid after homodimerization (2 MxB molecules bind to each other in antiparallel directions). The Arg-rich motif of this domain directly binds to the HIV-1 capsid and determines the ability of MxB to restrict the virus in [98]. It supposedly leads to the inhibition of virus “uncoating” similar to TRIM5α restriction in [99].
Thus, MxB influences HIV-1 replication in two cellular compartments and, accordingly, at the subsequent stages of virus replication cycle: “uncoating” in the cytoplasm, import of the pre-integration complex to the nucleus, and integration of the provirus into the cell genome. Clinical HIV-1 isolates carrying H87Q and G116A mutations in the capsid protein have lower sensitivity to MxB-mediated restriction and enhanced replicative activity compared to other circulating strains in [100, 101]. These data can be considered evidence in favor of the hypothesis of the inhibitory pressure of the MxB factor on HIV-1.
SCHLAFEN11 (SLFN11)
SLFN11 is a member of the Schlafen family of ISG-proteins regulating cell proliferation, immune response and viral replication in [102]. The involvement of SLFN11 in HIV restriction was shown in [19]. SLFN11 proved to have no effect on early stages of the cycle of retroviral infection, including reverse transcription, integration, and transcription. SLFN11 acts at the late stage of virus replication, selectively inhibiting the translation of viral proteins. SLFN11 binds tRNA and counteracts the changes in the cellular pool of tRNA caused by the presence of HIV. This is a new antiviral mechanism of innate immune response, where SLFN11 selectively inhibits the synthesis of viral proteins in HIV-infected cells.
The reduced translation of viral proteins under type-I IFN stimulation was observed 18 years before this discovery, as early as in 1994, but the regulation mechanism was not studied in [103]. SLFN11, under the conditions of type-I IFN stimulation, inhibits the translation of not only viral but also other codon-unoptimized transcripts in a cell in [104]; hence, this protein apparently should not be considered as a specific HIV restriction factor. Most likely, SLFN11 is involved in the total antiviral response of cells.
MARCH8 (c-MIR)
The involvement of MARCH8 (membrane-associated RING-CH8) protein in HIV-1 restriction has been shown lately in [105]. MARCH8 is an E3-ubiquitin ligase with a high level of expression in differentiated myeloid cells: macrophages and dendritic cells. At the moment of revealing the antiviral function of the protein, MARCH8 was known to reduce the expression of some cellular transmembrane proteins, in particular, MHC class II and TRAIL-R1 (TNF-related apoptosis-inducing ligand receptor 1) in [106, 107]. MARCH8 is supposed to recognize the three-dimensional structure of transmembrane domains of proteins but not specific motives; however, there is no reliable experimental evidence in favor of this fact.
Tada et al. have shown in [105] that the ectopic expression of MARCH8 in virus-producing cells does not influence the level of virus production but reduces the infectivity of viral particles. MARCH8 blocks the entry of viral envelope protein into virions by reducing the expression of Env at the cell surface, possibly by interacting with it. As a result, there is a substantial decrease in the efficiency of virus–cell fusion. At the moment, the exact strategy of HIV counteracting the MARCH8-mediated restriction HIV is unknown, however, it has been shown that accessory proteins Vpr, Vpu, and Nef do not interfere with MARCH8 in [105].
SERINC3 AND SERINC5
There was no understanding of how viral protein Nef increases the infectivity of viral particles in [106, 107]. Usami et al. in [108] supposed that Nef counteracted a certain restriction factor by decreasing its density at the cell surface and preventing incorporation into virions. The proteomic analysis of Nef+ HIV-1 and Nef- HIV-1 virions showed that they possessed membrane proteins SERINC3 and SERINC5 (SERin INCorporator). The incorporation of SERINC3 and SERINC5 into HIV virions actually decreased their ability to infect host cells in [108].
The SERINC family consists of five proteins characterized by the presence of 10–11 transmembrane domains. SERINC proteins are involved in the biosynthesis of sphingolipids and phosphatidylserine in [109]. However, only SERINC3 and SERINC5 are considered to be HIV-1 restriction factors, and SERINC5 has a more marked antiviral effect in [110, 111]. The supposed mechanism of action of SERINC5 consists in the formation of oligomers on the membranes of viral particles, which leads to the lower efficiency of viral–cell membrane fusion in [112]. In addition, the penetration of SERINC5 into viral particles results in their enhanced sensitivity to the broadly neutralizing antibodies recognizing conservative domain gp41 of the envelope protein in [112, 113].
The HIV-1 accessory protein Nef effectively removes SERINC3 and SERINC5 from the cell surface, preventing their penetration into virions in [108, 111, 114]. The mechanism of negative regulation of SERINC3 and SERINC5 is analogous to reduction of CD4 expression at the cell surface: in each case, Nef uses the cellular transport system to provide endolysosomal degradation of target cell proteins. The sensitivity of SERINC5 to Nef-mediated degradation is determined via the structure of its intracellular domain ICL4 (intracellular loop 4). With the substitution of a resistant variant of ICL4 for the Nef-sensitive variant, SERINC5 becomes resistant to Nef, which no longer prevents SERINC5-induced HIV restriction in [115].
BST-2 (CD317, TETHERIN)
One of the causes of why HIV-1 group M has become widespread in humans and became pandemic was the high resistance to cellular factor BST-2 acquired as a result of evolution and adaptation of viral protein Vpu to BST-2-induced restriction in [116]. Other HIV-1 groups, as well as HIV-2 and SIV, have not evolved in this direction and are supposed to protect themselves from the effect of BST-2 via a more ancient and less effective method: using accessory protein Nef in [117–120]. Accessory viral protein Vpu is encoded in the HIV-1 genome without being in HIV-2 and in most SIV strains. vpu-defective virus Δvpu-HIV-1 has definite characteristics that differentiate it from the wild-type virus: the smaller number of “spikes” at the virion surface and the agglomeration of viral particle “clusters” near the cell surface in [121–123]. The decreased number of spikes at the viral envelope is due to the interaction between CD4 molecule and viral envelope protein gp160 in the Golgi apparatus. Vpu binds to CD4, thereby releasing the envelope protein that can be easily transported to the cell plasma membrane in [124].
It was more difficult to understand the cause of the Δvpu-HIV-1 viral particle clustering near the cell surface. The site-directed mutagenesis in the vpu gene has shown that the release of gp160 and the clustering of virions near the cell surface involve different Vpu domains. Thus, the assembly of Δvpu-HIV-1 virions was observed only in particular types of cells and could be intensified with type-I IFN in [125, 126]. The Vpu-defective virus also proved more sensitive to the suppression of replication under the influence of IFN-α. In addition, these viral particles were mature and could be removed from the surface of infected cells via protease treatment. Consequently, there is a cellular protein capable of retaining Δvpu-HIV-1 virions on the cell surface, thereby preventing budding. According to data from electron microscopy, Δvpu-HIV-1 viral particles not only attach to the surface of infected cells but also “adhere” to each other in [23]. This means that the molecule involved in this process can be incorporated into a viral envelope and prevent virus distribution by retaining viral particles on the surface of infected cells. All the above gives grounds to believe that the Vpu protein originated and was established during the coevolution of HIV-1 with humans as a tool for counteracting IFN-mediated cell defense.
Neil et al. in [23] proposed the name “tetherin” (from “tether”: to tie, to restrict) for the cell surface protein responsible for this process. The specific molecule performing this function was identified by the microchip analysis of cell transcriptome before and after the stimulation with IFN-α. The candidates were BST-2 (bone marrow stromal antigen 2) and three IFN-induced transmembrane proteins: IFITM1, IFITM2, and IFITM3. The ability of BST-2 to “tether” the virus to the membrane was confirmed experimentally. BST-2 had no effect on the expression and processing of viral protein Gag, inhibiting only the release of formed viral particles from the cell.
BST-2 has a unique structure: the N-terminus with the cytoplasmic domain merges into the transmembrane domain; next there is an external coiled-coil domain and C-terminal glycosylphosphatidylinositol (GPI) anchor often referred to as second transmembrane domain in [127, 128] (Fig. 3). BST-2 is localized in lipid rafts of the plasma membrane, the trans-Golgi network, and endosomes in [127, 129, 130]. Two membrane-associated domains determine the ability of the protein to retain viral particles close to the membrane and bind them with each other: when the virions bud, one of the domains remains on the membrane, while the other is incorporated into the lipid envelope of the virus in [131–134] (Fig. 4). BST-2 functions as homodimers and homotetramers covalently bound via cysteine residues localized in the coiled-coil external domains of BST-2 in [129, 131, 135, 136] (Fig. 3).

The topology of BST-2, the interaction with Vpu, and the activation of the NF-κB pathway.

The scheme of BST-2-mediated retention of HIV-1 virions near the surface of infected cells.
The attachment of the virus to the membrane of infected cell reduces infection by the cell-free virus. HIV-1 is more effectively transmitted through intercellular junctions, especially through virological synapses attracting adhesion molecules, viral proteins, and cell receptors in the area of contact in [137, 138]. The question on the function of BST-2 in HIV cell-to-cell transmission is interesting and still open. There are contradictory data. The inhibitory effect of BST-2 on the cell-to-cell transmission of infection has been shown in [139–142]; contrariwise, BST-2 does not function as a restriction factor of HIV during its cell-to-cell transmission but even facilitates more effective infection of neighboring cells due to retaining viral particles in the infected/uninfected cell contact area in [143–145]. The development of methods for the quantitative measurement of cell-to-cell infection, making it possible to separate producer cells from infected cells in a single culture, can resolve this contradiction and elucidate the role of BST-2 in HIV-1 cell-to-cell transmission in [146, 147].
The cytoplasmic tail of BST-2 determines the protein involvement in intracellular signal transduction. BST-2 has been found among the proteins activating the NF-κB signaling pathway in [148]. Retention of the virus near the surface and activation of the NF-κB signaling pathway are two independent functions of the protein performed via different domains; at the same time, virion binding and BST-2 dimerization induce NF-κB-signaling in [149, 150]. TRAF2 and TRAF6 are attracted to the supposed TRAF-binding site at the BST-2 molecule, which leads to the activation of TAK1 and, accordingly, the triggering of the canonical NF-κB-signaling pathway, followed by the expression of proinflammatory cytokines IL-6, CXCL10, and IFN-β.
Accessory protein Vpu of the HIV-1 virus binds to the transmembrane domain of BST-2 in [151] and attracts SCF E3-ubiquitin ligase, which leads to ubiquitination and endolysosomal degradation of BST-2 in [151, 152]. SIV Nef protein effectively counteracts BST-2 in monkeys in [116, 117], while HIV Nef is not considered a factor interfering with BST-2 in humans.
METHODS FOR REVEALING NEW HIV RESTRICTION FACTORS
Most of the currently known (IFITM, BST-2, APOBEC3) HIV restriction factors have been found among ISGs. The triggering of IFN and NF-κB signaling pathways in the infected cell due to viral sensors (TLR, RLR, etc.) leads to the expression of proteins of cell protection against viral infection. It is not surprising that HIV restriction factors have been found among them.
One more efficient strategy for studying restriction factors consists in the search of partner proteins for accessory proteins of the virus. HIV is able to infect cells without expression of Vif, Vpr, Vpu, and Nef (Vpx in HIV-2); however, their presence in the viral genome is functionally justified. It is believed that the main function of accessory proteins is to counteract cell defense. The identification of SAMHD1 and SERINC3/5 restriction factors as the targets of Vpx and Nef, respectively, confirms this hypothesis.
The development of high-throughput sequencing techniques has opened up new prospects for searching restriction factors. Even now there is a large body of data accumulated as a result of siRNA-, shRNA-, and CRISPR/Cas9-screening for the search of restriction factors in [153] and HIV-1 replication factors in [154–158]. These data arrays still “wait in the wings”: they require systematization in [159] and interpretation in accordance with the results of experimental studies; but even now they stimulate and accelerate the search of new, clinically interesting cellular proteins involved in HIV infection.
ACKNOWLEDGMENTS
The work was supported by the Russian Science Foundation (Searching New HIV-1 Restriction Factors Affecting Viral Replication at Cell-to-Cell Transmission Settings via GeCKO Library Screening, project no. 15-15-00135) and the Russian Foundation for Basic Research (Role of KPNA1, CD82 and Other Cellular Factors Involved in HTLV-1 Replication and Selected after GeCKO Library Screening, project no. 18-34-00712).
COMPLIANCE WITH ETHICAL STANDARDS
The authors declare that they have no conflict of interest. This article does not contain any studies involving animals or human participants performed by any of the authors.
REFERENCES
Sharp P.M., Hahn B.H. 2011. Origins of HIV and the AIDS pandemic. Cold Spring Harb. Perspect. Med. 1 (1), a006841. https://doi.org/10.1101/cshperspect.a006841
Mourez T., Simon F., Plantier J.-C. 2013. Non-M variants of human immunodeficiency virus type 1. Clin. Microbiol. Rev. 26 (3), 448–461. https://doi.org/10.1128/CMR.00012-13
Vallari A., Holzmayer V., Harris B., Yamaguchi J., Ngansop C., Makamche F., Mbanya D., Kaptué L., Ndembi N., Gürtler L., Devare S., Brennan C.A. 2011. Confirmation of putative HIV-1 group P in Cameroon. J. Virol. 85 (3), 1403–1407. https://doi.org/10.1128/JVI.02005-10
Vallari A., Bodelle P., Ngansop C., Makamche F., Ndembi N., Mbanya D., Kaptué L., Gürtler L.G., McArthur C.P., Devare S.G., Simon F. 2010. Four new HIV-1 group N isolates from Cameroon: Prevalence continues to be low. AIDS Res. Hum. Retrov. 26 (9806), 109–115. https://doi.org/10.1016/S0140-6736(11)61457-8
Plantier J.C., Leoz M., Dickerson J.E., De Oliveira F., Cordonnier F., Lemée V., Damond F., Robertson D.L., Simon F. 2009. A new human immunodeficiency virus derived from gorillas. Nat. Med. 15 (8), 871–872. https://doi.org/10.1038/nm.2016
Soliman M., Srikrishna G., Balagopal A. 2017. Mechanisms of HIV-1 control. Curr. HIV/AIDS Rep. 14 (3), 101–109. https://doi.org/10.1007/s11904-017-0357-9
Karamov E.V., Petrov R.V. 2011. Sovereign immunity: 2. Cellular factors of antiretroviral defense: Tetherin, APOBEC3 family, cellular microRNA. CRISPR/Cas systems of prokaryotes. Fiziol. Patol. Immun. Sistemy. 15 (4), 3–23.
Karamov E.V., Petrov R.V. 2011. Sovereign immunity: 1. Specfic features of antiretroviral immune response. Cellular factors interacting with retroviral capsid proteins: TRIM5, cyclophilin. Fiziol. Patol. Immun. Sistemy. 15 (3), 3–22.
Brass A.L., Huang I.-C., Benita Y., John S.P., Krishnan M.N., Feeley E.M., Ryan B.J., Weyer J.L., van der Weyden L., Fikrig E., Adams D.J., Xavier R.J., Farzan M., Elledge S.J. 2009. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell. 139 (7), 1243–1254. https://doi.org/10.1016/j.cell.2009.12.017
Bishop K.N., Verma M., Kim E.-Y., Wolinsky S.M., Malim M.H. 2008. APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathog. 4 (12), e1000231. https://doi.org/10.1371/journal.ppat.1000231
Sheehy A.M., Gaddis N.C., Choi J.D., Malim M.H. 2002. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 418 (6898), 646–650. https://doi.org/10.1038/nature00939
Harris R.S., Bishop K.N., Sheehy A.M., Craig H.M., Petersen-Mahrt S.K., Watt I.N., Neuberger M.S., Malim M.H. 2003. DNA deamination mediates innate immunity to retroviral infection. Cell. 113 (6), 803–809. http://www.ncbi.nlm.nih.gov/pubmed/12809610.
Stremlau M., Owens C.M., Perron M.J., Kiessling M., Autissier P., Sodroski J. 2004. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature. 427 (6977), 848–853. https://doi.org/10.1038/nature02343
Sayah D.M., Sokolskaja E., Berthoux L., Luban J. 2004. Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature. 430 (6999), 569–573. https://doi.org/10.1038/nature02777
Stremlau M., Perron M., Lee M., Li Y., Song B., Javanbakht H., Diaz-Griffero F., Anderson D.J., Sundquist W.I., Sodroski J. 2006. Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5alpha restriction factor. Proc. Natl. Acad. Sci. U. S. A. 103 (14), 5514–5519. https://doi.org/10.1073/pnas.0509996103
Kane M., Yadav S.S., Bitzegeio J., Kutluay S.B., Zang T., Wilson S.J., Schoggins J.W., Rice C.M., Yamashita M., Hatziioannou T., Bieniasz P.D. 2013. MX2 is an interferon-induced inhibitor of HIV-1 infection. Nature. 502 (7472), 563–566. https://doi.org/10.1038/nature12653
Goujon C., Moncorgé O., Bauby H., Doyle T., Ward C.C., Schaller T., Hué S., Barclay W.S., Schulz R., Malim M.H. 2013. Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature. 502 (7472), 559–562. https://doi.org/10.1038/nature12542
Liu Z., Pan Q., Ding S., Qian J., Xu F., Zhou J., Cen S., Guo F., Liang C. 2013. The interferon-inducible MxB protein inhibits HIV-1 infection. Cell Host Microbe. 14 (4), 398–410. https://doi.org/10.1016/j.chom.2013.08.015
Li M., Kao E., Gao X., Sandig H., Limmer K., Pavon-Eternod M., Jones T.E., Landry S., Pan T., Weitzman M.D., David M. 2012. Codon-usage-based inhibition of HIV protein synthesis by human schlafen 11. Nature. 491 (7422), 125–128. https://doi.org/10.1038/nature11433
Hrecka K., Hao C., Gierszewska M., Swanson S.K., Kesik-Brodacka M., Srivastava S., Florens L., Washburn M.P., Skowronski J. 2011. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature. 474 (7353), 658–661. https://doi.org/10.1038/nature10195
Berger A., Sommer A.F.R., Zwarg J., Hamdorf M., Welzel K., Esly N., Panitz S., Reuter A., Ramos I., Jatiani A., Mulder L.C.F., Fernandez-Sesma A., Rutsch F., Simon V., König R., Flory E. 2011. SAMHD1-deficient CD14+ cells from individuals with Aicardi-Goutières syndrome are highly susceptible to HIV-1 infection. PLoS Pathog. 7 (12), e1002425. https://doi.org/10.1371/journal.ppat.1002425
Goldstone D.C., Ennis-Adeniran V., Hedden J.J., Groom H.C.T., Rice G.I., Christodoulou E., Walker P.A., Kelly G., Haire L.F., Yap M.W., de Carvalho L.P.S., Stoye J.P., Crow Y.J., Taylor I.A., Webb M. 2011. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature. 480 (7377), 379–382. https://doi.org/10.1038/nature10623
Neil S.J.D., Zang T., Bieniasz P.D. 2008. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature. 451 (7177), 425–430. https://doi.org/10.1038/nature06553
Sauter D., Specht A., Kirchhoff F. 2010. Tetherin: Holding on and letting go. Cell. 141 (3), 392–398. https://doi.org/10.1016/j.cell.2010.04.022
Francis M.L., Meltzer M.S. 1993. Induction of IFN-alpha by HIV-1 in monocyte-enriched PBMC requires gp120–CD4 interaction but not virus replication. J. Immunol. 151 (4), 2208–2216. http://www.ncbi.nlm. nih.gov/pubmed/8345204.
Jaffe E.A., Armellino D., Lam G., Cordon-Cardo C., Murray H.W., Evans R.L. 1989. IFN-gamma and IFN-alpha induce the expression and synthesis of Leu 13 antigen by cultured human endothelial cells. J. Immunol. 143 (12), 3961–3966. http://www.ncbi.nlm. nih.gov/pubmed/2512344.
Lu J., Pan Q., Rong L., He W., Liu S.-L., Liang C. 2011. The IFITM proteins inhibit HIV-1 infection. J. Virol. 85 (5), 2126–2137. https://doi.org/10.1128/JVI.01531-10
Weston S., Czieso S., White I.J., Smith S.E., Wash R.S., Diaz-Soria C., Kellam P., Marsh M. 2016. Alphavirus restriction by IFITM proteins. Traffic. 17 (9), 997–1013. https://doi.org/10.1111/tra.12416
Feeley E.M., Sims J.S., John S.P., Chin C.R., Pertel T., Chen L.-M., Gaiha G.D., Ryan B.J., Donis R.O., Elledge S.J., Brass A.L. 2011. IFITM3 inhibits influenza A virus infection by preventing cytosolic entry. PLoS Pathog. 7 (10), e1002337. https://doi.org/10.1371/journal.ppat.1002337
Li K., Markosyan R.M., Zheng Y.-M., Golfetto O., Bungart B., Li M., Ding S., He Y., Liang C., Lee J.C., Gratton E., Cohen F.S., Liu S.-L. 2013. IFITM proteins restrict viral membrane hemifusion. PLoS Pathog. 9 (1), e1003124. https://doi.org/10.1371/journal.ppat.1003124
Desai T.M., Marin M., Chin C.R., Savidis G., Brass A.L., Melikyan G.B. 2014. IFITM3 restricts influenza A virus entry by blocking the formation of fusion pores following virus-endosome hemifusion. PLoS Pathog. 10 (4), e1004048. https://doi.org/10.1371/journal.ppat.1004048
Amini-Bavil-Olyaee S., Choi Y.J., Lee J.H., Shi M., Huang I.-C., Farzan M., Jung J.U. 2013. The antiviral effector IFITM3 disrupts intracellular cholesterol homeostasis to block viral entry. Cell Host Microbe. 13 (4), 452–464. https://doi.org/10.1016/j.chom.2013.03.006
Compton A.A., Bruel T., Porrot F., Mallet A., Sachse M., Euvrard M., Liang C., Casartelli N., Schwartz O. 2014. IFITM proteins incorporated into HIV-1 virions impair viral fusion and spread. Cell Host Microbe. 16 (6), 736–747. https://doi.org/10.1016/j.chom.2014.11.001
Foster T.L., Wilson H., Iyer S.S., Coss K., Doores K., Smith S., Kellam P., Finzi A., Borrow P., Hahn B.H., Neil S.J.D. 2016. Resistance of transmitted founder HIV-1 to IFITM-mediated restriction. Cell Host Microbe. 20 (4), 429–442. https://doi.org/10.1016/j.chom.2016.08.006
Yu J., Li M., Wilkins J., Ding S., Swartz T.H., Esposito A.M., Zheng Y.-M., Freed E.O., Liang C., Chen B.K., Liu S.-L. 2015. IFITM proteins restrict HIV-1 infection by antagonizing the envelope glycoprotein. Cell Rep. 13 (1), 145–156. https://doi.org/10.1016/j.celrep.2015.08.055
Rajsbaum R., García-Sastre A., Versteeg G.A. 2014. TRIMmunity: The roles of the TRIM E3-ubiquitin ligase family in innate antiviral immunity. J. Mol. Biol. 426 (6), 1265–1284. https://doi.org/10.1016/j.jmb.2013.12.005
Versteeg G.A., Benke S., García-Sastre A., Rajsbaum R. 2014. InTRIMsic immunity: Positive and negative regulation of immune signaling by tripartite motif proteins. Cytokine Growth Factor Rev. 25 (5), 563–576. https://doi.org/10.1016/j.cytogfr.2014.08.001
Ozato K., Shin D.-M., Chang T.-H., Morse H.C. 2008. TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 8 (11), 849–860. https://doi.org/10.1038/nri2413
Uchil P.D., Hinz A., Siegel S., Coenen-Stass A., Pertel T., Luban J., Mothes W. 2013. TRIM protein-mediated regulation of inflammatory and innate immune signaling and its association with antiretroviral activity. J. Virol. 87 (1), 257–272. https://doi.org/10.1128/JVI.01804-12
Uchil P.D., Quinlan B.D., Chan W.-T., Luna J.M., Mothes W. 2008. TRIM E3 ligases interfere with early and late stages of the retroviral life cycle. PLoS Pathog. 4 (2), e16. https://doi.org/10.1371/journal.ppat.0040016
Versteeg G.A., Rajsbaum R., Sánchez-Aparicio M.T., Maestre A.M., Valdiviezo J., Shi M., Inn K.-S., Fernandez-Sesma A., Jung J., García-Sastre A. 2013. The E3-Ligase TRIM family of proteins regulates signaling pathways triggered by innate immune pattern-recognition receptors. Immunity. 38 (2), 384–398. https://doi.org/10.1016/j.immuni.2012.11.013
Reymond A., Meroni G., Fantozzi A., Merla G., Cairo S., Luzi L., Riganelli D., Zanaria E., Messali S., Cainarca S., Guffanti A., Minucci S., Pelicci P.G., Ballabio A. 2001. The tripartite motif family identifies cell compartments. EMBO J. 20 (9), 2140–2151. https://doi.org/10.1093/emboj/20.9.2140
Esposito D., Koliopoulos M.G., Rittinger K. 2017. Structural determinants of TRIM protein function. Biochem. Soc. Trans. 45 (1), 183–191. https://doi.org/10.1042/BST20160325
Napolitano L.M., Meroni G. 2012. TRIM family: Pleiotropy and diversification through homomultimer and heteromultimer formation. IUBMB Life. 64 (1), 64–71. https://doi.org/10.1002/iub.580
Sardiello M., Cairo S., Fontanella B., Ballabio A., Meroni G. 2008. Genomic analysis of the TRIM family reveals two groups of genes with distinct evolutionary properties. BMC Evol. Biol. 8 (1), 225. https://doi.org/10.1186/1471-2148-8-225
Han K., Lou D.I., Sawyer S.L. 2011. Identification of a genomic reservoir for new TRIM genes in primate genomes. PLoS Genet. 7 (12), e1002388. https://doi.org/10.1371/journal.pgen.1002388
Nisole S., Lynch C., Stoye J.P., Yap M.W. 2004. A Trim5-cyclophilin A fusion protein found in owl monkey kidney cells can restrict HIV-1. Proc. Natl. Acad. Sci. U. S. A. 101 (36), 13324–13328. https://doi.org/10.1073/pnas.0404640101
Lamichhane R., Mukherjee S., Smolin N., Pauszek R.F., Bradley M., Sastri J., Robia S.L., Millar D., Campbell E.M. 2017. Dynamic conformational changes in the rhesus TRIM5α dimer dictate the potency of HIV-1 restriction. Virology. 500, 161–168. https://doi.org/10.1016/j.virol.2016.10.003
Li Y.-L., Chandrasekaran V., Carter S.D., Woodward C.L., Christensen D.E., Dryden K.A., Pornillos O., Yeager M., Ganser-Pornillos B.K., Jensen G.J., Sundquist W.I. 2016. Primate TRIM5 proteins form hexagonal nets on HIV-1 capsids. Elife. 5. https://doi.org/10.7554/eLife.16269
Sastri J., Campbell E.M. 2011. Recent insights into the mechanism and consequences of TRIM5α retroviral restriction. AIDS Res. Hum. Retrov. 27 (3), 231–238. https://doi.org/10.1089/AID.2010.0367
Pertel T., Hausmann S., Morger D., Züger S., Guerra J., Lascano J., Reinhard C., Santoni F.A., Uchil P.D., Chatel L., Bisiaux A., Albert M.L., Strambio-De-Castillia C., Mothes W., Pizzato M., Grütter M.G., Luban J. 2011. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature. 472 (7343), 361–365. https://doi.org/10.1038/nature09976
Wagner J.M., Christensen D.E., Bhattacharya A., Dawidziak D.M., Roganowicz M.D., Wan Y., Pumroy R.A., Demeler B., Ivanov D.N., Ganser-Pornillos B.K., Sundquist W.I., Pornillos O. 2018. General model for retroviral capsid pattern recognition by TRIM5 proteins. J. Virol. 92 (4), e01563-17. https://doi.org/10.1128/JVI.01563-17
Yudina Z., Roa A., Johnson R., Biris N., de Souza Aranha Vieira D.A., Tsiperson V., Reszka N., Taylor A.B., Hart P.J., Demeler B., Diaz-Griffero F., Ivanov D.N. 2015. RING dimerization links higher-order assembly of TRIM5α to synthesis of K63-linked polyubiquitin. Cell Rep. 12(5), 788–797. https://doi.org/10.1016/j.celrep.2015.06.072
van Tol S., Hage A., Giraldo M., Bharaj P., Rajsbaum R. 2017. The TRIMendous role of TRIMs in virus–host interactions. Vaccines. 5 (3), e23. https://doi.org/10.3390/vaccines5030023
Rold C.J., Aiken C. 2008. Proteasomal degradation of TRIM5α during retrovirus restriction. PLoS Pathog. 4 (5), e1000074. https://doi.org/10.1371/journal.ppat.1000074
Kutluay S.B., Perez-Caballero D., Bieniasz P.D. 2013. Fates of retroviral core components during unrestricted and TRIM5-restricted infection. PLoS Pathog. 9 (3), e1003214. https://doi.org/10.1371/journal.ppat.1003214
Yap M.W., Nisole S., Stoye J.P. 2005. A single amino acid change in the SPRY domain of human Trim5alpha leads to HIV-1 restriction. Curr. Biol. 15 (1), 73–78. https://doi.org/10.1016/j.cub.2004.12.042
Barr S.D., Smiley J.R., Bushman F.D. 2008. The interferon response inhibits HIV particle production by induction of TRIM22. PLoS Pathog. 4 (2), e1000007. https://doi.org/10.1371/journal.ppat.1000007
Singh R., Gaiha G., Werner L., McKim K., Mlisana K., Luban J., Walker B.D., Karim S.S.A., Brass A.L., Ndung’u T., CAPRISA Acute Infection Study Team. 2011. Association of TRIM22 with the type 1 interferon response and viral control during primary HIV-1 infection. J. Virol. 85 (1), 208–216. https://doi.org/10.1128/JVI.01810-10
Kajaste-Rudnitski A., Marelli S.S., Pultrone C., Pertel T., Uchil P.D., Mechti N., Mothes W., Poli G., Luban J., Vicenzi E. 2011. TRIM22 inhibits HIV-1 transcription independently of its E3 ubiquitin ligase activity, Tat, and NF-kappaB-responsive long terminal repeat elements. J. Virol. 85 (10), 5183–5196. https://doi.org/10.1128/JVI.02302-10
Turrini F., Marelli S., Kajaste-Rudnitski A., Lusic M., Van Lint C., Das A.T., Harwig A., Berkhout B., Vicenzi E. 2015. HIV-1 transcriptional silencing caused by TRIM22 inhibition of Sp1 binding to the viral promoter. Retrovirology. 12 (1), 104. https://doi.org/10.1186/s12977-015-0230-0
Deeks S.G., Lewin S.R., Ross A.L., Ananworanich J., Benkirane M., Cannon P., Chomont N., Douek D., Lifson J.D., Lo Y.-R., Kuritzkes D., Margolis D., Mellors J., Persaud D., Tucker J.D., Barre-Sinoussi F., International AIDS Society Towards a Cure Working Group. 2016. International AIDS Society global scientific strategy: Towards an HIV cure 2016. Nat. Med. 22 (8), 839–850. https://doi.org/10.1038/nm.4108
Yuan T., Yao W., Huang F., Sun B., Yang R. 2014. The human antiviral factor TRIM11 is under the regulation of HIV-1 Vpr. PLoS One. 9 (8), e104269. https://doi.org/10.1371/journal.pone.0104269
Laguette N., Sobhian B., Casartelli N., Ringeard M., Chable-Bessia C., Ségéral E., Yatim A., Emiliani S., Schwartz O., Benkirane M. 2011. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature. 474 (7353), 654–657. https://doi.org/10.1038/nature10117
Descours B., Cribier A., Chable-Bessia C., Ayinde D., Rice G., Crow Y., Yatim A., Schwartz O., Laguette N., Benkirane M. 2012. SAMHD1 restricts HIV-1 reverse transcription in quiescent CD4+ T-cells. Retrovirology. 9 (1), 87. https://doi.org/10.1186/1742-4690-9-87
Ji X., Tang C., Zhao Q., Wang W., Xiong Y. 2014. Structural basis of cellular dNTP regulation by SAMHD1. Proc. Natl. Acad. Sci. U. S. A. 111 (41), E4305–E4314. https://doi.org/10.1073/pnas.1412289111
Ji X., Wu Y., Yan J., Mehrens J., Yang H., DeLucia M., Hao C., Gronenborn A.M., Skowronski J., Ahn J., Xiong Y. 2013. Mechanism of allosteric activation of SAMHD1 by dGTP. Nat. Struct. Mol. Biol. 20 (11), 1304–1309. https://doi.org/10.1038/nsmb.2692
Hansen E.C., Seamon K.J., Cravens S.L., Stivers J.T. 2014. GTP activator and dNTP substrates of HIV-1 restriction factor SAMHD1 generate a long-lived activated state. Proc. Natl. Acad. Sci. U. S. A. 111 (18), E1843–E1851. https://doi.org/10.1073/pnas.1401706111
Amie S.M., Bambara R.A., Kim B. 2013. GTP is the primary activator of the anti-HIV restriction factor SAMHD1. J. Biol. Chem. 288 (35), 25001–25006. https://doi.org/10.1074/jbc.C113.493619
Yan J., Kaur S., DeLucia M., Hao C., Mehrens J., Wang C., Golczak M., Palczewski K., Gronenborn A.M., Ahn J., Skowronski J. 2013. Tetramerization of SAMHD1 is required for biological activity and inhibition of HIV infection. J. Biol. Chem. 288 (15), 10406–10417. https://doi.org/10.1074/jbc.M112.443796
Beloglazova N., Flick R., Tchigvintsev A., Brown G., Popovic A., Nocek B., Yakunin A.F. 2013. Nuclease activity of the human SAMHD1 protein implicated in the Aicardi-Goutieres syndrome and HIV-1 restriction. J. Biol. Chem. 288 (12), 8101–8110. https://doi.org/10.1074/jbc.M112.431148
Ryoo J., Choi J., Oh C., Kim S., Seo M., Kim S.-Y., Seo D., Kim J., White T.E., Brandariz-Nuñez A., Diaz-Griffero F., Yun C.-H., Hollenbaugh J.A., Kim B., Baek D., Ahn K. 2014. The ribonuclease activity of SAMHD1 is required for HIV-1 restriction. Nat. Med. 20 (8), 936–941. https://doi.org/10.1038/nm.3626
Seamon K.J., Sun Z., Shlyakhtenko L.S., Lyubchenko Y.L., Stivers J.T. 2015. SAMHD1 is a single-stranded nucleic acid binding protein with no active site-associated nuclease activity. Nucleic Acids Res. 43 (13), 6486–6499. https://doi.org/10.1093/nar/gkv633
Antonucci J.M., St. Gelais C., de Silva S., Yount J.S., Tang C., Ji X., Shepard C., Xiong Y., Kim B., Wu L. 2016. SAMHD1-mediated HIV-1 restriction in cells does not involve ribonuclease activity. Nat. Med. 22 (10), 1072–1074. https://doi.org/10.1038/nm.4163
Ryoo J., Hwang S.-Y., Choi J., Oh C., Ahn K. 2016. Reply to SAMHD1-mediated HIV-1 restriction in cells does not involve ribonuclease activity. Nat. Med. 22 (10), 1074–1075. https://doi.org/10.1038/nm.4164
Ahn J., Hao C., Yan J., DeLucia M., Mehrens J., Wang C., Gronenborn A.M., Skowronski J. 2012. HIV/simian immunodeficiency virus (SIV) accessory virulence factor Vpx loads the host cell restriction factor SAMHD1 onto the E3 ubiquitin ligase complex CRL4DCAF1. J. Biol. Chem. 287 (15), 12550–12558. https://doi.org/10.1074/jbc.M112.340711
Wei W., Guo H., Han X., Liu X., Zhou X., Zhang W., Yu X.-F. 2012. A novel DCAF1-binding motif required for Vpx-mediated degradation of nuclear SAMHD1 and Vpr-induced G2 arrest. Cell. Microbiol. 14 (11), 1745–1756. https://doi.org/10.1111/j.1462-5822.2012.01835.x
Kyei G.B., Cheng X., Ramani R., Ratner L. 2015. Cyclin L2 is a critical HIV dependency factor in macrophages that controls SAMHD1 abundance. Cell Host Microbe. 17 (1), 98–106. https://doi.org/10.1016/j.chom.2014.11.009
Jarmuz A., Chester A., Bayliss J., Gisbourne J., Dunham I., Scott J., Navaratnam N. 2002. An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22. Genomics. 79 (3), 285–296. https://doi.org/10.1006/geno.2002.6718
Harris R.S., Petersen-Mahrt S.K., Neuberger M.S. 2002. RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Mol. Cell. 10 (5), 1247–1253. http://www.ncbi.nlm.nih.gov/pubmed/ 12453430.
Refsland E.W., Stenglein M.D., Shindo K., Albin J.S., Brown W.L., Harris R.S. 2010. Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: Implications for HIV-1 restriction. Nucleic Acids Res. 38 (13), 4274–4284. https://doi.org/10.1093/nar/gkq174
Desimmie B.A., Delviks-Frankenberrry K.A., Burdick R.C., Qi D., Izumi T., Pathak V.K. 2014. Multiple APOBEC3 restriction factors for HIV-1 and one Vif to rule them all. J. Mol. Biol. 426 (6), 1220–1245. https://doi.org/10.1016/j.jmb.2013.10.033
Mangeat B., Turelli P., Caron G., Friedli M., Perrin L., Trono D. 2003. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 424 (6944), 99–103. https://doi.org/10.1038/nature01709
Mariani R., Chen D., Schröfelbauer B., Navarro F., König R., Bollman B., Münk C., Nymark-McMahon H., Landau N.R. 2003. Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell. 114 (1), 21–31. http://www.ncbi.nlm.nih.gov/pubmed/12859895.
Suspène R., Rusniok C., Vartanian J.-P., Wain-Hobson S. 2006. Twin gradients in APOBEC3 edited HIV-1 DNA reflect the dynamics of lentiviral replication. Nucleic Acids Res. 34 (17), 4677–4684. https://doi.org/10.1093/nar/gkl555
Yu Q., König R., Pillai S., Chiles K., Kearney M., Palmer S., Richman D., Coffin J.M., Landau N.R. 2004. Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nat. Struct. Mol. Biol. 11 (5), 435–442. https://doi.org/10.1038/nsmb758
Holmes R.K., Malim M.H., Bishop K.N. 2007. APOBEC-mediated viral restriction: Not simply editing? Trends Biochem. Sci. 32 (3), 118–128. https://doi.org/10.1016/j.tibs.2007.01.004
Albin J.S., Brown W.L., Harris R.S. 2014. Catalytic activity of APOBEC3F is required for efficient restriction of Vif-deficient human immunodeficiency virus. Virology. 450–451, 49–54. https://doi.org/10.1016/j.virol.2013.11.041
Browne E.P., Allers C., Landau N.R. 2009. Restriction of HIV-1 by APOBEC3G is cytidine deaminase-dependent. Virology. 387 (2), 313–321. https://doi.org/10.1016/j.virol.2009.02.026
Yu X., Yu Y., Liu B., Luo K., Kong W., Mao P., Yu X.-F. 2003. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif–Cul5–SCF complex. Science. 302 (5647), 1056–1060. https://doi.org/10.1126/science.1089591
Sheehy A.M., Gaddis N.C., Malim M.H. 2003. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat. Med. 9 (11), 1404–1407. https://doi.org/10.1038/nm945
Kim E.-Y., Lorenzo-Redondo R., Little S.J., Chung Y.-S., Phalora P.K., Maljkovic Berry I., Archer J., Penugonda S., Fischer W., Richman D.D., Bhattacharya T., Malim M.H., Wolinsky S.M. 2014. Human APOBEC3 induced mutation of human immunodeficiency virus type-1 contributes to adaptation and evolution in natural infection. PLoS Pathog. 10 (7), e1004281. https://doi.org/10.1371/journal.ppat.1004281
Fourati S., Malet I., Binka M., Boukobza S., Wirden M., Sayon S., Simon A., Katlama C., Simon V., Calvez V., Marcelin A.-G. 2010. Partially active HIV-1 Vif alleles facilitate viral escape from specific antiretrovirals. AIDS. 24 (15), 2313–2321. https://doi.org/10.1097/QAD.0b013e32833e515a
Dicks M.D.J., Goujon C., Pollpeter D., Betancor G., Apolonia L., Bergeron J.R.C., Malim M.H. 2016. Oligomerization requirements for MX2-mediated suppression of HIV-1 infection. J. Virol. 90 (1), 22–32. https://doi.org/10.1128/JVI.02247-15
Haller O., Kochs G. 2011. Human MxA protein: An interferon-induced dynamin-like GTPase with broad antiviral activity. J. Interf. Cytokine Res. 31 (1), 79–87. https://doi.org/10.1089/jir.2010.0076
Matreyek K.A., Wang W., Serrao E., Singh P.K., Levin H.L., Engelman A. 2014. Host and viral determinants for MxB restriction of HIV-1 infection. Retrovirology. 11 (1), 90. https://doi.org/10.1186/s12977-014-0090-z
Fribourgh J.L., Nguyen H.C., Matreyek K.A., Alvarez F.J.D., Summers B.J., Dewdney T.G., Aiken C., Zhang P., Engelman A., Xiong Y. 2014. Structural insight into HIV-1 restriction by MxB. Cell Host Microbe. 16 (5), 627–638. https://doi.org/10.1016/j.chom.2014.09.021
Goujon C., Greenbury R.A., Papaioannou S., Doyle T., Malim M.H. 2015. A triple-arginine motif in the amino-terminal domain and oligomerization are required for HIV-1 inhibition by human MX2. J. Virol. 89 (8), 4676–4680. https://doi.org/10.1128/JVI.00169-15
Fricke T., White T.E., Schulte B., de Souza Aranha Vieira D.A., Dharan A., Campbell E.M., Brandariz-Nuñez A., Diaz-Griffero F. 2014. MxB binds to the HIV-1 core and prevents the uncoating process of HIV-1. Retrovirology. 11 (1), 68. https://doi.org/10.1186/s12977-014-0068-x
Nakayama E.E., Saito A., Sultana T., Jin Z., Nohata K., Shibata M., Hosoi M., Motomura K., Shioda T., Sangkitporn S., Loket R., Saeng-Aroon S. 2018. Naturally occurring mutations in HIV-1 CRF01_AE capsid affect viral sensitivity to restriction factors. AIDS Res. Hum. Retrov. 34 (4), 382–392. https://doi.org/10.1089/AID.2017.0212
Wei W., Guo H., Ma M., Markham R., Yu X.-F. 2016. Accumulation of MxB/Mx2-resistant HIV-1 capsid variants during expansion of the HIV-1 epidemic in human populations. EBioMedicine. 8, 230–236. https://doi.org/10.1016/j.ebiom.2016.04.020
Mavrommatis E., Fish E.N., Platanias L.C. 2013. The schlafen family of proteins and their regulation by interferons. J. Interferon Cytokine Res. 33 (4), 206–210. https://doi.org/10.1089/jir.2012.0133
Coccia E.M., Krust B., Hovanessian A.G. 1994. Specific inhibition of viral protein synthesis in HIV-infected cells in response to interferon treatment. J. Biol. Chem. 269 (37), 23087–23094. http://www.ncbi. nlm.nih.gov/pubmed/7521875.
Stabell A.C., Hawkins J., Li M., Gao X., David M., Press W.H., Sawyer S.L. 2016. Non-human primate schlafen11 inhibits production of both host and viral proteins. PLOS Pathog. 12 (12), e1006066. https://doi.org/10.1371/journal.ppat.1006066
Tada T., Zhang Y., Koyama T., Tobiume M., Tsunetsugu-Yokota Y., Yamaoka S., Fujita H., Tokunaga K. 2015. MARCH8 inhibits HIV-1 infection by reducing virion incorporation of envelope glycoproteins. Nat. Med. 21 (12), 1502–1507. https://doi.org/10.1038/nm.3956
Ohmura-Hoshino M., Matsuki Y., Aoki M., Goto E., Mito M., Uematsu M., Kakiuchi T., Hotta H., Ishido S. 2006. Inhibition of MHC class II expression and immune responses by c-MIR. J. Immunol. 177 (1), 341–354. http://www.ncbi.nlm.nih.gov/pubmed/16785530.
van de Kooij B., Verbrugge I., de Vries E., Gijsen M., Montserrat V., Maas C., Neefjes J., Borst J. 2013. Ubiquitination by the membrane-associated RING-CH-8 (MARCH-8. ligase controls steady-state cell surface expression of tumor necrosis factor-related apoptosis inducing ligand (TRAIL. receptor 1. J. Biol. Chem. 288 (9), 6617–6628. https://doi.org/10.1074/jbc.M112.448209
Usami Y., Wu Y., Göttlinger H.G. 2015. SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature. 526 (7572), 218–223. https://doi.org/10.1038/nature15400
Inuzuka M., Hayakawa M., Ingi T. 2005. Serinc, an activity-regulated protein family, incorporates serine into membrane lipid synthesis. J. Biol. Chem. 280 (42), 35776–35783. https://doi.org/10.1074/jbc.M505712200
Zhang X., Zhou T., Yang J., Lin Y., Shi J., Zhang X., Frabutt D.A., Zeng X., Li S., Venta P.J., Zheng Y.-H. 2017. Identification of SERINC5-001 as the predominant spliced isoform for HIV-1 restriction. J. Virol. 91 (10), e00137-17. https://doi.org/10.1128/JVI.00137-17
Rosa A., Chande A., Ziglio S., De Sanctis V., Bertorelli R., Goh S.L., McCauley S.M., Nowosielska A., Antonarakis S.E., Luban J., Santoni F.A., Pizzato M. 2015. HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature. 526 (7572), 212–217. https://doi.org/10.1038/nature15399
Sood C., Marin M., Chande A., Pizzato M., Meli-kyan G.B. 2017. SERINC5 protein inhibits HIV-1 fusion pore formation by promoting functional inactivation of envelope glycoproteins. J. Biol. Chem. 292 (14), 6014–6026. https://doi.org/10.1074/jbc.M117.777714
Beitari S., Ding S., Pan Q., Finzi A., Liang C. 2017. Effect of HIV-1 Env on SERINC5 antagonism. J. Virol. 91 (4), e02214-16. https://doi.org/10.1128/JVI.02214-16
Matheson N.J., Sumner J., Wals K., Rapiteanu R., Weekes M.P., Vigan R., Weinelt J., Schindler M., Antrobus R., Costa A.S.H., Frezza C., Clish C.B., Neil S.J.D., Lehner P.J. 2015. Cell surface proteomic map of HIV infection reveals antagonism of amino acid metabolism by Vpu and Nef. Cell Host Microbe. 18 (4), 409–423. https://doi.org/10.1016/j.chom.2015.09.003
Dai W., Usami Y., Wu Y., Göttlinger H. 2018. A long cytoplasmic loop governs the sensitivity of the anti-viral host protein SERINC5 to HIV-1 Nef. Cell Rep. 22 (4), 869–875. https://doi.org/10.1016/j.celrep.2017.12.082
Sauter D., Schindler M., Specht A., Landford W.N., Münch J., Kim K.-A., Votteler J., Schubert U., Bibollet-Ruche F., Keele B.F., Takehisa J., Ogando Y., Ochsenbauer C., Kappes J.C., Ayouba A., et al. 2009. Tetherin-driven adaptation of Vpu and Nef function and the evolution of pandemic and nonpandemic HIV-1 strains. Cell Host Microbe. 6 (5), 409–421. https://doi.org/10.1016/j.chom.2009.10.004
Jia B., Serra-Moreno R., Neidermyer W., Rahmberg A., Mackey J., Fofana I. Ben, Johnson W.E., Westmoreland S., Evans D.T. 2009. Species-specific activity of SIV Nef and HIV-1 Vpu in overcoming restriction by Tetherin/BST2. PLoS Pathog. 5 (5), e1000429. https://doi.org/10.1371/journal.ppat.1000429
Sauter D., Hué S., Petit S.J., Plantier J.-C., Towers G.J., Kirchhoff F., Gupta R.K. 2011. HIV-1 group P is unable to antagonize human tetherin by Vpu, Env or Nef. Retrovirology. 8 (1), 103. https://doi.org/10.1186/1742-4690-8-103
Sauter D., Unterweger D., Vogl M., Usmani S.M., Heigele A., Kluge S.F., Hermkes E., Moll M., Barker E., Peeters M., Learn G.H., Bibollet-Ruche F., Fritz J.V., Fackler O.T., Hahn B.H., Kirchhoff F. 2012. Human tetherin exerts strong selection pressure on the HIV-1 group N Vpu protein. PLoS Pathog. 8 (12), e1003093. https://doi.org/10.1371/journal.ppat.1003093
Zhang F., Wilson S.J., Landford W.C., Virgen B., Gregory D., Johnson M.C., Munch J., Kirchhoff F., Bieniasz P.D., Hatziioannou T. 2009. Nef proteins from simian immunodeficiency viruses are tetherin antagonists. Cell Host Microbe. 6 (1), 54–67. https://doi.org/10.1016/j.chom.2009.05.008
Strebel K., Klimkait T., Maldarelli F., Martin M.A. 1989. Molecular and biochemical analyses of human immunodeficiency virus type 1 vpu protein. J. Virol. 63 (9), 3784–3791. http://www.ncbi.nlm.nih.gov/ pubmed/2788224.
Terwilliger E.F., Cohen E.A., Lu Y.C., Sodroski J.G., Haseltine W.A. 1989. Functional role of human immunodeficiency virus type 1 vpu. Proc. Natl. Acad. Sci. U. S. A. 86 (13), 5163–5167. http://www.ncbi.nlm.nih.gov/ pubmed/2472639.
Klimkait T., Strebel K., Hoggan M.D., Martin M.A., Orenstein J.M. 1990. The human immunodeficiency virus type 1-specific protein vpu is required for efficient virus maturation and release. J. Virol. 64 (2), 621–629. http://www.ncbi.nlm.nih.gov/pubmed/2404139.
Willey R.L., Maldarelli F., Martin M.A., Strebel K. 1992. Human immunodeficiency virus type 1 Vpu protein regulates the formation of intracellular gp160-CD4 complexes. J. Virol. 66 (1), 226–234. http://www.ncbi.nlm.nih.gov/pubmed/1727486.
Neil S.J.D., Eastman S.W., Jouvenet N., Bieniasz P.D. 2006. HIV-1 Vpu promotes release and prevents endocytosis of nascent retrovirus particles from the plasma membrane. PLoS Pathog. 2 (5), 354–367. https://doi.org/10.1371/journal.ppat.0020039
Sakai H., Tokunaga K., Kawamura M., Adachi A. 1995. Function of human immunodeficiency virus type 1 Vpu protein in various cell types. J. Gen. Virol. 76 (11), 2717–2722. https://doi.org/10.1099/0022-1317-76-11-2717
Kupzig S., Korolchuk V., Rollason R., Sugden A., Wilde A., Banting G. 2003. Bst-2/HM1.24 is a raft-associated apical membrane protein with an unusual topology. Traffic. 4 (10), 694–709. http://www.ncbi. nlm.nih.gov/pubmed/12956872.
Sauter D. 2014. Counteraction of the multifunctional restriction factor tetherin. Front. Microbiol. 5, 1–14. https://doi.org/10.3389/fmicb.2014.00163
Goto T., Kennel S.J., Abe M., Takishita M., Kosaka M., Solomon A., Saito S. 1994. A novel membrane antigen selectively expressed on terminally differentiated human B cells. Blood. 84 (6), 1922–1930. http://www.ncbi.nlm.nih.gov/pubmed/8080996.
Masuyama N., Kuronita T., Tanaka R., Muto T., Hirota Y., Takigawa A., Fujita H., Aso Y., Amano J., Tanaka Y. 2009. HM1.24 is internalized from lipid rafts by clathrin-mediated endocytosis through interaction with α-adaptin. J. Biol. Chem. 284 (23), 15927–15941. https://doi.org/10.1074/jbc.M109.005124
Perez-Caballero D., Zang T., Ebrahimi A., McNatt M.W., Gregory D.A., Johnson M.C., Bieniasz P.D. 2009. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell. 139(3), 499–511. https://doi.org/10.1016/j.cell.2009.08.039
Hammonds J., Spearman P. 2009. Tetherin is as tetherin does. Cell. 139 (3), 456–457. https://doi.org/10.1016/j.cell.2009.10.011
Venkatesh S., Bieniasz P.D. 2013. Mechanism of HIV-1 virion entrapment by tetherin. PLoS Pathog. 9 (7), e1003483. https://doi.org/10.1371/journal.ppat.1003483
Bieniasz P.D. 2009. The cell biology of HIV-1 virion genesis. Cell Host Microbe. 5 (6), 550–558. https://doi.org/10.1016/j.chom.2009.05.015
Andrew A.J., Miyagi E., Kao S., Strebel K. 2009. The formation of cysteine-linked dimers of BST-2/tetherin is important for inhibition of HIV-1 virus release but not for sensitivity to Vpu. Retrovirology. 6 (1), 80. https://doi.org/10.1186/1742-4690-6-80
Ohtomo T., Sugamata Y., Ozaki Y., Ono K., Yoshimura Y., Kawai S., Koishihara Y., Ozaki S., Kosaka M., Hirano T., Tsuchiya M. 1999. Molecular cloning and characterization of a surface antigen preferentially overexpressed on multiple myeloma cells. Biochem. Biophys. Res. Commun. 258 (3), 583–591. https://doi.org/10.1006/bbrc.1999.0683
Jolly C., Kashefi K., Hollinshead M., Sattentau Q.J. 2004. HIV-1 cell to cell transfer across an Env-induced, actin-dependent synapse. J. Exp. Med. 199 (2), 283–293. https://doi.org/10.1084/jem.20030648
Sattentau Q.J. 2011. The direct passage of animal viruses between cells. Curr. Opin. Virol. 1 (5), 396–402. https://doi.org/10.1016/j.coviro.2011.09.004
Casartelli N., Sourisseau M., Feldmann J., Guivel-Benhassine F., Mallet A., Marcelin A.-G., Guatelli J., Schwartz O. 2010. Tetherin restricts productive HIV-1 cell-to-cell transmission. PLoS Pathog. 6 (6), e1000955. https://doi.org/10.1371/journal.ppat.1000955
Kuhl B.D., Sloan R.D., Donahue D.A., Bar-Magen T., Liang C., Wainberg M.A. 2010. Tetherin restricts direct cell-to-cell infection of HIV-1. Retrovirology. 7 (1), 115. https://doi.org/10.1186/1742-4690-7-115
Blanchet F.P., Stalder R., Czubala M., Lehmann M., Rio L., Mangeat B., Piguet V. 2013. TLR-4 engagement of dendritic cells confers a BST-2/tetherin-mediated restriction of HIV-1 infection to CD4+ T cells across the virological synapse. Retrovirology. 10 (1), 6. https://doi.org/10.1186/1742-4690-10-6
Giese S., Marsh M. 2014. Tetherin can restrict cell-free and cell-cell transmission of HIV from primary macrophages to T cells. PLoS Pathog. 10 (7). https://doi.org/10.1371/journal.ppat.1004189
Jolly C., Booth N.J., Neil S.J.D. 2010. Cell-cell spread of human immunodeficiency virus type 1 overcomes tetherin/BST-2-mediated restriction in T cells. J. Virol. 84 (23), 12185–12199. https://doi.org/10.1128/JVI.01447-10
Coleman C.M., Spearman P., Wu L. 2011. Tetherin does not significantly restrict dendritic cell-mediated HIV-1 transmission and its expression is upregulated by newly synthesized HIV-1 Nef. Retrovirology. 8 (1), 26. https://doi.org/10.1186/1742-4690-8-26
Zhong P., Agosto L.M., Ilinskaya A., Dorjbal B., Truong R., Derse D., Uchil P.D., Heidecker G., Mothes W. 2013. Cell-to-cell transmission can overcome multiple donor and target cell barriers imposed on cell-free HIV. PLoS One. 8 (1), e53138. https://doi.org/10.1371/journal.pone.0053138
Mazurov D., Ilinskaya A., Heidecker G., Lloyd P., Derse D. 2010. Quantitative comparison of HTLV-1 and HIV-1 cell-to-cell infection with new replication dependent vectors. PLoS Pathog. 6 (2), e1000788. https://doi.org/10.1371/journal.ppat.1000788
Shunaeva A., Potashnikova D., Pichugin A., Mishina A., Filatov A., Nikolaitchik O., Hu W.-S., Mazurov D. 2015. Improvement of HIV-1 and human T cell lymphotropic virus type 1 replication-dependent vectors via optimization of reporter gene reconstitution and modification with intronic short hairpin RNA. J. Virol. 89 (20), 10591–10601. https://doi.org/10.1128/JVI.01940-15
Matsuda A., Suzuki Y., Honda G., Muramatsu S., Matsuzaki O., Nagano Y., Doi T., Shimotohno K., Harada T., Nishida E., Hayashi H., Sugano S. 2003. Large-scale identification and characterization of human genes that activate NF-κB and MAPK signaling pathways. Oncogene. 22 (21), 3307–3318. https://doi.org/10.1038/sj.onc.1206406
Galão R.P., Le Tortorec A., Pickering S., Kueck T., Neil S.J.D. 2012. Innate sensing of HIV-1 assembly by tetherin induces NFκB-dependent proinflammatory responses. Cell Host Microbe. 12 (5), 633–644. https://doi.org/10.1016/j.chom.2012.10.007
Tokarev A., Suarez M., Kwan W., Fitzpatrick K., Singh R., Guatelli J. 2013. Stimulation of NF-κB activity by the HIV restriction factor BST2. J. Virol. 87 (4), 2046–2057. https://doi.org/10.1128/JVI.02272-12
Kobayashi T., Ode H., Yoshida T., Sato K., Gee P., Yamamoto S.P., Ebina H., Strebel K., Sato H., Koyanagi Y. 2011. Identification of amino acids in the human tetherin transmembrane domain responsible for HIV-1 Vpu interaction and susceptibility. J. Virol. 85 (2), 932–945. https://doi.org/10.1128/JVI.01668-10
Douglas J.L., Viswanathan K., McCarroll M.N., Gustin J.K., Fruh K., Moses A.V. 2009. Vpu directs the degradation of the human immunodeficiency virus restriction factor BST-2/Tetherin via a TrCP-dependent mechanism. J. Virol. 83 (16), 7931–7947. https://doi.org/10.1128/JVI.00242-09
Liu L., Oliveira N.M., Cheney K.M., Pade C., Dreja H., Bergin A.-M.H., Borgdorff V., Beach D.H., Bishop C.L., Dittmar M.T., McKnight Á. 2011. A whole genome screen for HIV restriction factors. Retrovirology. 8 (1), 94. https://doi.org/10.1186/1742-4690-8-94
Zhou H., Xu M., Huang Q., Gates A.T., Zhang X.D., Castle J.C., Stec E., Ferrer M., Strulovici B., Hazuda D.J., Espeseth A.S. 2008. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe. 4 (5), 495–504. https://doi.org/10.1016/j.chom.2008.10.004
Brass A.L., Dykxhoorn D.M., Benita Y., Yan N., Engelman A., Xavier R.J., Lieberman J., Elledge S.J. 2008. Identification of host proteins required for HIV infection through a functional genomic screen. Science. 319 (5865), 921–926. https://doi.org/10.1126/science.1152725
Park R.J., Wang T., Koundakjian D., Hultquist J.F., Lamothe-Molina P., Monel B., Schumann K., Yu H., Krupzcak K.M., Garcia-Beltran W., Piechocka-Trocha A., Krogan N.J., Marson A., Sabatini D.M., Lander E.S., Hacohen N., Walker B.D. 2017. A genome-wide CRISPR screen identifies a restricted set of HIV host dependency factors. Nat. Genet. 49 (2), 193–203. https://doi.org/10.1038/ng.3741
König R., Zhou Y., Elleder D., Diamond T.L., Bonamy G.M.C., Irelan J.T., Chiang C.-Y., Tu B.P., De Jesus P.D., Lilley C.E., Seidel S., Opaluch A.M., Caldwell J.S., Weitzman M.D., Kuhen K.L., et al. 2008. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell. 135 (1), 49–60. https://doi.org/10.1016/j.cell.2008.07.032
Nguyen D.G., Yin H., Zhou Y., Wolff K.C., Kuhen K.L., Caldwell J.S. 2007. Identification of novel therapeutic targets for HIV infection through functional genomic cDNA screening. Virology. 362 (1), 16–25. https://doi.org/10.1016/J.VIROL.2006.11.036
Gélinas J.-F., Gill D.R., Hyde S.C. 2018. Multiple inhibitory factors act in the late phase of HIV-1 replication: A systematic review of the literature. Microbiol. Mol. Biol. Rev. 82 (1), e00051-17. https://doi.org/10.1128/MMBR.00051-17
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated by E. Makeeva
Rights and permissions
About this article

Cite this article
Zotova, A.A., Atemasova, A.A., Filatov, A.V. et al. HIV Restriction Factors and Their Ambiguous Role during Infection. Mol Biol 53, 212–226 (2019). https://doi.org/10.1134/S0026893319020171
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0026893319020171