
Abstract
Wild varieties of plants have stronger stress resistances than their cultivated relatives, and rhizosphere bacteria play an important role in improving the environmental adaptabilities of plants. However, the responses and adaptations of bacterial communities to wild soybean (Glycine soja Sieb. et Zucc) and cultivated soybean (Glycine max L.) have rarely been studied. Thus, the aim of this study was to investigate the differences in the rhizosphere bacterial communities of wild and cultivated soybeans under field and potted conditions using metagenomic analysis. The results showed that the rhizosphere bacterial diversity was higher in wild soybean than that in cultivated soybean in field samples, indicating that domestication leads to a decrease in the rhizosphere bacterial diversity of cultivated soybean. In addition, the higher RAs of beneficial and plant growth-promoting bacteria such as Acidobacteria, Gemmatimonadetes,Bradyrhizobium and Bacillus were in the wild soybean rhizosphere, illustrating that wild soybean has a stronger environmental resistance and adaptation than cultivated soybean. Meanwhile, soil pH, soil organic carbon, total nitrogen, total phosphorus, available phosphorus, and available potassium were significantly correlated with rhizosphere bacteria. Collectively, the rhizosphere bacteria of wild and cultivated soybean were different, wild soybeans increase the numbers of beneficial microbes in the rhizosphere to improve their environmental adaptability, and the utilization of wild resources might be an effective way to improve crop stress resistance.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Soybean, one of the most important oilseed crops in the world (Fan et al., 2015), provides high-quality proteins and edible oil to humans (Wang et al., 2006), and occupies an important position in agricultural production and rural incomes (Sulieman et al., 2015). However, with increasing populations and changing climate, soybean production is not only threatened by biotic and abiotic stresses (Manavalan et al., 2009), but also cannot meet the demands of the growing population. Understanding how to improve the stress resistance and yield of soybean is important to the countries and scientists.
Wild varieties of plants, which survive in natural conditions without any human intervention such as irrigation, pesticide spraying and fertilizer application, inevitably have stronger stress resistance and environmental suitability than their cultivated relatives (Tripathy et al., 2018; Wu et al., 2018). The genetic diversity of wild species is an important resource for the improvement of crops, and the reasonable protection and sustainable use of wild varieties are highly significant for global food security (Mammadov et al., 2018). Currently, a good way to improve the quality of cultivated crops is by breeding with wild resources worldwide (Jitendra et al., 2016; Chandrasekhar et al., 2017; Dwivedi et al., 2017). It is well known that present widely cultivated soybean (Glycine max L.) was domesticated from wild soybean (Glycine soja Sieb. et Zucc) to meet human needs such as higher yield thousands of years ago (Li et al., 2008). However, studies have found that many good traits of wild soybean have been lost over the evolution of wild species to cultivated species (Chang et al., 2018). To clarify the causes of these changes is an important issue that needs to be addressed.
Soil microbes play a key role in the domestication process. Microbial community is an essential part of the plant rhizosphere and participates in the functioning of plants (Yu et al., 2013; Zhang et al., 2018). Bacteria account for the majority of the microbial community and play critical roles in nutrient exchange and metabolism in the soil ecosystem. Different genotypes of plants have different rhizosphere bacterial communities, and rhizosphere bacteria have significant influence on host growth and metabolism. Thus, rhizosphere bacteria should be an important part of the domestication process. Some studies have noted that the long-term domestication process often leads to a decrease in the diversity of rhizosphere bacterial communities in cultivated crops, and the rhizosphere diversities of wild wheat (Germida and Siciliano, 2001) and beet (Zachow et al., 2014) were significantly higher than their cultivated relatives were reported. However, the effects of different genotype crops on bacterial community structure and the correlations between bacterial community and soil chemical properties have rarely been studied.
To better understand how rhizosphere bacteria are affected by genotypes, the rhizosphere bacterial communities of wild and cultivated soybeans in field and potted conditions were conducted using metagenomics analysis. Thus, in this study, we aimed to: (1) evaluate the differences in soil bacterial communities in wild and cultivated soybean rhizosphere under field and potted condition, (2) investigate the relationship between bacterial communities and soil properties, and (3) gain deeper insight into utilization of wild resources.
MATERIALS AND METHODS
Experimental design. The materials used for this experiment were four groups of black soil samples, including three groups of field samples and one group of potted samples. Each of these three groups of field samples included wild soybean rhizosphere soil (FW), cultivated soybean rhizosphere soil (FC) and bare soil (FB) with four replications. The group of potted samples also included wild soybean rhizosphere soil (PW), cultivated soybean rhizosphere soil (PC) and bare soil (PB) with four replications, except for PB which had with three replications.
Plant growth and sample collection. The cultivars of wild soybean 01289 and cultivated soybean Zhonghuang were selected for this experiment. The soils used for planting were collected at the Changchun Agricultural Experimental Station of Northeast Institute of Geography and Agroecology, Chinese Academy of Sciences in Jilin Province, China (43°59′ N, 125°23′ E), in April 2017. The main soil properties were as follows: 16.75 g kg–1 soil organic carbon (SOC), 1.02 g kg–1 total nitrogen (TN), 1.26 g kg–1 total phosphorous (TP), 33.90 mg kg–1 available phosphorous (AP), 102.68 mg kg–1 available potassium (AK) and pH 7. Soybean was planted in pots in May 2017 with the method described by Shi et al. (2018). The three groups of field samples were collected in Fuyuan of Heilongjiang Province, China. The sampling sites were A (47°91′ N, 134°47′ E), B (47°97′ N, 134°30′ E), and C (47°99′ N, 134°08′ E). Three contiguous sampling areas (wild soybean, W; cultivated soybean, C; and bare soil, B) were selected at each sampling site. In general, the sites were characterized by an annual mean air temperature of 2.2°C, a mean annual rainfall of 600 mm, and an annual accumulated temperature of 2200°C. In addition, the soybean samples cultivated under field conditions were planted in May 2017 at the same time as the potted samples.
After the soybean plants had grown under potted and field conditions for approximately three months, the rhizosphere soils were collected at a distance of 0–1 cm from the plant roots. Potted plants were manually uprooted, and roots were vigorously shaken to remove loose soil. For each field sample, five sampling plots (1 m2 each) were randomly selected within an area of approximately 100 m2 in each of the three contiguous areas (W, C and B). Both field and potted soil samples were sealed in airtight plastic bags and placed on ice for transport to the laboratory. Soil samples were subsequently homogenized and subdivided for a variety of analyses. The soil used for DNA extraction was kept at –80°C, and the soil used for determination of chemical properties was air dried at room temperature.
Determination of soil chemical properties. Air-dried subsamples were used to determine the pH, SOC, TN, TP, AP and AK. Subsamples were passed through a 0.15 mm sieve to evaluate SOC, TN, TP and were passed through a 1 mm sieve to evaluate AP and AK. The soil pH was determined using a 1 : 5 soil-to-water suspension after 3 min of shaking. SOC and TN were analyzed by dichromate oxidation (Mebius, 1960) and the Kjeldahl method (Bremner and Mulvaney, 1982), respectively. The TP content was determined by digestion with HClO4-H2SO4 and measured using spectrophotometry (UV2300, Shimadzu, Japan). The contents of AP and AK were determined using the Bray-1 method and the ammonium acetate extraction method, respectively (Lu et al., 1999).
DNA extraction and high-throughput sequencing. Bacterial DNA samples were extracted from 0.5 g wet soil using the FastDNATM SPIN Kit for soil (MP Biomedicals, CA, USA, code No. 116560200) according to the manufacturer’s instructions. The quantity and quality of extracted DNAs were measured using a NanoDrop 2000 spectrophotometer (NanoDrop Technologies, Inc., Wilmington, USA) and agarose gel electrophoresis, respectively. The bacterial 16S rRNA genes of the V3 and V4 regions were amplified using the primer pair 338F (5'-ACTCCTACGGGAGGCAGCA-3') and the reverse primer 806R (5'-GGACTACHVGGGTWTCTAAT-3'). Sample-specific 7-bp barcodes were incorporated into the primers for multiplex sequencing. The PCR components contained 5 μL of Q5 reaction buffer (5×), 5 μL of Q5 High-Fidelity GC buffer (5×), 0.25 μL of Q5 High-Fidelity DNA Polymerase (5 U/μL), 2 μL (2.5 mM) of dNTPs, 1 μL (10 μM) of each forward and reverse primer, 2 μL of the DNA template, and 8.75 μL of ddH2O. Thermal cycling consisted of an initial denaturation at 98°C for 2 min, followed by 25 cycles consisting of denaturation at 98°C for 15 s, annealing at 55°C for 30 s, and extension at 72°C for 30 s, with a final extension of 5 min at 72°C. PCR amplicons were purified with Agencourt AMPure Beads (Beckman Coulter, Indianapolis, IN) and quantified using the PicoGreen dsDNA Assay Kit (Invitrogen, Carlsbad, CA, USA). After the individual quantification step, amplicons were pooled in equal amounts, and paired-end 2 × 300 bp sequencing was performed using the Illumina MiSeq platform with the MiSeq Reagent Kit v3 at Shanghai Personal Biotechnology Co., Ltd. (Shanghai, China).
Bacterial sequence processing. As previously described (Caporaso et al., 2010), the Quantitative Insights Into Microbial Ecology (QIIME, v. 1.8.0) pipeline was employed to process the sequencing data. Briefly, raw sequencing reads with exact matches to the barcodes were assigned to respective samples and identified as valid sequences. The low-quality sequences were removed using the following criteria (Gill et al., 2006; Chen and Jiang, 2014): sequences that were shorter than 150 bp, sequences that had average Phred scores lower than 20, sequences that contained ambiguous bases, and sequences that contained mononucleotide repeats more than 8 bp were removed. Paired-end reads were assembled using FLASH (Magoc and Salzberg, 2011). The remaining high-quality sequences were clustered into operational taxonomic units (OTUs) at 97% sequence identity by UCLUST after chimaera detection (Edgar, 2010). A representative sequence was selected from each OTU using default parameters. OTU taxonomic classification was conducted by a BLAST search of the representative sequences set against the Greengenes Database (DeSantis et al., 2006) using the best hit criteria (Altschul et al., 1997). An OTU table was further generated to record the abundance of each OTU in each sample and the taxonomy of the OTUs. OTUs containing less than 0.001% of the total sequences across all samples were discarded. To minimize sequencing depth differences across samples, an averaged, rounded rarefied OTU table was generated by averaging 100 evenly resampled OTU subsets under 90% of the minimum sequencing depth for further analysis. The bacterial Illumina raw sequence data are available at the National Center for Biotechnology Information Sequence Read Archive (NCBI SRA) database (http://www.ncbi.nlm.nih.gov/sra) under the accession number SRP158197.
Statistical analysis. The significance of the relative abundances (RAs) of bacteria at different taxonomic levels, bacterial alpha diversity and soil chemical properties was analyzed using SPSS 22.0 with a significance threshold of p < 0.05. Nonmetric multidimensional scaling (NMDS) and the unweighted pair-group method with arithmetic means (UPGMA) hierarchical clustering were performed (Ramette, 2007). Redundancy analysis (RDA) was used to determine the correlation between soil chemical properties and bacterial communities with R (v. 3.5.0).
RESULTS
Relative abundances of bacterial phyla and plant growth-promoting bacteria. In total, we obtained 2,816,280 quality sequences from all samples, and 40,586 to 92,091 bacterial sequences were obtained per sample (average = 59,921) (Table 1). The average read length was 437 bp. The dominant bacterial phyla (>5% of all the DNA sequences) across all soil samples were Proteobacteria, Acidobacteria, Gemmatimonadetes, Chloroflexi and Actinobacteria, with relative abundances (RAs) ranging from 22.1 to 48.3%, 9.0 to 34.7%, 6.2 to 17.7%, 4.3 to 20.7%, and 5.1 to 13.3%, respectively (Fig. 1).

The relative abundances (RAs) of bacterial phyla. F and P indicate field samples and potted samples, respectively. W, C, and B indicate wild soybean, cultivated soybean and bare soil, respectively.
The RAs of bacterial phyla among the potted samples were similar, but very different from those of the field samples (Fig. 1). The RAs of Proteobacteria, Verrucomicrobia and Tenericutes in the field samples were significantly less than those in the potted samples, but the RAs of Acidobacteria and Chloroflexi in the field samples were significantly higher than those in the potted samples, except for the RAs of Chloroflexi in cultivated soybean rhizosphere (Table S1). Under field conditions, the RAs of Acidobacteria,Chloroflexi and Gemmatimonadetes in the rhizosphere of wild soybean and bare soil were significantly higher than those in the rhizosphere of cultivated soybean; reversely, Bacteroidetes in cultivated soybean rhizosphere were significantly higher than that in wild and bare soil. The same trend was observed in the potted samples but not significant (Fig. 2, Table S1).

The relative abundances (RAs) of distinct changes in bacterial phyla between wild soybean and cultivated soybean. F indicates field samples, and P indicates potted samples. W, C and B indicate wild soybean, cultivated soybean and bare soil, respectively. Designations: 1—Acidobacteria; 2—Gemmatimonadetes; 3—Chloroflexi; 4—Bacteroides.
Further analyses of Bradyrhizobium andBacillus were performed. The RAs of Bradyrhizobium and Bacillus ranged from 0.17 to 3.91%, and 0.07 to 0.47%, respectively (Fig. 3). In field conditions, the RAs of Bradyrhizobium (Fig. 3a) and Bacillus (Fig. 3b) in the wild soybean were significantly higher than those in cultivated soybean. In potted conditions, these two grown promoting bacteria had the same trend as the field conditions in wild and cultivated soybean rhizosphere, but the difference were not significant (Fig. 3).

The relative abundances (RAs) of Bradyrhizobium (a) and Bacillus (b). F indicates field samples, and P indicates potted samples. W, C, and B indicate wild soybean, cultivated soybean and bare soil, respectively.
Bacterial Community Diversity
To estimate bacterial diversity and richness of different samples, OTU numbers, Chao 1 and Shannon indexes were calculated. The sequence analysis shows the existence of 8589 OTUs for bacterial communities at the 97% sequence identity. In field conditions, The OTU numbers and Chao1 index value of the wild soybean rhizosphere bacteria were significantly higher than those of the cultivated soybean and bare soil except for site A, but there were no obvious differences between the potted samples (Table 1). The Shannon’s index results were not significantly different for the wild and cultivated soybean samples. In addition, the coverage of all samples was more than 95%, indicating that the sequencing depth in this study was sufficient to capture the diversity of the entire bacterial community (Table 1).
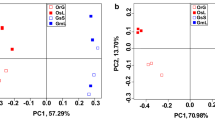
Bacterial community structure and its correlation with soil properties. The relationship among all soil bacterial community compositions was illustrated by NMDS analysis (Fig. 4). The NMDS plot shows that the four replicates from each treatment clustered closely, underscoring the reproducibility of these bacterial community profiles (Fig. 4). The plot also shows that the bacterial communities sampled from field sites A, B, C were entirely different from those sampled from pots. In addition, the wild soybean, cultivated soybean and bare soil rhizosphere bacteria separated from each other (Fig. 4). These findings indicate that although location is the main factor affecting microbial compositions, the soil bacterial community is also influenced by genotypes.

Nonmetric multidimensional scaling (NMDS) plot of all soil bacterial communities. The first capital letters A, B, C, and P indicate the different sampling sites. The second capital letters, W, C and B, indicate wild soybean, cultivated soybean and bare soil, respectively.
Soil chemical properties influence the distribution of microorganisms, so we measured the chemical properties of the soil in each sample (Table S2), and Redundancy analysis (RDA) was performed in order to determine the relationship between soil chemical properties and bacterial community (Fig. 5). The results showed that the two axes explained 15.36 and 12.35% of the variance in the bacterial communities, respectively. The rhizosphere bacteria were significantly correlation with SOC (p < 0.01), pH (p < 0.001), TN (p < 0.001), TP (p < 0.001), AP (p < 0.001), and AK (p < 0.001) (Fig. 5).

Redundancy analysis (RDA) of the relationship between soil chemical properties and bacterial community. The variables followed by asterisks indicate significant influences on the bacterial community (* P < 0.01; ** P < 0.001). The first capital letters A, B, C and P indicate the different sampling sites. The second capital letters, W, C, and B, indicate wild soybean, cultivated soybean and bare soil, respectively.
DISCUSSION
Effect of wild and cultivated soybeans on soil bacterial abundances. In recent years, the comparative study of different genotypes has drawn increasing attention from researchers. Karamac et al. (2018) researched the phenolic contents and antioxidant capacities of wild and cultivated white lupin seeds. Roleda et al. (2018) investigated the iodine content in the bulk biomass of wild-harvested and cultivated edible seaweed. Several studies have suggested that many bacterial groups are highly correlated with plant genotypes. Khan et al. (2017) compared the differences of rhizospheric microbial communities associated with wild and cultivated Boswellia sacra tree and reported a significantly higher abundance of Ascomycota and Actinobacteria in the wild population than in the cultivated population, while Basidiomycota, Acidobacteria and Proteobacteria were highly abundant in cultivated trees.
In this study, we compared the differences of bacteria in wild soybean rhizosphere, cultivated soybean rhizosphere and bare soil. The results showed that the RAs of Acidobacteria and Gemmatimonadetes in wild samples were significantly higher than those in cultivated samples under field conditions (Fig. 2). Researchers found that Acidobacteria has extensive metabolic diversity and significantly effect on nutrient cycling in soils (Hugenholtz et al., 1998; Eichorst et al., 2007). The wild soybean samples had a higher Acidobacteria content than the cultivated samples under field conditions, which illustrates that wild soybean have a positive influence on nutrient cycling in soil environments. DeBruyn et al. (2011) found that Gemmatimonadetes can adapt to dry soils, and in our study, the RA of Gemmatimonadetes in wild soybean was significantly higher than that in cultivated soybean under field conditions, explaining why wild soybean has a stronger drought resistance than cultivated soybean. The phylum Bacteroidetes is a sensitive biological indicator of agricultural soil usage (Wolińska et al., 2017), and the relative abundance of Bacteroidetes in the cultivated soybean was significantly higher than that in the wild soybean under field conditions (Fig. 2), which verified that the changes caused by human activities increased the content of Bacteroidetes in the soil. Studies have shown that, Bradyrhizobium and Bacillus are potential plant growth promoting rhizobacteria inhabiting soybean rhizospheres (Sugiyama et al., 2014). Under field conditions, these two promoting bacteria in wild soybean were significantly higher than these in cultivated soybean (Fig. 3), indicated that wild soybean can promote its growth by recruiting more beneficial bacteria in its rhizosphere. These results all explain why wild soybean could grow well without any human intervention under field conditions. Research have found that the rhizosphere community is selected based on functional cores, which are related to benefits to the plant (Mendes et al., 2014), and root exudates have a certain effect on the number of rhizosphere bacteria in soil (Liang et al., 2014). The differences of rhizosphere bacteria may be caused by the different rhizosphere secretions produced by different genotypes of crops.
Effect of wild and cultivated soybeans on soil bacterial diversity. Soil microbial diversity represents the stability of the microbial community and reflects soil ecology mechanisms and the influence of soil stress on the community (Wang et al., 2016). A large number of studies have shown that different genotypes have different effects on soil microbial diversity (Khan et al., 2017; Gregory et al., 2018). In this study, the OTU numbers and Chao 1 index of wild populations were significantly higher than those of cultivated populations and bare soils in field condition except for site A which was higher but not significantly (Table 1). These results indicate that the bacterial community diversity of the wild soybean rhizosphere was higher than that of the cultivated soybean rhizosphere and bare soil. Thus, domestication results in a decrease in the rhizosphere bacterial diversity of cultivated soybean. High soil bacterial diversity and richness are beneficial to the improvement and maintenance of the soil ecological environment (Mendes et al., 2013); therefore, compared with cultivated soybean, wild soybean is more conducive to the improvement and maintenance of the rhizosphere ecological environment. However, in the potted samples, there was no significant difference in bacterial diversity, perhaps because a short experimental period will not result in changes in the bacterial diversity of small pots.
Effect of wild and cultivated soybeans on soil bacterial community structure. Soil microbes are affected by many factors (Luo et al., 2018; Robledo-Mahon et al., 2018; Sun et al., 2018). In this study, we found that the bacterial communities sampled at the same sites clustered closely in the NMDS plot (Fig. 4), a result caused by soil properties. Studies have noted that the physical and chemical properties of soil are important factors influencing the bacterial community composition (Berg and Smalla, 2009; Chang et al., 2018; Luo et al., 2018). Through the RDA analysis, we found that the rhizosphere bacteria were significantly correlated with SOC (p < 0.01), pH (p < 0.001), TN (p < 0.001), TP (p < 0.001), AP (p < 0.001), and AK (p < 0.001).
Other scholars also found that different genotypes of Silene vulgaris (Moench) Garcke influence the root organic acid composition and rhizosphere bacterial communities (Garcia-Gonzalo et al., 2017), and tree genotypes have been shown to affect the microbe-mediated soil ecosystem functions in a subtropical forest (Purahong et al., 2016). In this study, at each sampling sites, the wild soybean, cultivated soybean and bare soil samples were separate from each other (Fig. 4), although sampling site had a greater effect on the soil bacterial communities, these findings indicate that genotypes are another important factor affecting the bacterial community composition.
In the present study, Proteobacteria, Acidobacteria, Gemmatimonadetes, Chloroflexi and Actinobacteria were dominant bacterial phyla across all soil samples. In field samples, the rhizosphere bacterial diversity was higher in wild soybean than in cultivated soybean, indicating that domestication leads to a decrease in the rhizosphere bacterial diversity of cultivated soybean. In addition, the higher RAs of beneficial and plant growth promoting bacteria such as Acidobacteria, Gemmatimonadetes,Bradyrhizobium and Bacillus in the wild soybean rhizosphere illustrate that wild soybean has a stronger environmental resistance and adaptation than cultivated soybean. At the same time, soil pH, soil organic carbon (SOC), total nitrogen (TN), total phosphorus (TP), available phosphorus (AP), and available potassium (AK) were significantly correlated with rhizosphere bacteria. Collectively, the rhizosphere bacteria of wild and cultivated soybean were different, and wild soybeans increase the numbers of beneficial microbes in the rhizosphere to improve their environmental adaptability.
REFERENCES
Altschul, S., Madden, T., Schäffer, A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D., Gapped BLAST and PSI-BLAST: a new generation of protein database search programs, Nucleic Acids Res., 1997, vol. 25, pp. 3389–3402.
Berg, G. and Smalla, K., Plant species and soil type cooperatively shape the structure and function of microbial communities in the rhizosphere, FEMS Microbiol. Ecol., 2009, vol. 68, pp. 1–13.
Bremner, J. and Mulvaney, C., Chemical and microbiological properties, in Methods of Soil Analysis, Madison: Amer. Soc. Agron., Soil Sci. Soc. Amer., 1982, pp. 595–624.
Caporaso, J.G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F.D., Costello, E.K., Fierer, N., Pena, A.G., Goodrich, J.K., Gordon, J.I., Huttley, G.A., Kelley, S.T., Knights, D., Koenig, J.E., Ley, R.E., et al., QIIME allows analysis of high-throughput community sequencing data, Nat. Methods, 2010, vol. 7, pp. 335–336.
Chandrasekhar, K., Nashef, K., and Ben-David, R., Agronomic and genetic characterization of wild emmer wheat (Triticum turgidum subsp. dicoccoides) introgression lines in a bread wheat genetic background, Genet. Resour. Crop Ev., 2017, vol. 64, pp. 1917–1926.
Chang, C., Chen, W., Luo, S., Ma, L., Li, X., and Tian, C., Rhizosphere microbiota assemblage associated with wild and cultivated soybeans grown in three types of soil suspensions, Arch. Agron. Soil Sci., 2019, vol. 65, pp. 74–87.
Chen, H. and Jiang, W., Application of high-throughput sequencing in understanding human oral microbiome related with health and disease, Front. Microbiol., 2014, vol. 5, pp. 1–6.
DeBruyn, J.M., Nixon, L.T., Fawaz, M.N., Johnson, A.M., and Radosevich, M., Global biogeography and quantitative seasonal dynamics of Gemmatimonadetes in soil, Appl. Environ. Microbiol., 2011, vol. 77, pp. 6295–6300.
DeSantis, T.Z., Hugenholtz, P., Larsen, N., Rojas, M., Brodie, E.L., Keller, K., Huber, T., Dalevi, D., Hu, P., and Andersen, G.L., Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB, Appl. Environ. Microbiol., 2006, vol. 72, pp. 5069–5072.
Dwivedi, S.L., Scheben, A., Edwards, D., Spillane, C., and Ortiz, R., Assessing and exploiting functional diversity in germplasm pools to enhance abiotic stress adaptation and yield in cereals and food legumes, Front. Plant Sci., 2017, vol. 8, pp. 1–30.
Edgar, R.C., Search and clustering orders of magnitude faster than blast, Bioinformatics, 2010, vol. 26, pp. 2460–2461.
Eichorst, S.A., Breznak, J.A., and Schmidt, T.M., Isolation and characterization of soil bacteria that define Terriglobus gen. nov., in the phylum Acidobacteria,Appl. Environ. Microbiol., 2007, vol. 73, pp. 2708–2717.
Fan, S., Li, B., Yu, F., Han, F., Yan, S., Wang, L., and Sun, J., Analysis of additive and epistatic quantitative trait loci underlying fatty acid concentrations in soybean seeds across multiple environments, Euphytica, 2015, vol. 206, pp. 689–700.
Garcia-Gonzalo, P., Del Real, A.E.P., Lobo, M.C., and Perez-Sanz, A., Different genotypes of Silene vulgaris (Moench) Garcke grown on chromium-contaminated soils influence root organic acid composition and rhizosphere bacterial communities, Environ. Sci. Pollut. Res. Int., 2017, vol. 24, pp. 25713–25724.
Gill, S.R., Pop, M., Deboy, R.T., Eckburg, P.B., Turnbaugh, P.J., Samuel, B.S., Gordon, J.I., Relman, D.A., Fraser-Liggett, C.M., and Nelson, K.E., Metagenomic analysis of the human distal gut microbiome, Science, 2006, vol. 312, pp. 1355–1359.
Gregory, A.S., Dungait, J.A.J., Shield, I.F., Macalpine, W.J., Cunniff, J., Durenkamp, M., White, R.P., Joynes, A., and Richter, G.M., Species and genotype effects of bioenergy crops on root production, carbon and nitrogen in temperate agricultural soil, BioEnerg. Res., 2018, vol. 11, pp. 382–397.
Hugenholtz, P., Goebel, B., and Pace, N., Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity, J. Bacteriol., 1998, vol. 180, pp. 4765–4774.
Germida, J.J. and Siciliano, S.D., Taxonomic diversity of bacteria associated with the roots of modern, recent and ancient wheat cultivars, Biol. Fert. Soils, 2001, vol. 33, pp. 410–415.
Jitendra, K., Sunanda, G., Priyanka, G., Sonali, D., Singh, T.R.S., and Shiv, K., Breeding strategies to improve lentil for diverse agro-ecological environments, Indian J. Genet. Pl. Br., 2016, vol. 74, pp. SI530–549.
Karamac, M., Orak, H.H., Amarowicz, R., Orak, A., and Piekoszewski, W., Phenolic contents and antioxidant capacities of wild and cultivated white lupin (Lupinus albus L.) seeds, Food Chem., 2018, vol. 258, pp. 1–7.
Khan, A.L., Asaf, S., Al-Rawahi, A., Lee, I.J., and Al-Harrasi, A., Rhizospheric microbial communities associated with wild and cultivated frankincense producing Boswellia sacra tree, PLoS One, 2017, vol. 12, pp. 1–18.
Li, Y., Guan, R., Liu, Z., Ma, Y., Wang, L., Li, L., Lin, F., Luan, W., Chen, P., Yan, Z., Guan, Y., Zhu, L., Ning, X., Smulders, M.J., Li, W., et al., Genetic structure and diversity of cultivated soybean (Glycine max (L.) Merr.) landraces in China, Theor. Appl. Genet., 2008, vol. 117, pp. 857–871.
Liang, J., Sun, S., Ji, J., Wu, H., Meng, F., Zhang, M., Zheng, X., Wu, C., and Zhang, Z., Comparison of the rhizosphere bacterial communities of Zigongdongdou soybean and a high-methionine transgenic line of this cultivar, PLoS One, 2014, vol. 9, pp. 1–10.
Lu, R.K., Methods of Soil Agricultural Chemical Analysis, Beijing: Agric. Sci. Technol. China, 1999.
Luo, S., Tian, L., Chang, C., Wang, S., Zhang, J., Zhou, X., Li, X., Tran, L.-S.P., and Tian, C., Grass and maize vegetation systems restore saline-sodic soils in the Songnen Plain of northeast China, Land Degrad. Dev., 2018, vol. 29, pp. 1107–1119.
Magoc, T. and Salzberg, S.L., Flash: Fast length adjustment of short reads to improve genome assemblies, Bioinformatics, 2011, vol. 27, pp. 2957–2963.
Mammadov, J., Buyyarapu, R., Guttikonda, S.K., Parliament, K., Abdurakhmonov, I.Y., and Kumpatla, S.P., Wild relatives of maize, rice, cotton, and soybean: Treasure troves for tolerance to biotic and abiotic stresses, Front. Plant Sci., 2018, vol. 9, pp. 1–21.
Manavalan, L.P., Guttikonda, S.K., Tran, L.S., and Nguyen, H.T., Physiological and molecular approaches to improve drought resistance in soybean, Plant Cell Physiol., 2009, vol. 50, pp. 1260–1276.
Mendes, L.W., Kuramae, E.E., Navarrete, A.A., van Veen, J.A., and Tsai, S.M., Taxonomical and functional microbial community selection in soybean rhizosphere, ISME J., 2014, vol. 8, pp. 1577–1587.
Mendes, R., Garbeva, P., and Raaijmakers, J.M., The rhizosphere microbiome: Significance of plant beneficial, plant pathogenic, and human pathogenic microorganisms, FEMS Microbiol. Rev., 2013, vol. 37, pp. 634–663.
Mebius, L.J., A rapid method for the determination of organic carbon in soil, Anal. Chim. Acta, 1960, vol. 22, pp. 120–124.
Purahong, W., Durka, W., Fischer, M., Dommert, S., Schops, R., Buscot F., and Wubet, T., Tree species, tree genotypes and tree genotypic diversity levels affect microbe-mediated soil ecosystem functions in a subtropical forest, Sci. Rep., 2016, vol. 6, pp. 1–11.
Ramette, A., Multivariate analyses in microbial ecology, FEMS Microbiol. Ecol., 2007, vol. 62, pp. 142–160.
Robledo-Mahon, T., Aranda, E., Pesciaroli, C., Rodriguez-Calvo, A., Silva-Castro, G.A., Gonzalez-Lopez, J., and Calvo, C., Effect of semi-permeable cover system on the bacterial diversity during sewage sludge composting, J. Environ. Manage., 2018, vol. 215, pp. 57–67.
Roleda, M.Y., Skjermo, J., Marfaing, H., Jonsdottir, R., Rebours, C., Gietl, A., Stengel, D.B., and Nitschke, U., I-odine content in bulk biomass of wild-harvested and cultivated edible seaweeds: inherent variations determine species-specific daily allowable consumption, Food Chem., 2018, vol. 254, pp. 333–339.
Shi, S., Tian, L., Ma, L., and Tian, C., Community structure of rhizomicrobiomes in four medicinal herbs and its implication on growth management, Microbiology (Moscow), 2018, vol. 87, pp. 425–436.
Spleck, E. and Bock, E., The Proteobacteria, Part A Introductory Essays, Bergey’s Manual of Systematic Bacteriology, Garrity, G., Ed., New York: Springer, 2004, vol. 2, pp. 149–153.
Sugiyama, A., Ueda, Y., Zushi, T., Takase, H., and Yazaki, K., Changes in the bacterial community of soybean rhizospheres during growth in the field, PLoS One, 2014, vol. 9, pp. 1–9.
Sulieman, S., Van Ha, C., Nasr Esfahani, M., Watana-be, Y., Nishiyama, R., Pham, C.T., Van Nguyen, D., and Tran, L.S., Dt2008: a promising new genetic resource for improved drought tolerance in soybean when solely dependent on symbiotic N2 fixation, Biomed. Res. Int., 2015. https://doi.org/10.1155/2015/687213
Sun, Y., Xu, S., Zheng, D., Li, J., Tian, H., and Wang, Y., Effects of haze pollution on microbial community changes and correlation with chemical components in atmospheric particulate matter, Sci. Total Environ., 2018, vol. 637–638, pp. 507–516.
Tripathy, K., Singh, B., Singh, N., Rai, V., Misra, G., and Singh, N.K., A database of wild rice germplasm of Oryza rufipogon species complex from different agro-climatic zones of India, Database (Oxford), 2018, pp. 1–6.
Wang, L., Zhang, T., and Ding, S., Effect of drought and rewatering on photosynthetic physioecological characteristics of soybean, Acta Ecol. Sin., 2006, vol. 26, pp. 2073–2078.
Wang, P., Zhang, H., Zuo, J., Zhao, D., Zou, X., Zhu, Z., Jeelani, N., Leng, X., and An, S., A hardy plant facilitates nitrogen removal via microbial communities in subsurface flow constructed wetlands in winter, Sci. Rep., 2016, vol. 6, pp. 1−11.
Wolińska, A., Kuźniar, A., Zielenkiewicz, U., Izak, D., Szafranek-Nakonieczna, A., Banach, A., and Błaszczyk, M., Bacteroidetes as a sensitive biological indicator of agricultural soil usage revealed by a culture-independent approach, Appl. Soil Ecol., 2017, vol. 119, pp. 128–137.
Wu, L., Yu, J., Shen, Q., Huang, L., Wu, D., and Zhang, G., Identification of microRNAs in response to aluminum stress in the roots of Tibetan wild barley and cultivated barley, BMC Genomics, 2018, vol. 19, p. 560.
Yu, H., Wang, C., and Tang, M., Fungal and bacterial communities in the rhizosphere of Pinus tabulaeformis related to the restoration of plantations and natural secondary forests in the Loess Plateau, Northwest China, The Sci. World J., 2013, pp. 1–12.
Zachow, C., Muller, H, Tilcher, R, and Berg, G., Differences between the rhizosphere microbiome of Beta vulgaris ssp. maritima-ancestor of all beet crops-and modern sugar beets, Front. Microbiol., 2014, vol. 5, p. 415.
Zhang, B., Zhang, J., Liu, Y., Shi, P., and Wei, G., Co-occurrence patterns of soybean rhizosphere microbiome at a continental scale, Soil Biol. Biochem., 2018, vol. 118, pp. 178–186.
Funding
This research was financially supported by the Special Foundation for State Major Basic Research Program of China (2016YFC0501202), Special Foundation for Basic Research Program in Soil of CAS (XDB15030103), Key Research Program of CAS (KFZD-SW-112-05-04), National Natural Science Foundation of China (41571255 and 41701332), Key Laboratory Foundation of Mollisols Agroecology (2016ZKHT-05), 135 Project of Northeast Institute of Geography and Agroecology (Y6H2043001), and Jilin Provincial Natural Science Foundation (20180520048JH and 20180519002JH).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflicts of interest. The authors declare that there are no conflicts of interest. Statement on the welfare of animals. This article does not contain any studies involving animals performed by any of the authors.
Rights and permissions
About this article

Cite this article
Ma, L., Luo, S., Xu, S. et al. Different Effects of Wild and Cultivated Soybean on Rhizosphere Bacteria. Microbiology 88, 720–728 (2019). https://doi.org/10.1134/S0026261719060109
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0026261719060109