
Abstract—
Molecular genetic study of 22 methanol-assimilating Komagataella strains maintained at the UCDFST collection (United States) was carried out. Multigene phylogenetic analysis revealed that strains previously assigned to “Pichia pastoris” based on the standard taxonomic tests actually belong to four different species: K. pastoris, K. phaffii, K. mondaviorum, and K. ulmi. Seven Komagataella species are practically indistinguishable by the standard morphological and physiological tests, and have similar D1/D2 sequences. Multigene phylogenetic analysis should be applied for reliable delineation of all seven Komagataella species. The biogeography of Komagataella yeasts is noteworthy. K. mondaviorum, K. kurtzmanii, K. phaffii, K. populi, and K. ulmi have been found only in North America, while K. pastoris and K. pseudopastoris are characteristic of Europe.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Methylotrophic yeasts Komagataella (previously assigned to the polyphyletic genus Pichia) are characterized by ability to grow on methanol as the sole carbon source. They possess strong methanol-inducible promoters and are widely used in biotechnology to produce high-quality recombinant proteins for medical and microbiological purposes (Cregg et al., 1988, 1993; Chen et al., 2012; Love et al., 2016; Zahrl et al., 2017). The “Pichia pastoris” expression system was commercialized by Invitrogen and is widely used for scientific and industrial purposes (Cregg et al., 1993).
The great applied value of these yeasts contributed to their careful classification and identification. This yeast was first described as Zygosaccharomyces pastori based on a single strain isolated from chestnut in France (Guilliermond, 1919). In the 1950s, Hermann Phaff isolated several related black oak strains in California and renamed the species Pichia pastoris (Phaff, 1956). The next revision of the heterogeneous genus Pichia led to description of a new monotypic genus Komagataella, to which the species Pichiapastoris was assigned (Yamada et al., 1995).
After discovery of the European species Komagataella pseudopastoris, originally described as Pichia pseudopastoris (Dlauchy et al., 2003), the genus Komagataella became generally recognized (Kurtzman, 2005, 2011). Based on comparative analysis of the 26S rRNA gene D1/D2 nucleotide sequences, a third species of K. phaffii was described, to which American isolates were assigned (Kurtzman, 2005). It was found that the biotechnologically important yeasts “Pichiapastoris” actually belonged to two species: K. pastoris and K. phaffi, and the strain “Pichiapastoris” NRRL Y-48124 used in the commercially available Invitrogen expression kit belonged to K. phaffi (Kurtzman, 2009). Most industrial strains belong to K. phaffii (Gasser and Mattanovich, 2018). Three new Komagataella species were described based on individual strains: K. populi, K. ulmi, and K. kurtzmanii (Kurtzman, 2012; Naumov et al., 2013). Recently, we described the seventh species of the genus Komagataella: K. mondaviorum (Naumov et al., 2018). The yeast K. kurtzmanii, which is superior to the K. phaffii and K. pastoris commercial strains in a number of characteristics, is also important from the biotechnological point of view (Tyurin et al., 2013). Recently, sequencing of the full-length genomes of the type cultures of K. pastoris, K. phaffii, K. populi, and K. pseudopastoris was performed (Love et al., 2016; Valli et al., 2016).
In this work, we conducted a molecular genetic study and reidentification of the Komagataella strains maintained in the UCDFST collection (Phaff Yeast Culture Collection, Department of Food Science and Technology, University of California, Davis, United States) in order to expand the scientific and applied use of the natural gene pool of these yeasts.
MATERIALS AND METHODS
Strains and media. The strains used in the work and their origin are presented in Table 1. Most strains were obtained from the UCDFST yeast collection (http://phaffcollection.ucdavis.edu/) and were isolated from different natural sources in different years. The yeasts were grown at 28°C on complete YPD medium containing the following (g/L): bacto-agar (Difco, United States), 20; glucose (Merck, Germany), 20; yeast extract (Difco), 10; bacto-peptone (Difco), 20.
The polymerase chain reaction was carried out on a Bio-Rad DNA thermal cycler (United States). Yeast DNA was isolated using the Genomic DNA Purification Kit (Fermentas, Lithuania). The 26S rDNA D1/D2 domain, the 5.8S-ITS fragment (5.8S RNA gene and the internal transcribed spacers ITS1/ITS2), the translation elongation factor EF-1α, and the RNA polymerase II RPB1 subunit gene were amplified using the standard primers (Kurtzman and Robnett, 1998, 2003; Kurtzman, 2009). PCR was performed in 30 μL of the buffer containing 2.5 mM MgCl2, 0.1 mM of each dNTP, 50 pmol of each primer, 2.5 U of Taq polymerase (Syntol, Russia), and 20–200 ng of DNA. Initial denaturation was carried out at 94°C for 3 min, with 30 subsequent cycles in the following mode: denaturation at 94°C for 45 s; annealing of primers, at 52°C for 30 s; DNA synthesis, at 72°C for 120 s; with the final elongation stage at 72°С for 10 min. The amplification products were separated by electrophoresis in 1% agarose gel at 60–65 V in 0.5× TBE buffer (45 mM Tris, 10 mM EDTA, 45 mM boric acid, pH 8.0) for 2–3 h. The gel was stained with ethidium bromide, washed in distilled water, and photographed under ultraviolet light on a Vilber Lourmat transilluminator (France). The 1kb DNA Ladder (Fermentas, Lithuania) was used as a molecular weight marker.
Sequencing. Amplified fragments were eluted from the gel using the DNA Extraction Kit (Fermentas, Lithuania) according to the manufacturer’s protocol. The nucleotide sequences of the D1/D2 domain, 5.8S-ITS region, EF-1α, and RPB1 were determined with two strands using direct sequencing according to Sanger on an Applied Biosystems 3730 automatic sequencer (United States).
Phylogenetic analysis. The nucleotide sequences obtained were analyzed using the SeqMan software package (DNA Star Inc., United States). Homology searches with known nucleotide sequences were performed in the GenBank database (http://www.ncbi. nlm.nih.gov/genbank/) using the BLAST software. Multiple alignments of the studied nucleotide sequences were carried out using the BioEdit software (http://www.mbio.ncsu.edu/BioEdit/bioedit.html). Phylogenetic trees were constructed using the Neighbor-Joining method in the MEGA 6 software package (Tamura et al., 2013). The type cultures of Ogataea glucozyma NRRL YB-2185 and Pichia membranifaciens NRRL Y-2026 were used as outgroups. Bootstrap indices, which determine the statistical significance of group identification, were determined for 1000 pseudo replicas.
RESULTS
Current classification of ascomycetous yeasts is based on phylogenetic analysis of a number of molecular markers, primarily the 26S rRNA D1/D2 domain (Kurtzman and Robnett, 1998; Kurtzman, 2011). We performed D1/D2 sequencing for 22 Komagataella collection strains, which, on the basis of the standard taxonomic tests, were previously assigned to Komagataella (Pichia) pastoris (Table 1). The obtained nucleotide sequences were compared with the D1/D2 sequences of the type cultures of seven known Komagataella species: K. pastoris (NRRL Y-1603), K. phafii (NRRL Y-7556), K. kurtzmanii (VKPM Y‑727), K. ulmi (NRRL Y-407), K. populi (NRRL YB-455), K. pseudopastoris (NRRL Y-27603), and K. mondaviorum (UCDFST 71-1024). Four more K. mondaviorum strains were included in the analysis: UCDFST 54-11.16, UCDFST 54-11.141, UCDFST 68-967.1, and UCDFST 74-1030 (Table 1).
Seven strains isolated in California (UCDFST: 67-1008, 71-1016, 71-1036, 72-1017, 72-256, 96-151.1, and 96-151.2) and UCDFST 49-54 isolated from the elm exudate in Missouri, had the D1/D2 sequences identical with those of the type culture of K. ulmi NRRL Y-407. According to our analysis, five strains (UCDFST: 54-11.214, 54-11.229, 56-57, 52-155, and 68-1033.1) belong to K. phaffii (Table 1). Identification of four strains (UCDFST 60-58, UCDFST 68-773.2, LE2B88, and LU2T88) as belonging to the species K. pastoris was confirmed. The strains UCDFST 54-11.16, UCDFST 54-11.141, and UCDFST 68-967.1 had identical D1/D2 sequences that differed from the corresponding sequences of the yeasts UCDFST 71-1024 (type culture of K. mondaviorum), UCDFST 74-1030, UCDFST 72-1033, and UCDFST 77-1019 by one nucleotide substitution. The D1/D2 sequences of strains UCDFST 68-692.1, UCDFST 68-974.1, and UCDFST 68-891.2 differed from the corresponding sequences of the indicated seven strains by one or two nucleotide substitutions. Previously, we showed that the species K. populi, K. pseudopastoris, and K. mondaviorum cannot be differentiated solely on the basis of D1/D2 sequences (Naumov et al., 2018).
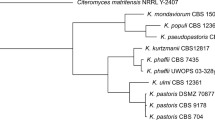
To establish the phylogenetic relationship between the 22 Komagataella strains studied and to determine the taxonomic status of the strains UCDFST 68-692.1, UCDFST 68-974.1, UCDFST 68-891.2, UCDFST 72-1033, and UCDFST 77-1019, we performed comparative analysis of the nucleotide sequences of the D1/D2, ITS-region, and EF-1α and RPB1 genes. Based on the nucleotide sequences obtained, the phylogenetic tree was constructed (Fig. 1).

Phylogenetic analysis of the nucleotide sequences of the D1/D2 domain, 5.8S-ITS, elongation factor EF-1α, and RNA polymerase II RPB1 subunit gene of Komagataella yeasts. The type cultures of Ogataea glucozyma NRRL YB-2185 and Pichia membranifaciens NRRL Y-2026 were used as outgroups. The scale corresponds to 20 nucleotide substitutions per 1000 nucleotide positions. The bootstrap values >70% are given. T is a type culture.
The studied Komagataella strains fell into two main clusters. In turn, the first cluster included three subclusters. The type culture of K. ulmi NRRL Y-407 and eight more strains with identical D1/D2 sequences and very similar sequences of the EF-1α and RPB1 nuclear genes, formed the first subcluster. The main differences between the strains were the ITS sequences. The second subcluster combined K. pastoris strains of various origins with 100% statistical support (Table 1). Strains LU2T88, LE2B82, and UCDFST 60-58 did not differ in the D1/D2, ITS, and RPB1 gene sequences from the NRRL Y-1603 type culture isolated from chestnut exudate in France. The third subcluster included the type culture of K. phaffii NRRL Y-7556 and five other strains isolated in California. Six strains had identical D1/D2 and EF-1α sequences. Single nucleotide substitutions were revealed only in the ITS sequences and the RPB1 gene. Five strains of this subcluster were isolated from the Quercus kellogii black oak, and UCDFST 52-155, from Drosophilapseudoobscura. Adjacent to this sub-cluster was the type culture of K. kurtzmanii VKPM Y-727, which differed significantly from K. phaffii strains in the sequences of all the four molecular markers analyzed.
The second cluster consisted of two subclusters. The first one includes four K. pseudopastoris strains isolated from decaying Salix alba willow wood in Hungary. The type culture of K. populi NRRL YB-455 isolated from poplar sap flow in Illinois was very close to this subcluster.
The second subcluster (99% of statistical support) combined ten K. mondaviorum strains (Fig. 1). Most strains of this subcluster were isolated in California from the poplar and black oak exudate. The strains UCDFST 68-967.1 and UCDFST 68-974.1 were isolated in the state of Washington; UCDFST 68-692.1, in Alaska; and UCDFST 68-891.2, in Canada (Table 1). This is the most heterogeneous subcluster. The strains UCDFST 54-11.16, UCDFST 54-11.141, and UCDFST 68-967.1 had identical D1/D2, ITS, EF-1α, and RPB1 nucleotide sequences. The type culture of UCDFST 71-1024 and strains UCDFST 74-1030 and UCDFST 77-1019 had almost identical sequences of all the four molecular markers. The remaining four strains (UCD72-1033, UCD68-692.1, UCDFST 68-974.2, and UCDFST 68-891.2) differed from each other and from the rest of K. mondaviorum strains in the ITS-region, EF-1α, and RPB1 sequences. The differences between ten K. mondaviorum strains in the EF-1α and RPB1 gene sequences constituted 0–4 and 0–9 nucleotide substitutions, respectively. At the same time, the EF-1α and RPB1 sequences of K. mondaviourum strains differed from the corresponding sequences of the species K. populi and K. pseudopastoris by more than 10 and 28 nucleotide substitutions.
DISCUSSION
Using multigene phylogenetic analysis, we carried out a cardinal reidentification of Komagataella strains that were isolated in different years (from 1919 to 1996) from various natural sources (Table 1). Of the 22 strains designated as K. pastoris, this species identification was confirmed only for UCDFST 60-58, UCDFST 68-773.2, LU2T88, and LE2B88 (Table 1, Fig. 1). Eight strains were reidentified as K. ulmi, five each as K. phaffii and K. mondaviorum (Table 1). It should be noted that among the studied yeasts, we were unable to detect new K. kurtzmanii, K. populi, and K. pseudopastoris strains.
Our study revealed a close phylogenetic relationship between Komagataella yeasts of different origin with similar D1/D2 sequences. At the same time, the difference in the D1/D2 domain with the phylogenetically closest genus Phaffomyces exceeds 80 nucleotide substitutions (Naumov, 2015). Significant differences between the strains of seven Komagataella species were revealed in the ITS region, EF-1α, and RPB1 sequences. The ITS1 sequences of the studied strains were more variable, while the ITS2 sequences were characterized by intraspecies conservatism. For example, all the strains assigned to K. pastoris, K. phaffii, K. ulmi, and K. mondaviorum had identical ITS2 sequences with the corresponding type cultures. The molecular markers EF-1α and RPB1 were also characterized by low intraspecies polymorphism.
It should be noted that the seven Komagataella species are almost indistinguishable based on the standard morphological and physiological tests. An exception is K. kurtzmanii, which, unlike the other six species, is unable to assimilate trehalose (Naumov et al., 2013, 2018). Taking into account the variability of physiological properties and the fact that K. kurtzmanii and K. populi are represented by single strains, it is impossible to differentiate between all seven species using the standard taxonomic tests. Therefore, reliable species identification of Komagataella strains requires application of the multigene phylogenetic analysis.
The genus Komagataella established by phylogenetic analysis is fully consistent with the concept of the genetic genus in ascomycetous fungi: its constituent species have a common system of mating types that allows them to cross (Naumov, 1978, 2015). Komagataella species possess postzygotic isolation and form sterile hybrids with nonviable ascospores (Naumov et al., 2016). Biogeography of the Komagataella yeasts is noteworthy. Strains of five species (K. mondaviorum, K. kurtzmanii, K. phaffii, K. populi, and K. ulmi) are found in North America, while K. pastoris and K. pseudopastoris are characteristic of Europe (Dlauchy et al., 2003; Kurtzman, 2011; current study). The strains LU2T88 (Taiwan) and UCDFST 68-773.2 (Canada) identified by us as K. pastoris are apparently invasive from Europe. This agrees well with the fact that, despite years of yeast research, only single Komagataella isolates were found in Japan (Phaff et al., 1972; Kodama, 1974; Banno and Mikata, 1981).
We should also note the ecological characteristics of Komagataella species—its association with broad-leaved trees (Table 1). An exception is the type culture of K. kurtzmanii isolated from a coniferous tree exudate—a fir in Southern Arizona (United States). Our study did not confirm the uniqueness of the species epithet of the K. ulmi yeast, for which the type culture NRRL Y-407 was isolated from elm. Among the strains that we assigned to this species, isolates from the exudates of various species of oaks and maple were also present (Table 1). All four known K. pseudopastoris strains were isolated in Hungary from decaying willow (Salix alba) wood (Dlauchy et al., 2003). Interestingly, the yeast K. pseudopastoris was not detected in any of the fifty oak samples, while K. pastoris strains were isolated with high frequency. Apparently, this is due to the higher sensitivity of K. pseudopastoris to tannic acid contained in Quercus spp., as distinct from K. pastoris (Dlauchy et al., 2003; Peter et al., 2019).
Taking into account the great biotechnological and scientific importance of Komagataella yeasts, it is advisable to continue the study of environmental genetics and biogeography of this genus. The molecular genetic study of Komagataella (Pichia) pastoris strains from various yeast collections and the isolation of new natural strains will make it possible to discover new species and additional strains of the already-known species of this genus.
REFERENCES
Banno, I. and Mikata, K. Ascomycetous yeasts isolated from forest materials in Japan, IFO Res. Comm., 1981, vol. 10, pp. 10‒19.
Chen, M.T., Lin, S., Shandil, I., Andrews, D., Stadheim, T.A., and Choi, B.K. Generation of diploid Pichia pastoris strains by mating and their application for recombinant protein production, Microb. Cell Fact., 2012, vol. 11, p. 91.
Cregg, J.M. and Madden, K.R., Development of the methylotrophic yeast, Pichia pastoris, as a host system for the production of foreign proteins, Dev. Ind. Microbiol., 1988, vol. 29, pp. 33–41.
Cregg, J.M., Vedvick, T.S., and Raschke, W.C., Recent advances in the expression of foreign genes in Pichia pastoris,Biotechnol., 1993, vol. 11, pp. 905–910.
Dlauchy, D., Tornai-Lehoczki, J., Fülöp, L., and Péter, G., Pichia (Komagataella) pseudopastoris sp. nov., a new yeast species from Hungary, Antonie van Leeuwenhoek, 2003, vol. 83, pp. 327–332.
Gasser, B. and Mattanovich, D., A yeast for all seasons ‒ is Pichia pastoris a suitable chassis organism for future bioproduction?, FEMS Microbiol. Lett., 2018, vol. 365, pp. 1–4.
Guilliermond, A., Sur une nouvelle levure à copulation hétérogamique, C.R. Soc. Biol., 1919, no. 82, pp. 466–470.
Kodama, K., Ascosporogenous yeasts isolated from tree exudates in Japan, Ann. Microbiol. Enzimol., 1974, no. 24, pp. 215–231.
Kurtzman, C.P. and Robnett, C.J., Identification and phylogeny of ascomycetous yeasts from analysis of nuclear large subunit (26S) ribosomal DNA partial sequences, Antonie Van Leeuwenhoek, 1998, vol. 73, pp. 331–371.
Kurtzman, C.P. and Robnett, C.J., Phylogenetic relationships among yeasts of the “Saccharomyces complex” determined from multigene sequence analyses, FEMS Yeast Res., 2003, vol. 3, pp. 417–432.
Kurtzman, C.P., Biotechnological strains of Komagataella (Pichia) pastoris are Komagataella phaffii as determined from multigene sequence analysis, J. Ind. Microbiol. Biotechnol., 2009, vol. 36, pp. 1435–1438.
Kurtzman, C.P., Description of Komagataella phaffii sp. nov. and the transfer of Pichia pseudopastoris to the methylotrophic yeast genus Komagataella,Int. J. Sys. Evol. Microbiol., 2005, vol. 55, pp. 973–976.
Kurtzman, C.P., Komagataella Y. Yamada, Matsuda, Maeda & Mikata 1995, in The Yeast. A Taxonomic Study, Kurtzman, C.P., Fell, J., and Boekhout, T., Eds., 5th ed. Amsterdam: Elsevier, 2011, vol. 2, pp. 491–495.
Kurtzman, C.P., Komagataella populi sp. nov. and Komagataella ulmi sp. nov., two new methanol assimilating yeasts from exudates of deciduous trees, Antonie van Leeuwenhoek, 2012, vol. 101, pp. 859–868.
Love, K.R., Shah, K.A., Whittaker, C.A., Wu, J., Bartlett, M.C., Ma, D., Leeson, R.L., Priest, M., Borowsky, J., Young, S.K., and Love, J.C., Comparative genomics and transcriptomics of Pichia pastoris,BMC Genomics, 2016, vol. 17, pp. 1–17.
Naumov, G.I., Genetic concept of genus in fungi, Doklady Biol. Sci., 1979, vol. 241, pp. 345‒347.
Naumov, G.I., Kondratieva, V.I., Meshcheryakova, E.V., and Naumova, E.S., Taxonomic genetics of methylotrophic yeast genus Komagataella: new biological species K. kurtzmanii,Russ. J. Genet., 2016, vol. 52, pp. 378–382.
Naumov, G.I., Naumova, E.S., and Boundy-Mills, K.L., Description of Komagataella mondaviorum sp. nov., a new sibling species of Komagataella (Pichia) pastoris,Antonie van Leeuwenhoek, 2018, vol. 111, pp. 1197–1207.
Naumov, G.I., Naumova, E.S., Tyurin, O.V., and Kozlov, D.G., Komagataella kurtzmanii sp. nov., a new sibling species of Komagataella (Pichia) pastoris in accordance with multigene sequence analysis, Antonie van Leeuwenhoek, 2013, vol. 104, pp. 339‒347.
Naumov, G.I., The yeast Komagataella: a genetic genus in accordance with interspecies hybridization, Microbiology (Moscow), 2015, vol. 84, pp. 538–543.
Péter, G., Nagy, E.S., and Dlauchy, D., Systematics, diversity and ecology of the genus Yarrowia and the methanol-assimilating yeasts, in Non-Conventional Yeasts: From Basic Research to Application, Sibirny, A., Ed., Springer Nature, 2019, pp. 297–339.
Phaff, H.J., Miller, M.W., Yoneyama, M., and Soneda, M., A comparative study of the yeast florae associated with trees from the Japanese islands and on the west coast of North America, Fermentation Technology Today: Proceedings IV IFS, Terui, G., Ed., Osaka: Soc. Ferm. Technol., 1972, pp. 759–774.
Phaff, H.Y., A proposal for amendment of the diagnosis of the genus Pichia Hansen, Antonie van Leeuwenhoek, 1956, vol. 22, pp. 113–116.
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S., MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0, Mol. Biol. Evol., 2013, vol. 30, pp. 2725–2729.
Tyurin, O., Gubaidullin, I., Cheperegin, S., Efremov, B., Naumova, E., Naumov, G., and Kozlov, D., The new expression system based on a novel yeast species of the genus Komagataella,FEBS J., 2013, vol. 280, suppl. 1, p. 602.
Valli, M., Tatto, N.E., Peymann, A., Gruber, C., Landes, N., Ekker, H., Thallinger, G.G., Mattanovich, D., Gasser, B., and Graf, A.B., Curation of the genome annotation of Pichia pastoris (Komagataella phaffii) CBS 7435 from gene level to protein function, FEMS Yeast Res., 2016, vol. 16. pii: fow051.
Yamada, Y., Matsuda, M., Maeda, K., and Mikata, K., The phylogenetic relationships of methanol-assimilating yeasts based on the partial sequences of 18S and 26S ribosomal RNAs: the proposal of Komagataella gen. nov. (Saccharomycetaceae), Biosci. Biotechnol. Biochem., 1995, vol. 59, pp. 439–444.
Zahrl, R.J, Peña, D.A, Mattanovich, D., and Gasser, B., Systems biotechnology for protein production in Pichia pastoris,FEMS Yeast Res., 2017, vol. 17, pp. 1–15. https://doi.org/10.1093/femsyr/fox068
Funding
The study was carried out as part of state assignment no. 595-00003-19 PR. K.L. Boundy-Mills was supported by the National Science Foundation (United States), project no. 1349395.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflict of interest. This article does not contain any studies involving animals or human participants performed by any of the authors.
Additional information
Translated by E. Babchenko
Rights and permissions
About this article

Cite this article
Naumova, E.S., Boundy-Mills, K.L. & Naumov, G.I. Phylogenetics and Biogeography of Biotechnologically Important Methylotrophic Yeasts Komagataella. Microbiology 89, 294–300 (2020). https://doi.org/10.1134/S002626172003011X
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S002626172003011X