
Abstract
Methods for carrying out coupling reactions in the chemistry of alkynes to form C–C bonds (oxidative dehydrocondensation reactions, Cadiot–Chodkiewicz reaction, and Sonogashira reaction) are analyzed and generalized. Protocols of synthesis of products of these reactions are also presented, including homogeneous and heterogeneous catalytic systems. In all cases, emphasis is placed on the kinetics and mechanisms of reactions with the discussion of the results of kinetic and spectrometric studies of the mechanisms of coupling reactions involving Cu(I, II, III), Au(I, III), Pd(0, I, II), and Fe (0, I, II, III) complexes. Particular attention is paid to the heterogeneous catalysis of oxidation reactions of alkynes with the participation of nanoparticles and nanoclusters of Pd, Au, Ag, and other metals. The nature of the intermediates containing these metals and the relationships between various oxidative and nonoxidative transformations of alkynes are discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
In the previous review [1], advances in the theory and practice of alkyne dimerization reactions were considered. Over a century and a half of research on the processes of alkyne oxidation, a huge amount of information has appeared that allowed one to make interesting generalizations regarding the mechanisms of such reactions in various catalytic systems.
Problems associated with the attitude of specialists in synthetic organic chemistry to redox reactions (RRs), or to the so-called oxidative reactions, and to the application of the concept of oxidation state (OS) of atom were discussed in our review [2]. A common opinion among organic chemists is to deny the usefulness of the definition and use of OS in organic and organoelement chemistry (see, e.g., chapter 19 of March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, which discusses this problem; see also our review [2]). This point of view was refuted in our review [2], however, the specifics of this review require some additional explanations to facilitate the perception of the main body of the text. When studying ORR or when choosing the conditions for carrying out inorganic, organic, and organoelement syntheses, it is necessary to know the stoichiometry of reactions. This is important in evaluating the selectivity of reactions, checking the material balance, and determining the required amount of an oxidizing agent or a reducing agent. Although several methods are known for determining the stoichiometry of reactions, the use of OS of atom and the electron balance based on it is the most general approach for all types of chemical reactions; it is the approach that is demonstrated in this review.
When determining the OS of the A atom in A–X compound, it is necessary to know the sign and magnitude of the OS of the X atom (or a group of atoms). For this purpose, the fundamental concept of electronegativity (EN) of an element is used, specifically, the Pauling, Allred–Rochow, or Batsanov EN scale (averaged values [2]). For example, in the case of C–H, the electronegativities of the C and H atoms are very different (the EN of the H atom is 2.2, and the EN of the C atom is 2.6); therefore, it is assumed that, in the heterolysis of the polar A–X bond, the electron pair of this bond is completely transferred to the atom with higher EN, i.e., to the C atom in the C–H bond. Therefore, the OS of the H atom is +I, or simply I, and the OS of the C atom is –I. The same situation occurs in the case of X = N, Hal, and Te. Since OS is a formal tool, in case of equality or proximity of ENs (C–I, P–H, C–S), it is necessary to use the existing agreement that, in these cases, the heterolysis leads to C+ and I–, P–, and S–. These assumptions used on the left and right sides of the stoichiometric equations give correct information about the electronic balances and RR stoichiometry. For example, in the CH3X molecule, where X = Hal, OH, NR2, SR, or PR2, the OS of the X atom or group of atoms is –I, and that of the carbon atom is +II. Specifically, if it is assumed that, in the compound K3W(CH3)6, the OS of the CH3 group is –I, then the OS of tungsten is III, and the reduction of K3W(CH3)6 to W(0) requires three electrons (3 mol of one-electron reducing agents or 1 mol of three-electron ones).
In the electrophilic substitution of a hydrogen ion in benzene with metal cations in the oxidation states II, III, and IV, the OSs of the carbon atom and the Ph group, equal to –I, do not change, but after the interaction with the electrophile I+ (oxidizing agent I2), their OSs increase from –I to I, and the OS of iodine decreases from I to –I. For example, in ArI, the OS of iodine is –I. If we want to replace a hydrogen atom with a Cl– ion, then we need an oxidizing agent that can oxidize the C(–I) atom to C(I) in phenyl and make it an electrophile that adds the chloride ion. It is also possible to oxidize the chloride ion to a Cl2 molecule, which, as an electrophile, leads to the replacements of a proton, and therefore, e.g., in ArI, the OS of iodine is –I. Thus, the definition of OS of atom is applicable to any covalent compounds.
Let us show the usefulness of OS values for writing stoichiometric equations by two more examples. In the oxidation of an ammonium ion by sodium hypochlorite, six electrons should be withdrawn from the N(–III) atom to form a nitrite anion, in which the OS of the N atom is III:
Let us oxidize CH4 to CO2 by the oxidizing agent \({\text{Fe(CN)}}_{6}^{{3 - }}\). Because the OSs of the H, C (CO2), and O atoms are I, IV, and –II, respectively, it is obvious that eight electrons should be withdrawn from the C atom to oxidize methane; i.e., it is necessary to use, e.g., eight anions of the one-electron oxidizing agent \({\text{Fe(CN)}}_{6}^{{3 - }}\) or 4 mol of a two-electron oxidizing agent. As a result, according to the electronic, charge, and material balances, the following equation is obtained:
Obviously, at least two reagents (substrates) should participate in an RR: an oxidizing agent and a reducing agent. If the reducing agent is a substrate to be oxidized (ethylene) and the oxidizing agent is reduced, such an ORR is typically called an oxidation process. If the oxidizing agent is a substrate to be reduced (ethylene), which accepts electrons in the course of the RR, and the reducing agent is oxidized, this reaction is called a reduction process.
In the case of organic compounds being oxidized by an oxygen molecule, there are no doubts about the definition of reducing agent and oxidizing agent; however, it is not always easy to understand whether or not the reaction belongs to RRs. It is the calculation of the total OSs of carbon atoms (or other elements) in the reactants and products that makes it possible to identify which substrate is oxidized and which is reduced, and whether or not the process belongs to ORRs.
Let us briefly consider this issue. If an oxidizing agent and a reducing agent are within the same molecule that isomerizes with the redistribution of OSs between atoms, such reactions should hardly be considered ORRs. Among examples are the Meyer–Schuster rearrangement of alkynols:
the Rupe rearrangement; and the Favorskii isomerization of alkynes; however, the Meyer–Schuster reaction is for some reason called redox isomerization [3]. The reactions of addition of HX molecules to alkynes and olefins, in which the OSs of carbon atoms in the reactant and product are redistributed, are not considered RRs, and they are also not oxidation reactions because the total OS of carbon atoms does not change when passing from alkyne to the product in these processes:
In contrast to the reactions of addition of HX molecules to unsaturated molecules, the reaction of alkyne with methyl iodide,
has all the features of an RR: the total OSs of carbon atoms in alkyne increases from –II to –I and the OS of a carbon atom in the methyl group decreases from –II to –III; i.e., alkyne is oxidized, and methyl iodide as an external oxidizing agent and a substrate included in the product is reduced.
This reaction is no different from the reaction of iodination of acetylene:
In view of the above, all RRs can be divided into two large groups, A and B. Group A comprises ORRs in which an oxidizing agent is external to the substrates, i.e., is not a product of substrate oxidation:
The oxidizing agents in reaction (IV) are O2, benzoquinone (BQ), MXn, I2, R3NO, TEMPO, ClCH2COCH3, and other compounds [3–5]. For example, in reaction
the oxidizing agent accepts 2е and 2Н+, turning into HCl and acetone, and the total OS of carbon atoms in two alkynes increases from –II to zero.
Group B comprises reactions with an oxidizing agent that, according to stoichiometry, is completely or partially contained in the RR products:


In reaction (VI), the total OS of carbon atoms in acetylene molecules increases from –IV to IV, and in reaction (IX), this parameter in two substrates (RC2H and CO) increases from I to III. In reactions (VII)–(IX), the oxygen molecule is both an oxidizing agent and a substrate, which enters the oxidation product of the hydrocarbon molecule. In this case, the total OS of atoms in the substrates, of course, does not change when passing to the product due to the conservation of the electronic balance, but the total OS of carbon atoms in ethylene, CO, and RC2H + CO decreases.
To ORRs involving alkynes, we also assign processes in which the total OS of carbon atoms in the oxidized alkyne changes when passing to the product as a result of the breaking of one or two π-bonds or triple bonds, and the ≡С–Н bond with the formation of ≡С–X, ≡С‒С≡, and С–О bonds.
The Cadiot–Chodkiewicz coupling
and the Sonogashira coupling (acetylene condensation)
are usually not considered as oxidative processes [3–7], and only in chapter 8 of Lu and Zhou’s monograph [8], which considers the oxidation of the ≡C–H bond, they are discussed as alkyne oxidation reactions.
Indeed, the OS of the terminal carbon atom in the alkyne in reaction (XI) increases from –I to 0, whereas the haloalkyne acts as an oxidizing agent (and a substrate), and the OS of the carbon atom in the ≡C(I)X fragment decreases from I to 0. In reaction (XII), \({\text{R}'}{\kern 1pt} {\text{X}}\) is also both an oxidizing agent and a substrate, partly included in the reaction product of alkyne oxidation. Formally, the carbenium ions \({\text{R}'}{\kern 1pt} {\text{C}}{\equiv} {{{\text{C}}}^{{\text{ + }}}}\) and \({\text{R}'}{{{\kern 1pt} }^{ + }}\) accept one electron each from the carbon atom ≡C(‒I)H of the alkyne. Note that the famous Heck reaction
which is no different from the reaction
as well as reaction (XII), can be attributed to RRs. Reactions (X) and (XII) are similar in OS: R+ is used instead of I+. Note that the Heck reduction reaction is also known [9]. The OS of silicon (equal to IV) remains constant during the reaction, but the OS of the H atom changes from –I to I:
Features of the mechanisms of various reactions of alkyne oxidation, depending on the nature of the oxidizing agent and the catalyst and the type of reactions, are advisable to consider within the framework of individual types of processes. As a result, it is possible to identify the “genetic” relationship between the mechanisms of reactions of different types through the general structures of intermediates.
The review presents the reactions of dehydrocondensation (oxidative coupling, oxidative dimerization) (IV), Cadiot–Chodkiewicz C‒C coupling (XI), and Sonogashira C‒C coupling (XII). The role of Cu(I, II, III), Pd(0, I, II), Au(I, III), and Fe(0, I, II, III) complexes in the oxidative transformations of alkynes is analyzed in detail.
The reactions of oxidative halogenation, amination, and oxidative carbonylation of alkynes, triple bond oxidation to form oxygen-containing products, including some processes of oxidative cyclization, will probably be considered in the next review.
CHAPTER 1. OXIDATIVE DEHYDROCONDENSATION (OD) OF ALKYNES
This reaction, which is very useful for organic synthesis and has very complex and interesting mechanisms, is the subject of many reviews [3–8, 10–16]. The OD reaction is used for several purposes: the synthesis of symmetrical and unsymmetrical diynes, as well as cyclic diynes in the case of intramolecular OD of ethynyl substituents:

and the synthesis of macrocycles:
where Ox is an oxidizing agent and Red is a reducing agent.
In addition, linear oligomers H(C≡C–Z–C≡C)nH and, in the case of acetylene, polyynes H(C≡C)nH are also formed [10].
1.1. OD Involving Cu(I, II) Complexes
Let us consider very briefly the main stages in the history of this reaction, which began in 1869 with the research of Carl Glaser, a well-known German academic and industrial chemist. Working since 1864 in the laboratories of Adolph Strecker and August Kekulé at the University of Tübingen in the field of the chemistry of benzene and its derivatives, he, together with Kekulé, moved to Bonn (Friedrich Wilhelm Rhine University) in 1867 and continued research on synthesis of cinnamic acid (trans-PhCH=CHCOOH), during which he discovered the reaction for the synthesis of diacetylenic compounds by oxidation of ethynyl derivatives of Cu(I):
The oxidizing agents were O2, K3Fe(CN)6, etc. Already in 1882, Adolf von Baeyer (see the review [14]) showed the synthetic utility of this reaction, which later received the name of Glaser, in a three-step synthesis of the indigo dye from m-nitrophenylpropiolic acid.
In 1936, Zal’kind and Fundyler studied the Newland catalyst for the synthesis of vinylacetylene from acetylene (CuCl–NH4Cl–HCl (0.2%)–H2O) in order to carry out the dimerization of terminal alkynes. At the boiling point of the solution, phenylacetylene was introduced into the reactor in portions in a flow of CO2! Diphenyldiacetylene(!) C16H10 was isolated from the solid precipitate, the selectivity of the formation of which was about 50% [17]. The expected phenylacetylene dimer (C16H12) was also found in the solution, and so was a small amount of C16 hydrocarbons with a higher hydrogen content. Further, it was shown that tertiary alkynols also form diacetylenic alcohols under the same conditions [18]. Possibly, copper chloride was not purified to remove the CuCl2 impurity, the solution was not degassed, and the CO2 flow contained oxygen. Having made the wrong conclusion about the release of two atoms or a molecule of hydrogen during the reaction, Zal’kind and Fundyler decided to use oxygen as an oxidizing agent [5] and, when studying the reactions of substituted phenylacetylenes at 55–65°C, air was passed through the reaction mixture [19]. In this case, e.g., di(p-tolyl)diacetylene was obtained with a yield of 90% of the taken alkyne. Thus, these researchers discovered a new catalytic reaction of oxidative dimerization of alkynes without using ethynyl derivatives of Cu(I) as reagents.
A similar situation has already been encountered in the chemistry of alkynes. Studying the Glaser reaction with various oxidizing agents, Straus in 1905 determined that PhC≡CCu in boiling acetic acid in the absence of oxygen transforms into precipitate PhC≡CCu(CuOAc) and hydrocarbon C16H12 (1,4-diphenylbutenyne) by the stoichiometric equation [20]
Thus, phenylacetylene formed by the hydrolysis of copper phenylacetylide dimerizes in the reaction catalyzed by copper(I) acetate. This is how the catalytic dimerization of alkynes was discovered even before the research of Newland (1932). Later, in a 1959 article [21], Straus’ observation was confirmed, and alkyne dimerization products were obtained from RC≡CCu in hot acetic acid even when aerating the solution for the oxidation of metallic copper formed by the dismutation of CuOAc (with the formation of only 1% diacetylene derivative). This system was revisited in 1997 [22]: Z- and E-1,4-dialkylbutenynes were synthesized and the formation of RC≡CCu and Cu(OAc)2 (dismutation product) was also determined. Advances in catalytic chemistry concerning dimerization, cross-dimerization, and oligomerization of alkynes were discussed in the monograph [7] and the review [1].
Zal’kind et al.’s works [17–19, 23, 24] were followed by numerous studies of the discovered catalytic reaction in the CuCl–NH4Cl–H2O system, which were described in detail in Shvartsberg and Fisher’s review in the collective monograph [5]. Just note 1947–1957 works [25–33], in which the conditions, compositions of the catalytic system, pH values, and solvents (water, methanol, ethanol, acetone, dioxane) were varied. Mkryan and Papazyan [28] studied the effect of preliminary oxidation of CuCl by oxygen. When studying the reaction of oxidative dimerization of propargyl alcohol, the technology of which was brought to the level of a pilot plant [32, 33], Reppe showed that, at [CuCl] = const, an increase in the [alkyne] : [CuCl] ratio in the range of 1 to 7 leads to a drop in the reaction rate, and at a ratio of 4, the oxygen consumption versus time curve is S-shaped. These observations led to an important conclusion that the binding of CuCl to alkyne (to form an acetylide compound) inhibits the reaction by decreasing the concentration of Cu(II), which oxidizes the intermediate copper acetylide (RC≡CCu) more rapidly than O2. As the acetylide is formed, there is an increase in the concentration of the hydrogen ion, which also inhibits the process. The important role of pH in the process of oxidative dimerization of alkynes was noted by Zal’kind and Aizikovich [18], who showed that, in the Cu2O‒NH4OH‒H2O system, the reaction occurs rapidly during the oxidation by oxygen even at 20°C.
Various copper(I, II) complexes in catalytic and stoichiometric OD reactions. Apparently, the first use of amines in copper chloride catalytic systems was described by Klebanskii et al. (CuCl–Py–H2O) [30] and Cameron and Bennett (CuCl–amine–amine·HCl–H2O) [31]. Cameron and Bennett [31] used NH3, tBuNH2, Py, Et3N, and ethylenediamine (EDA), specifically, EDA in tetrahydrofuran (THF). In the same year of 1957, Franke and Meister’s patent (CuCl in pyridine (Py) and cyclohexylamine) was published [34], and a little later, Hay’s studies were published [35, 36], in which the amine was used as a solvent or as a ligand in the form of small amounts of CuCl·amine complex in an organic solvent (alcohols, acetone). The most popular was the CuCl complex with tetramethylethylenediamine (TMEDA) in methanol. This technique became most widely used in organic synthesis and is known as the Hay method (or catalytic system) (more precisely, the Franke–Meister–Hay method).
In 1957–1959, another system for the oxidative dimerization of alkynes was developed, in which Cu(II) complexes were used as a stoichiometric oxidizing agent. Eglinton and Galbraith showed [37, 38] that, in Cu(OAc)2–Py or Cu(OAc)2–Py–CH3OH solutions, various alkynes are oxidized under homogeneous conditions (without formation of insoluble ethynyl copper compounds RC≡CCu) at a high rate and very selectively. The use of terminal dialkynes at low concentrations makes it possible to carry out intramolecular and intermolecular reactions of oxidative cyclization and oxidative polycondensation to form cyclic polyynes and macrocyclic enynes [3, 11–15]. This is how the Eglinton oxidative dimerization method or the Eglinton system appeared. It should be noted that, in the Zal’kind catalytic system, Sondheimer et al. [39–41] also obtained polyyne macrocycles, but the product yields and oxidation selectivity were very low. In 1961 [42], they applied the Eglinton method and synthesized cyclic enediynes, and in 1962 [43], from 1,5-hexadiyne (HC≡CCH2CH2C≡CH) in solutions of Cu(OAc)2 in Py, they confidently synthesized macrocycles containing n-mers of this alkyne with n = 3, 4, 5, and 6, the hydrogenation of which gives saturated cycles C18, C24, C30, and C36 (Scheme 1)!

Scheme 1 . Unsaturated macrocycles.
Thus, copper(I, II) complexes participate in the reaction of oxidative dimerization (oxidative coupling, oxidative dehydrocondensation) of alkynes in various systems.
I. The catalytic oxidative dimerization of alkynes, discovered by Zal’kind et al., is performed in two systems:
Ia. Zal’kind (CuCl–NH4Cl–HCl–H2O) with variations;
Ib. Franke–Meister–Hay (CuCl–amine–organic solvent).
II. The stoichiometric reaction of OD of alkynes occurs in the Eglinton system (Cu(OAc)2–Py–organic solvent). Studies showed that, in this system, the dimerization reaction is autocatalytic in Cu(I), and the addition of Cu(I) compounds can significantly accelerate the oxidation reaction, depending on the conditions.
Specific conditions and results of oxidative dimerization in systems Ia, Ib, and II (with various variations) were in the 1967 monograph [5]. More than 360 syntheses were described, and in Shvartsberg and Fisher’s review in this collection, the catalytic reaction of oxidative dimerization of alkynes was proposed to be called the Glaser–Zal’kind reaction (at least for systems Ia and Ib).
Over the past 50 years, many studies have been made aimed at modifying the Glaser–Zal’kind and Eglinton methods for the synthesis of dialkynes and at expanding the scope of the OD reaction for the preparation of linear and cyclic diynes, oligomers, polymers, and macrocycles. For example, the review [16] presented tables describing 68 procedures for the synthesis of various diynes and 17 procedures for the preparation of oligomers and polymers in Hay systems with fantastically complex structures of products. Varying the nature of bases, ligands, oxidizing agents, solvents, and conditions made it possible to choose fairly universal systems efficient at room temperature for alkynes of various nature. Let us give several examples of such processes in copper-containing systems. OD reactions are carried out both in ionic liquids [44], and in polyethylene glycols [45], and even in amine alone (CuI–amine) [46] with the choice of optimal bases and ligands for Cu(I) [47]. The use of PhI(OAc)2 as an oxidizing agent in the CuCl–Et3N–AN (AN is acetonitrile) system enables one to perform the OD process with high yields at room temperature for 10 min for aromatic and aliphatic alkynes [48]. Variants of the Eglinton system are also used very efficiently in synthetic practice for the synthesis of diynes with substituents having many reactive functional groups, e.g., to obtain (bis-β-lactam)-1.3-diynes by of OD of N‑ and C-alkynyl-2-azetidinones [49]. In that work, Cu(OAc)2 in AN in the presence of Et3N or solid K2CO3 at room temperature was used. Interestingly, in oxygen or argon atmospheres with both bases, the yields of products reached 98–100% in the same time intervals (from 4 to 20 h). A comparison of the catalytic properties of various copper(I, II) compounds in CH2Cl2 in the synthesis of bis-β-lactam)-1,3-diynes in the presence of various bases and air at 25°C for 3 h showed [50] that the highest yields (93–98%) were obtained using pyrrolidine and piperidine with yields of 93–98% in CuCl, Cu2O, Cu(OAc)2∙H2O, Cu2SO4, CuCl2∙2H2O, and Cu(OTf)2 solutions. Cu(OAc)2∙H2O was recommended as the most available (!) salt that catalyzes the OD of various alkynes with the participation of oxygen as an oxidizing agent. In this system, the cross-coupling of alkynes also occurs successfully with yields of 75–99%.
Collins and colleagues [51–53] proposed an interesting variant of oxidative macrocyclization of diethynyl compounds in the synthesis of cycles containing conjugated 1,3-diynes in a two-phase system of CuCl or CuCl2 with the addition of Ni(II) salts. It was shown [51] that the presence of two phases—polar and nonpolar ones—gives rise to macrocycles with high yields at relatively high substrate concentrations. For example, the formation of a macrocycle from HC≡C(CH2)7OCO(CH2)3C≡CH in an amount of 0.36 mmol in a two-phase system (CuCl2–Ni(NO3)2–Et3N–Py, 60°C) requires only 15 mL of the 2 : 1 PEG400–methanol solvent, whereas in a homophase system, this requires as much as 1800 mL. An even more efficient system was that containing PEG1900 modified with tetramethylethylenediamine (TMEDA): (Т–PEG1900), or (Me2NCH2CH2N(Me)–(CH2CH2O)40), which ensures better solubility of Cu(I, II) and Ni(II) complexes in the polar phase. The two-phase strategy allows one to perform the catalytic cyclization reaction (with O2) at the interface between two phases (or in the polar phase) at a relatively small dilution and virtually without the formation of linear polymers. When T–PEG1900 is used, dialkyne cyclization with a concentration of 0.03 M is carried out in 5 mL of solution (instead of 600 mL of a solution with a substrate concentration of 0.0002 M). In this case, the product yield increases from 11 to 65%. This macrocyclization technique is called the Collins strategy. It was shown [52] that its efficiency noticeably increases when microwave radiation is used. The reaction time reduces by a factor of 4–24 at 100°C and 1 atm O2 with maintaining high yields of macrocycles of 61–81%, depending on the nature of the dialkyne. This Collins technique was successfully applied to the synthesis of a macrocyclic precursor in the production of the hepatitis C virus protease inhibitor vaniprevir [53].
OD reaction mechanisms. Let us consider the development of concepts of mechanisms of the OD reaction in various systems containing Cu(I) and Cu(II) complexes. In the pioneering works of Zal’kind et al. [23, 24, 27], Mkryan and Papazyan [28], and Reppe [33], it was determined that the OD reaction occurs over a wide pH range in CuCl solutions with oxygen as an oxidizing agent and is accelerated by the addition of copper(II) salts. Studying the kinetics of oxidative dimerization of alkynes in aqueous solutions of CuSO4 in a buffer system at pH 6, Baxendale and Westcott (see [11]) showed that the reaction rate is described by equations of the first order in the concentrations of Cu(II) and alkyne and depends on 1/[H+]. In this case, the reaction rate is determined by the stationary concentration of Cu+. Thus, the process is autocatalytic.
Klebanskii et al. [30] studied the OD reaction in CuCl–NH4Cl–H2O solutions with oxygen as an oxidizing agent. The role of oxygen, in their opinion, is the oxidation of CuCl to CuCl2; it is CuCl2 oxidizes the alkyne, but only in the presence of bases (NaOH, NH3, Py, Et3N). In the CuCl2–NH3–H2O system, the reaction rate is described by the equation
where k is the observed rate constant.
Klebanskii et al. [30] gave no explanation of the role of CuCl in acidic solutions in the presence of oxygen (conditions of Zal’kind, Reppe, and other authors). The presented scheme of the mechanism [30] includes the formation of RC≡C– anions and their oxidation by a copper(II) complex to RC≡C• radicals, the rapid recombination of which gives dialkyne.
The works of Eglinton and Galbraith [11, 37, 38] and Sondheimer [42] on the OD of alkynes in Cu(OAc)2–Py solutions stimulated kinetic studies of this and related systems [13, 54, 55]. Clifford and Waters [54] studied the oxidation of propargyl alcohol by Cu(II) acetate in pyridine in the presence of piperidine buffer (PiPy–AcOH). The process was determined to be autocatalytic in CuOAc, in excess of which the order of the reaction in [Cu(OAc)2] becomes zero. The reaction rate is described by an equation of fractional orders in alkyne and in [CuOAc] and increases with increasing [PiPy]. The derivation of the kinetic equation [54] ignored the material balances for the concentrations of alkyne and compounds of copper(I); the role of Cu(I) was reduced to the formation of RC≡CCu from the anion RC≡C– and Cu(I) (!).
Bohlmann et al. [55] compared the activities of two systems in the OD reaction: in acidic solutions at pH 3 in the system CuCl–CuCl2–HOCH2CH2NH∙HCl–HCl–CH3OH (80%) similar to those studied by Zal’kind, and in the Eglinton system Cu(OAc)2–Py–CH3OH–H2O. It was determined that an increase in the acidity of the alkyne with an increase in the number of conjugated triple bonds in R(C≡C)n–C≡CH leads to an increase in the OD rate in a basic medium and to a decrease in the rate in acidic media (pH 3). In Bohlmann et al.’s opinion [55], an increase in the degree of conjugation in alkyne is accompanied by a decrease in its ability to form Cu(I) π-complexes and, consequently, a decrease in the concentration of copper acetylide compound R(C≡C)nC≡CCu, which is oxidized by Cu(II) complexes in an acidic medium . In systems with a fivefold excess of the concentration of Cu(II) complexes over [RC≡СH], the second order in alkyne was obtained in the reaction rate equation, which contradicts the previous schemes of the reaction mechanism with the formation of RC≡CCu oxidized by Cu(II) complexes or other oxidizing agents up to RC≡C•. Bohlmann et al. [55] proposed a mechanism involving two molecules of ethynyl Cu(II) complexes with structure 1.

It is shown below that this mechanism can also occur without the participation of dimeric Cu(I) complexes.
In a series of papers by Fedenok, Berdnikov, and Shvartsberg [56–63], the concepts of the mechanisms and kinetic models of OD in Cu(I)- and Cu(II)-containing systems were significantly expanded. The “stoichiometric” and catalytic oxidation of alkynes in acidic (with amine–AcOH buffer) and basic (without acid) media were studied.
System (CuCl + O 2 )–Et 3 N–AcOH–Py (I)
The reaction of OD of PhC≡CH (A) was studied in an inert atmosphere (N2) with the preliminary oxidation of CuCl by oxygen [56]. The reaction rate was moni-tored by the change in the optical density D of a Cu(II)* solution at 680 nm (Cu(II)* are copper complexes obtained by the oxidation of CuCl, and [A]0 >> [CuCl]0, where [A]0 is the initial PhC≡CH concentration). Divalent copper formed by the complete oxidation of CuCl in the presence of acetic acid in pyridine is virtually indistinguishable spectrophotometrically from Cu(OAc)2. The studies determined the second order in [Cu(II)*]. The normalized quantity \(\left[ {{\text{A}'}} \right]\)0/[Py] was used as the concentration of phenylacetylene, and the second order in \(\left[ {{\text{A}'}} \right]\) was determined. The dependence of the reaction rate on [AcOH] is described by an equation of the minus second order (1/[AcOH]2). These results are described by the empirical kinetic equation
where a is an empirical parameter, B is a base, D is the optical density, and t is time.
The use of the normalized concentration of the alkyne was hardly necessary, since the concentrations of pyridine (0.5 M) and the alkyne (0.5–5.0 M) are much higher than the concentrations of CuCl and Cu(II)* (10–3 M), and [Py] remains almost constant after replacing Py by the alkyne in Cu(II) complexes in the formation of π-complexes. The order in [CuCl] turned out to be zero.
The second order in alkyne concentration, which was determined by Bohlmann [13] and by Fedenok and colleagues, excludes the appearance of RC≡C• radicals as intermediates of OD of alkynes: their recombination is an incomparably faster process in comparison with the steps of transformation of alkyne into this radical. Fedenok et al. [56] estimated the enthalpy of the formation of the RC≡C• radical from alkynes by the reaction
involving bases B and showed that \(\Delta H_{{298}}^{ \circ }\) of this reaction is ≥60 kcal/mol, which also excludes the formation of RC≡C• by OD of alkynes. Note that the oxidation of ethynyl carbanions in ethynyl compounds of alkali metals (MC≡CR) by strong oxidizing agents \(({\text{Cu(N}}{{{\text{H}}}_{{\text{3}}}}{\text{)}}_{4}^{{2 + }},\) KMnO4) can also give rise to the radical RC≡C• [7].
The zero order in Cu(I) in this system is probably due to the rapid formation of ethynyl Cu(II) complexes with the participation of the buffer, CuCl2, and other oxidized forms of Cu(II)*. The kinetic data suggest that the process in system I occurs with a rate-determining step, the transition state of which includes two RC≡C– anions and two Cu(II) ions (complexes). Therefore, the following reaction mechanism was proposed:
Taking into account the material balance of [Cu(II)]∑, an empirical kinetic equation with rate-determining step (XIX) and an expression for the effective rate constant keff (Eq. (3)) were obtained. From the dependence of this constant on temperature (20–50°C), the observed activation energy was determined to be 21 kcal/mol.
where l is the thickness of the solution layer, εs is the extinction coefficient, and \(\sum {{K}_{{i - 1}}}\left[ {{{L}_{{i - 1}}}} \right]\) characterizes Cu(II) complexes with other ligands (CH3COO–, Cl–, Et3N) and is a constant value at high concentrations of ligands and similar extinction coefficients for complexes with different ligands.
The observed facts of inhibition of the reaction by the chloride ion and acceleration by the acetate ion can be correctly explained after determining the forms of Cu(II)* and the compositions of the Cu(I) and Cu(II) complexes. It is known [64, 65] that such forms in a buffer system can be ClCuOOH, ClCuOH, CuCl2, and Cu(OAc)2. The main kinetic laws found in system I are also retained when passing to the 2 : 1 Py–H2O solvent, and the reaction rate in the aqueous system is an order of magnitude higher than that in the anhydrous one. The study of the kinetics of oxidation of 13 alkynes in the similar system CuCl–PiPy–AcOH–Py at 50°C confirmed the quadratic dependences of the rate on [Cu(II)*] and on the concentration of alkyne [62]. When passing to an aqueous ammonia solution of CuCl2 in the oxidation of propargyl alcohols, the kinetic laws described above for CuCl solutions and in the Py–H2O solvent are also retained [57]. It was also found that the values of keff in acidic media (BH+) are correlated for substituted phenylacetylenes with the Hammett σ-constant (ρ = 2.2) [58].
System (CuCl + O 2 )–Py (II)
Two systems were compared: one with buffer (I) and the other without buffer (II) [62]. After preliminary oxidation of CuCl by oxygen, phenylacetylene was introduced into the reactor. In this system, in the absence of additional amines and AcOH, an induction period appeared in the time curves of the formation of the reaction product, which was due to the formation of Cu(I) and, consequently, to autocatalysis. The addition of CuCl to the initial (oxidized) solution removed the induction period. These observations indicated the participation of Cu(I) complexes in the process with the formation of PhC≡CCu. In the oxidation of copper phenylacetylide in the same system with Cu(II)*, diyne was formed rapidly with a yield of approximately 50% [62]. Since of the two oxidized forms of copper, e.g., CuCl2 and Cu(II)*, chloride complexes oxidize copper acetylides rapidly at a rate of controlled diffusion, it can be concluded that the oxygen-containing forms of Cu(II)* are less active in this process. The addition of Et4NCl to the same system was accompanied by the rapid formation of 100% PhC4Ph. These observations confirmed that RC≡CCu intermediates can also participate in the catalytic process with oxygen as an oxidizing agent.
System CuCl–Et 3 N–AcOH–Py (III)
Let us now consider the results of the study of the kinetics of the catalytic syntheses of diynes with oxygen by Fedenok et al. [59, 62] under conditions close to those for studying the kinetics in systems I and II.
The oxygen consumption rate was monitored with a pressure-measuring setup, and the initial reaction rates were used to describe kinetic laws. It was found that the CuCl oxidation rate is quite high, and Cu(I) at \({{P}_{{{{{\text{O}}}_{2}}}}}\) > 450 mm Hg is completely converted into Cu(II). In the region of zero order in \({{P}_{{{{{\text{O}}}_{2}}}}}\), the same quadratic dependences in \(\left[ {{\text{A}'}} \right]\) and [Cu(II)]∑ were observed; i.e., the reaction mechanism is the same as that in system I (with preliminary oxidation of CuCl). The well-known Hay catalytic system (CuCl–amine–methanol) used no buffer; therefore, system IV in the catalytic version was also studied [61, 62].
System CuCl–Py (IV)
The study showed that the kinetics of phenylacetylene oxidation by oxygen without a buffer is much more complicated than that in system III:
– there is an extreme dependence of the initial reaction rate R0 on \({{P}_{{{{{\text{O}}}_{2}}}}}\);
– the reaction order in [A] is variable from zero in the region of increase in R0 with increasing \({{P}_{{{{{\text{O}}}_{2}}}}}\) to ~1.5 in the region of decrease in R0 with \({{P}_{{{{{\text{O}}}_{2}}}}}\);
– the order in [CuCl]0 is ~2 at low \({{P}_{{{{{\text{O}}}_{2}}}}}\).
Because the rate of oxidation of CuCl by oxygen without a buffer is much lower than that with it; an acid (BH+) is needed for the oxidation of CuCl. The rate of oxidation of phenylacetylene with Cu(II) complexes without AcOH is higher due to the 1/[ВН+]2 term in Eq. (2). When phenylacetylene is oxidized by oxygen under conditions of complete oxidation of CuCl, an induction period is observed, which disappears if CuCl is added at the beginning of the reaction. In system IV, a quasi-stationary concentration of Cu(I) is established during the reaction, which leads to catalysis of the process by copper(I) chloride to form acetylide complexes RC≡CCu [62]. The zero order in [Cu(II)] in the bufferless system is probably due to a change in the limiting step—slow formation of RC≡CCu because of the absence of an amine (PiPy, Et3N)—and to the rapid oxidation of acetylides by such strong oxidizing agents as CuCl2, ClCuOOH, or ClCu=O. Empirical kinetic equation
obtained in system IV for the initial reaction rates describes the entire set of experimental data (α1 and α2 are empirical parameters).
The high orders in \({{\left[ {{\text{A}}{\kern 1pt} '} \right]}_{0}}\) and [CuCl]0 may be due to the participation of dimeric ethynyl copper (I) complexes (RC≡CCu)2 in the OD reaction under these conditions. It was assumed [52] that the decrease in R0 at high \({{P}_{{{{{\text{O}}}_{2}}}}}\) is related to the appearance of the free chloride ion, which prevents the formation of dimeric Cu(I) acetylides. Indeed, in system IV, phenylacetylene is not oxidized catalytically after the addition of Et4NCl at a ratio to CuCl of 1 : 1. Thus, it was assumed that, in the formation of PhC≡CCu and its dimer, the oxidized forms of the Cu(II)* type are converted into \({\text{CuCl}}_{n}^{{2 - n}}\) due to the release of chloride ions with subsequent rapid oxidation of the dimer by this chloride complex to [RC≡CCu(II)]2 and rapid formation of PhC4Ph and \({\text{CuCl}}_{n}^{{1 - n}}.\) Within this hypothesis, it turns out that the chloride ion in this system has negative and positive effects. Experiments showed that the rate of oxidation of Cu(I) acetylides by copper chloride is so high that it is controlled by the diffusion rate. For example, when pyridine solutions of Cu(I) phenylacetylide and CuCl2 are poured together, the reaction of the formation of diphenylbutadiyne ends already during stirring. Just as rapid is the process when mixing pyridine solutions of Cu(I) phenylacetylide and oxygen-oxidized CuCl, with the difference that it occurs to 50% completion, which is due to the amount of CuCl2 in the composition of Cu(II)*. But if chlorine anions in the form of tetraethylammonium chloride are introduced into CuCl preliminarily oxidized by oxygen, then the reaction occurs rapidly and to 100% completion.
System Cu(OAc) 2 –PiPy–AcOH–Py (V)
This system is similar to the Eglinton system and to the system studied by Clifford and Waters [54] in the oxidation of propargyl alcohol. A more detailed study of this reaction in system V by Fedenok and colleagues [60, 62, 63] showed that the initial reaction rate R0 in the absence of CuOAc strongly depends on the water concentration, and in the absence of H2O, it is close to zero. The quasi-stationary rate of the decrease in [Cu(II)] with the appearance of CuOAc is virtually independent of [H2O] and increases with an increase in the concentration of piperidine and [BH+] (at a constant AcO– concentration). To explain this phenomenon, it was assumed that the acid shifts the equilibrium of the formation of Cu(I) alkoxide, which is inactive in the process.

where P are the reaction products.
The ionization of the π-complex (X1) to form an acetylide compound (X2) occurs, in Fedenok and colleagues’ opinion, irreversibly due to its rapid dimerization and further rapid oxidation of the dimer by Cu(II) acetate. The orders of the reaction in \(\left[ {{\text{A}'}} \right]\) and [CuOAc] under quasi-stationary conditions are less than 1, and the order in [PiPy] is equal to 1 at [B]/[BH+] = const. Zero order was obtained in the concentration of the oxidizing agent.
According to Fedenok and colleagues, in this case, too, the reaction occurs by the formation of the (RC≡CCuOAc)2 dimer from dimer X3 with Cu(I).

Kinetic equation (5) for the mechanism of the oxidation of propargyl alcohol (where \(X_{1}^{'}\) in the scheme of the mechanism is the alkoxide CuOCH2C≡CH) with the rate-determining step in the synthesis of copper(I) acetylide X2 was obtained taking into account the material balance of [Cu(I)]:
Although the existence of π-complexes of Cu(II) with olefins and alkynes has not yet been established, the appearance of RC≡CCuOAc at the initial stage of the reaction in the absence of Cu(I) can occur with the participation of even a very weak π-complex of alkyne with Cu(II) in the presence of amine bases. In connection with the above facts and hypotheses, it is interesting to compare the results of studying the kinetics of the catalytic syntheses of diynes with oxygen in basic media [59, 62] with the kinetics of the OD reaction in acidic Zal’kind systems in the oxidation of alkynes by Cu(II) chloride [66–68].
System CuCl 2 –CuCl–MCl–HCl–H 2 O (VI)
The kinetics of the reaction of OD of methylacetylene (MA) was studied in the CuCl2–CuCl–MCl–HCl–H2O system, where [MCl] = 5 M and M = Li, Na, K, under conditions of the simultaneous formation of chloromethylacetylene (CMA) [66–68]. Under these conditions, at [CuCl2]0 = 0.1–0.5 M and [CuCl]0 = 0.01–0.5 M, only mononuclear chloride complexes of Cu(I) and Cu(II) are present in the solution [7, 69, 70]. The [Cu(II)]/[Cu(I)] ratio was determined from the results of measurements of the platinum electrode potential, which was described by the Nernst equation due to the preservation of the constancy of all activity coefficients under conditions of a high cation background and a high concentration of chloride ions. The concentration of H3O+ ions was monitored by an empirical linear equation relating the pH values to log[H3O+] in the systems under study.
The kinetics was studied by measuring the initial reaction rates in a closed thermostated reactor (30–60°C) with an efficient precessional stirrer ensuring the kinetic control. The course of the reaction was monitored by analyzing gas samples taken from the reactor [65, 67]. Since chloroalkynes are obtained through the oxidation of ethynyl copper(I) compounds [69, 70], the study of the kinetics of two parallel routes (XXI) and (XXII) with an obvious species that couples them [70] allows answering a number of fundamental questions related to the mechanisms of oxidative transformations of organometallic copper compounds:
The experimental dependences of the rates of the formation of dimethyldiacetylene (RDMDA) and chloromethylacetylene (RCMA) on the concentrations of the reagents and components of the catalytic system are expressed by Eqs. (6) and (7) with the notation of the rate constants as in Hoan et al. [67]. The dependence on [Cl–]0 was determined by replacing LiCl by LiClO4 at [Li+]∑ = 5 M. It turned out that an increase in [Cl–]0 slows down the OD process and accelerates the process of oxidative chlorination of MA.
where PMA is the partial pressure of MA.
It follows from the form of the obtained equations that both processes occur with rate-determining steps, and that the material balances of [CuCl2]0 and [CuCl]0 do not include π- and σ-complexes of Cu(I) and Cu(II) with alkynes and products reactions. Thus, the degrees \({{F}_{{{\text{C}}{{{\text{u}}}^{{\text{ + }}}}}}}\) and \({{F}_{{{\text{C}}{{{\text{u}}}^{{{\text{2 + }}}}}}}}\) of complexation of Cu(I) and Cu(II), respectively, are independent of the concentrations of MA, DMDA, and CMA. An analysis of the states of Cu(I) and Cu(II) in solutions of NH4Cl and alkali metal chlorides [7, 67, 71] suggested that, in a solution with [LiCl] = 5 M, more than 80% Cu(I) is in the form of complexes \({\text{CuCl}}_{3}^{{2 - }}\) and \({\text{CuCl}}_{4}^{{3 - }},\) whereas the fraction of complexes \({\text{CuCl}}_{3}^{ - }\) and \({\text{CuCl}}_{4}^{{2 - }}\) is ≤30% of Cu(II). A complete analysis of kinetic models (6) and (7), taking into account the degrees of complexation \({{F}_{{{\text{C}}{{{\text{u}}}^{{\text{ + }}}}}}}\) and \({{F}_{{{\text{C}}{{{\text{u}}}^{{{\text{2 + }}}}}}}},\) and the activities of Cu+, Cu2+, and Cl– ions, was made by Hoan et al. [66, 67].
The first question that arises when analyzing the mechanisms corresponding to the kinetic model of DMDA synthesis is the sequence of participation of CuCl and CuCl2 molecules in the steps of the mechanism. To explain the second order in [CuCl]0, one can propose two hypotheses (A and B) with different sequences of participation of CuCl and CuCl2 molecules at different steps of the process:
A: (1) CuCl, (2) CuCl2, and (3) CuCl;
B: (1) CuCl, (2) CuCl, and (3) CuCl2.
Let the possible hypotheses about the mechanisms of the formation of dialkynes be considered using simple forms of chloride complexes \({\text{CuCl}}_{2}^{ - }\) and \({\text{CuCl}}_{3}^{ - }.\)
Variant A. The first two steps are quite obvious (in a simplified version) and are quasi-equilibrium:
Ethynyl complex X1 is naturally formed through an intermediate π-complex [7, 70, 71]. Intermediate X2 is a mixed complex with possible fast electron transfer between the Cu(I) and Cu(II) forms. The second-order rate constant for the electron transfer between \({\text{CuCl}}_{2}^{ - }\) and \({\text{CuCl}}_{3}^{ - }\) is 5 · 108 L mol–1 s–1 [72, 73]. Therefore, complex X2 can be represented by electronic mesomers 2:

Intermediate X2 due to the fast intramolecular electron exchange cannot probably lead to the formation of RC≡CCuCl (X3) and dimer (RC≡CCuCl)2, which is necessary for oxidative transformation to dialkyne, but if intermediate X2 is a π-complex with structure 3, which is highly likely, its interaction with \({\text{CuCl}}_{2}^{ - }\) can lead to the disappearance of the π-complex in the rate-determining step (A3) and the appearance of a dimer capable of converting to dialkyne in the fast step.

Variant B. The participation of the second CuCl molecule in the OD process is also possible at an earlier step. Ethynyl Cu(I) compounds in the absence of strong ligands (R3N, Py, DiPy, PR3) are known to be poorly soluble in aqueous solutions. It is for this reason that most reactions of addition of HX molecules and oxidation involving alkynes (dimerization, hydrocyanation, diene synthesis, oxidative chlorination at the ≡С–Н bond) occur in more concentrated CuCl solutions than those used in the study of OD kinetics [66, 67]. In concentrated solutions, organometallic compounds of copper(I) are highly soluble due to the formation of polynuclear complexes [7, 70, 71]. In accordance with these considerations, this variant of the mechanism can contain the following sequence of steps with the first two quasi-equilibrium steps:
where intermediate \({\mathbf{X}}_{2}^{'}\) has the structure of π-com-plex 4:

The redox transformation of two molecules of the ethynyl complex RC≡CCuCl (X3) with the simultaneous formation of a C–C bond is considered, starting with Bohlmann [13], through the dimeric Cu(II) π‑complex with structure 5 (similar to complex 1), which is formed in the quasi-equilibrium step to steps (A4) and (B4):

Dimeric π-complexes of this type for metals that do not form stable π-complexes are extremely rare. For example, a dimeric π-complex consisting of two ethynyl Zn(II) complexes (Ph3P)ZnEt(C≡CPh) was isolated [74], and its structure 6 was determined by X‑ray powder diffraction analysis (XRD):

Bicyclic complexes of copper and other metals in the chemistry of alkynes. Since the transition state of the step of conversion of dimer 5 to dialkyne seems to be very complex (breaking of two C–Cu bonds and the simultaneous formation of a C–C bond), one can assume the existence of one more bicyclic intermediate, dicuprabicyclohexadiene (X5), with structure 7 [7, 70] (known for Ti(IV) complexes [75–80]), which is an organometallic compound of Cu(III).

The formation of two additional Cu–C bonds and a C–C bond compensates for the energy consumption for the breaking of two π-bonds in the dimer (RC≡CCuCl)2. Organometallic compounds of Cu(III) have now been synthesized, and their reactivity has been fairly well studied [81] (see below). In this regard, steps (A4) and (B4) can be detailed by the following sequence of fast elementary acts, where X4 is dimeric π-complex 5 and X5 is bicyclic complex 7:
The study of the transformations of ethynyl complexes Cp2Ti(C≡CR)2 and their reactions with dialkynes RC4R [75–80] determined that, depending on the nature of R in alkynes, dimeric ethynyl titanium(III) π-complexes of the type of complexes 1, 5, and 6 (R = SiMe3) or bicyclic titanium(IV) complexes of type 7 (R = Ph, Me, tBu). In this case, the reactions of π-complexes Cp2Ti(RC≡CR) (or \(''{\text{C}}{{{\text{p}}}_{{\text{2}}}}{\text{Ti}''}\)) with dialkynes also gives dimeric ethynyl titanium(III) π-complexes (R = SiMe3) or bicyclic titanium complexes (without breaking the C–C bond in dialkyne) in case R = Ph, Me, tBu (scheme 2)!

Scheme 2 . Mechanisms of the formation of bicyclic titanium(IV) complexes and dimeric ethynyl titanium(III).
Alkyne zirconium π-complexes Cp2Zr(RC≡CR) [82] in reactions with dialkynes are converted only into dimeric ethynyl zirconium(III) π-complexes of the type of complexes 1. Dimeric η1-η2-ethynyl π-complexes of type 1 can also contain different metals Cp2Ti (or Cp2Zr) with Ni(PPh3)2 [83]. The interaction of [Cp2Ti(C≡CR)]2 or the cumulene complex of Cp2Ti and 1,4-substituted butadiyne with NiL2 is a way to break the C–C bond in diyne [84]. Breakage of C–C bonds in diynes was also observed in the case of cumulene complexes of diynes with Cp2Ln and Cp2Ce [85]. The issues of mutual transformations of diethynyl π‑complexes into diynes and of diynes into diethynyl π-complexes were discussed in the reviews [86, 87], and the 2022 review [88] encompassed the synthesis, structure, and functions of titanium complexes, including dimeric ones, which are shown in Scheme 2. The review [87] noted that bicyclic dimetallahexadiene complexes can also contain different metals, e.g., Ti(IV) and Si(IV), or Zr(IV) and Si(IV) (7а), where X = SiR2 (see also [83]).

Two monoethynyl complexes of Sm(III), Ce(III), and Nd(III) easily form bimetallic complexes with the cumulene ligand \({\text{Cp}}_{2}^{*}\)M(Ph)C=C=C=C(Ph)M\({\text{Cp}}_{2}^{*}\) and cumulene products by the reactions of these complexes with alkynes [89]. Transformations of bicyclic Cu(III) complex 7 can also lead to Cu(II) cumulene intermediates and cumulene products.
Let us note another possible mechanism of the transformation of the intermediate RC≡CCuCl (X3) into products of the OD reaction, specifically, the reaction of dismutation of two X3 molecules to form diethynyl complex X6 with structure 8:

The redox decomposition of X6 with the transfer of two electrons to copper(II) atoms gives dialkyne.
The mechanism of the formation of CMA obviously includes the sequential oxidation of the ethynyl Cu(I) complex by two CuCl2 molecules:
C: (1) CuCl, (2) CuCl2, and (3) CuCl2.
Thus, the first two steps of this mechanism correspond to steps (A1) and (A2). The transformation of the RC≡CCu2Cl3– (X2) (A2) complex requires one more molecule of the oxidizing agent.
Intermediate X2 is a species that couples two routes (reactions (XXI) and (XXII)) with the following ratio of rates along the routes:
In the mechanism (С), the route-coupling species is intermediate X1 with the same ratio (8) of rates along the routes. The observed dependences of RDMDA and RCMA on [Cl–]0 do not follow from the considered steps of the mechanism, but are the result of approximating the complex dependences of the rates RDMDA and RCMA on the free concentration of Cl– by the following equations [67]:
where \({{F}_{{{\text{C}}{{{\text{u}}}^{{\text{ + }}}}}}}\) and \({{F}_{{{\text{C}}{{{\text{u}}}^{{{\text{2 + }}}}}}}}\) are the degrees of complexation of Cu(I) and Cu(II), respectively; and n and q are the numbers of chloride ligands in complexes \({\text{CuCl}}_{n}^{{\left( {n - 1} \right)}}\) and \({\text{CuCl}}_{q}^{{\left( {q - 2} \right)}}\), respectively.
The activation energy of DMDA synthesis in the range of 30–60°C according to Hoan et al. [67] is 67.7 ± 0.3 kJ/mol, i.e., below the Eact value 87.8 kJ/mol, which was obtained by Fedenok et al. [56]. The kinetics of the reaction of OD of acetylene under the same conditions obeys Eq. (6) for diacetylene (DA) and Eq. (7) for chloroacetylene (CA). The activation energy of DA synthesis is 53.9 ± 0.4 kJ/mol (25–50°C) [67].
Since the participation of two RC≡CCuCl molecules in the steps giving rise to a C–C bond after the rate-determining step is not confirmed by the kinetics in the studied chloride system (first orders in methylacetylene and acetylene and first orders in [CuCl2]0), it was necessary to study the cross coupling of methylacetylene and acetylene [68]. Cross coupling of two different alkynes makes it possible to have two route-coupling species (Scheme 3), specifically, intermediates X3 (CH3C≡CCuCl) and \(X_{3}^{'}\) (HC≡CCuCl), with the step of the formation of methyldiacetylene (MDA) and to test the hypothesis of the interaction of two ethynyl Cu(II) complexes at the step of C–C bond formation.

Scheme 3 . Route-coupling species in the three-route mechanism of the synthesis of DMDA, MDA, and DA.
Under the same conditions as in Hoan et al. [67], in the temperature range 25–45°C, we studied the kinetics of the formation of DMDA, MDA, and DA from a mixture of two alkynes, as well as MCA and chloroacetylene (CA) and a small amount of trans-1,2- dichloroethylene (DCE). The selectivity for the sum of diynes at 20°C is ~90%, and the maximum selectivity for MDA can reach 50%.
First, due to the coupling of three routes (Scheme 3), the reaction orders in C2H2 (PA) and MA (PMA) in the kinetic equations for the rates of the reactions of the formation of DA and DMDA, respectively, turned out to be lower than 1. This observation is explained by the routes presented in Scheme 3, which implies that the rates of the formation of the coupling and cross-coupling products are described by the following nonlinear equations:
The rates of the formation of X3 and \(X_{3}^{'}\) in the reactions of individual reagents are described by the following equations [66] at constant Cl– concentration:
Under the conditions of quasi-stationarity in [X3] and \([X_{3}^{'}]\), let us obtain, in accordance with Scheme 3, dependences of the first order in PMA and PA in the form of the equation
which satisfactorily describes all experimental results for j = DA, i = A and for j = DMDA, i = MA. At different pairs of indices, j = DA, i = A and j = DMDA, i = MA, different equations relating the reaction rates with \(~~{{k}_{i}}{{P}_{i}}.\)
Second, Eqs. (11)–(13) imply the equation
All experimental results at three temperatures (26, 30, and 45°C) in a solution with [LiCl] = 5 M are described by Eq. (17), and the β values are virtually constant within this temperature range at the initial and current rates (β = 1.92 ± 0.02).
Thus, there is no doubt that there are steps of coupling of ethynyl Cu(II) complexes in the mechanism of OD of alkynes in acidic solutions of chloride complexes of Cu(I) and Cu(II), even at the first orders of the reaction in the alkyne and CuCl2 concentrations. It follows from the kinetic results that the formation of the C–C bond and the transfer of two electrons occur simultaneously, whereas steps (XXIII)–(XXV) with single-electron transfer (SET) [81] in the case of OD of alkynes also contradict both the kinetic data, and the thermodynamic estimates [58]:
The variant of the OD mechanism with the step of alkyne insertion into the ≡C–Cu bond,
followed by the oxidation of the resulting enyne by the CuCl3– complex is not described by Eq. (16) and (17), whereas the oxidation of the enyne intermediate by Cu(II) chloride under the conditions of acetylene dimerization in the CuCl–NH4Cl–HCl–H2O system gives of 2-chlorovinylacetylene [1, 7] (see chapter 4):
It follows from the analysis of the kinetic models of the reactions of OD of alkynes that the dependences of the reaction rates on [CuCl2]n in various systems describe the experiment at n = 2, 1, and 0. What reasons lead to such a variety of reaction orders?
(1) The value n = 2 occurs in systems in which the rate-determining step is the interaction of two molecules of ethynyl complexes RC≡CCuX to form dimers that rapidly decompose to dialkynes.
(2) The value n = 1 arises if the rate-determining step is the formation of RC≡CCuХ by the oxidation of RC≡CCu or RC≡CCuL2 by Cu(II) chloride with possible subsequent dimerization (any form of dimeric ethynyl complexes of Cu(II) or even Cu(III)) .
(3) The value n = 0 occurs in the case of efficient catalysis by Cu(I) complexes with a slow step of the formation of copper acetylide RC≡CCu and its rapid oxidation.
Let us briefly consider some aspects of the technology of OD of alkynes.
The catalytic oxidation of lower alkynes (C2H2, C3H4) by oxygen (air) can be carried out in a two-reactor system with separate supply of gaseous alkynes (A) and air for the regeneration of CuCl2. If the Zal’kind catalytic system is used, it is easy to choose the conditions for the equality of the rates of diyne formation and Cu(I) oxidation by oxygen, since the kinetic equation
was derived for the CuCl oxidation rate at [LiCl] = 10 M [66], which does not depend on the Н3О+ ion concentration in the pH range 0.8–6.2.
The synthesis of DMDA in a synthesis reactor or an electrolyzer should be carried out in a two-phase system, e.g., with CCl4, to extract DMDA from the aqueous catalytic system [66]. The second version of the technology is the process in the anode chamber of a two-chamber electrolyzer with controlling the [CuCl2]/[CuCl] ratio. The high rate of C2H2 oxidation makes it possible to perform the process with a productivity of 30–50 g L–1 h–1 in a solution with 5 M LiCl at 0–5°C.
Since the OD mechanism of alkynes in the Zal’kind catalytic system involves two CuCl molecules, it was interesting to study the behavior of solid copper(I) methylacetylide in the same solution, i.e., to study the products and kinetics of the Glaser reaction with CuCl2 as an oxidizing agent [90].
Oxidative transformations of ethynyl compounds of copper and other metals. The kinetics of these reactions was studied in a closed reactor with intense stirring of three phases in an argon flow by measuring the initial rates of accumulation of reaction products at 20 and 30°C in a 5 M LiCl solution at [CuCl2] = 0.1 ± 0.5 M and a total amount of copper methylacetylide (MAM) of 0.3 M. The following facts were established:
(1) the only primary product is DMDA;
(2) chloromethylacetylene (CMA) is formed from MA released from MAM at pH ~ 4;
(3) in the oxidation of MAM by copper(II) bromide, that the amount of bromomethylacetylene (BMA) was twice as large as that of DMDA, the rate of formation of which is independent of MA (PMA) introduced into the reactor. The rate of BMA formation is described by the equation RBMA = kPMA;
(4) the activity of oxidizing agents decreases in their series (according to the values of the first-order rate constants in [Ox]): FeCl3 > CuCl2 > Li2Cr2O7 > KMnO4 > p-BQ;
(5) the absence of CMA and BMA among the primary products probably indicates that the OD reaction occurs on the surface of solid MAM;
(6) the redox decomposition of surface complexes of MAM with CuCl2 occurs without the participation of CuCl.
The participation of the CuCl molecule in MAM transformations under homogeneous conditions is probably due to not only the reasons considered above, but also the possible delocalization of the electron transferred from the Cu atom of methylacetylide to the Cu(II) atom of the oxidizing agent, which facilitates this transfer. On the surface of solid MAM, which is a coordination polymer, each copper atom is bonded to at least two ethynyl groups acting as η1- and η2-ligands [91–94]. In such a polymer, there is a high degree of conjugation, which is sufficient for electron delocalization. Therefore, the surface intermediate (MeC≡CCu∙CuCl2)s rapidly detaches a CuCl molecule and, interacting with the neighboring (MeC≡CCuCl)s, transforms into DMDA and does not give CMA.
To complete the picture, let us consider the redox reactions (RRs) of ethynyl complexes of other metals. Solid silver(I) methylacetylde [90] under the same conditions in the reaction with CuCl2 (and other oxidizing agents) turns into DMDA, apparently, also through the formation of complexes (MeC≡CAg∙CuCl2)s and (MeC≡CAgCl)s. However, it is possible that, in the case of silver complexes, a transmetalation process occurs on the surface with the formation of (MeC≡CCuCl)s and (AgCl)s.
When considering the mechanism of the oxidation of ethynyl compounds of Cu(I) and Ag(I), it should be kept in mind that, for ethynyl complexes of these metals, there are no thermodynamic constraints on single-electron electron transfer processes, and (MeC≡CCuCl)s and (MeC≡CAgCl)s can also be converted homolytically to form the MeC≡C• radical. This problem requires additional research.
Symmetrical solid Hg(II) methylacetylide Hg(C≡CMe)2 under the same conditions is not oxidized by such oxidizing agents as FeCl3, CuSO4, Na2Cr2O7, and KMnO4. At the same time, this compound easily interacts with CuCl2 to form CMA as the primary product. Obviously, it is the transmetalation to form MeC≡CCuCl in solution that leads to the synthesis of CMA by the reaction with the second CuCl2 molecule.
Interestingly, chloride complexes of Pd(II) and Rh(III) catalyze the oxidation of mercury methylacetylide by FeCl3 to DMDA, apparently via intermediate ethynyl compounds MeC≡CPdCl (or (MeC≡C)2Pd) and similar rhodium complexes. When studying the catalytic redox transformations of Hg(C≡CR)2 in solutions of hydride complexes of Ru and Os [95],
a new type of intermediates and an original mechanism for the transformation of ethynyl anions into OD products were discovered. For example, in the case of the initial complexes HRu(Cl)(CO)L3, stable complex ClRu{C(=CHR)(C≡CR)}(CO)L3 (9) was isolated, which is an intermediate in the synthesis of RC4R. In the case of a similar initial osmium complex, no intermediate with such a structure was isolated, but within 2 min a tenfold excess of Hg(C≡CR)2 in boiling toluene was converted into RC4R and Hg(0) to form the initial complex HOs(Cl)(CO)L3. The following mechanism of reaction (XXIX) was proposed:
Another variant of the mechanism of the formation of intermediate X8 with a carbene fragment can also be presented:
Obtained complex X7, just as in steps 4 and 5 in the previous mechanism, is converted into a dialkyne. Interestingly, complex X8 is also an intermediate in the nonoxidative dimerization of alkynes that is catalyzed by hydride complexes of Ru(II) and Os(II): HMX(CO)L2 [1]. An intriguing mechanism of the catalysis of Hg(C≡CR)2 demercuration by a rhodium(I) complex was proposed by Collman and Kang [96]. The possibility of oxidative addition of Rh(I) to the Hg–C bond was considered:
Intermediate of type X9 was synthesized and studied by Collman and Kang using Ir(I) complexes [96].
Reactions of stable di- and polyethynyl complexes are known for both Cu(I) and other metals, e.g., zirconium. Polyethynyl copper complexes have been obtained quite a long time ago: (RC≡C)2Cu–M+, (RC≡C)3CuM2, and even (RC≡C)3CuK3 with Cu(0) [94]. The [(PhC≡C)2Cu–]2 (NH3)x complex synthesized by Nast and Pfab [97] decomposes on heating to form RC≡C–C≡CR as a result of the oxidation of the [(PhC≡C)2Cu]– anion by the Cu(II) ammine complex to (PhC≡C)2Cu. Decomposition of the ethynyl complex (PhC≡C)2Cu thermally or under the action of the second Cu2+ ion gives dialkyne. This observation confirms to some extent the possibility of dismutation of two RC≡CCuCl molecules to form complex X6 and its participation in the OD reaction. Such an intermediate does not contradict the kinetic model obtained for a mixture of MA and A (Eq. (17)). The decomposition of the ethynyl Zr(IV) complex (the product of the oxidative addition of diyne to the Zr(II) complex) leads to a π-complex with diyne (or to a metallacyclopropene intermediate), which is oxidized by iodine to RC4R [98] (Scheme 4). In addition to the kinetic results indicating the participation of dimeric ethynyl Cu(II) complexes of types 1, X3, and 5 in the OD reaction, interesting data were obtained by Mizuno and colleagues [99, 100] by the catalysis of the OD reaction directly by dimeric Cu(II) complexes.

Scheme 4 . Mechanism of the formation of diynes with the participation of the ethynyl zirconium(IV) complex.
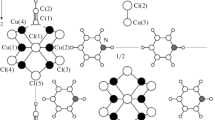
UV–Vis [99], and NMR and CSI-MS [100] spectroscopy studies convincingly showed that the monomeric complex [TBA]4[γ-H2SiW10O36Cu2(μ-1,1-N3)2] (a) in benzonitrile is an excellent catalyst for OD of alkynes in the reaction with oxygen. Complex a was obtained by the reaction of K4[γ-H2SiW10O36] with two equivalents of CuCl2 in excess of NaN3 in water at 25°С, followed by the addition of TBABr (TBA is tetrabutylammonium). The structure of the crystalline complex was determined by XRD [100]. Dimeric complexes of type a containing four Cu(II) atoms each were also synthesized earlier [101]. Figure 1 presents the structure of the azide fragment Cu2(μ-1,1-N3)2 of complex a.

Fralgment of the structure of complex a: [TBA]4[γ-H2SiW10O36Cu2(μ-1,1-N3)2].
The oxygen atoms belong to the silicotungstate anion. The azide ligand occurs as one of the electronic mesomers. Note that LCu(N3)2 complexes with macrocyclic ligands, e.g., 1,4,7-triazacyclononane, have long been studied in detail [102]. Complexes with trimethyl-substituted ligands [\({\text{L}}{\kern 1pt} '\)Cu(N3)2]2 and [\({\text{L}}{\kern 1pt} '\)Cu2(μ-N3)(N3)2]ClO4·H2O are also known [102]. It would be interesting to carry out the catalytic reaction of OD of alkynes with these complexes. The OD with complex a in an argon atmosphere occurs with an induction period, during which the azide anions are substituted by the reaction
Then, the stoichiometric (in Cu(II)) synthesis of dialkyne to form [Cu(I)]2 complex begins. It was shown that, under the same conditions, the use of CuCl2, CuCl, CuI, or Cu(AN)4PF6 leads to PhC4Ph with an (average) yield of <1%. In the catalytic variant with oxygen, the induction period is about 10 min, and the [Cu(I)]2 complex bound to silicotungstate is oxidized to form an ethynyl dimeric complex:
The amount of water formed over time was monitored by the change in the concentration of benzamide produced from benzonitrile. The catalytic process occurs very selectively in alkyne and in oxygen consumption. The yield of dialkynes for 31 substrates in benzonitrile at 100°C and 1 atm O2 is mainly 90 to 97% in 3 h at a catalyst turnover number of up to 470. A study of the kinetics of the reaction of OD with catalytic complex a showed [100] that the initial rate of diyne accumulation is described by the equation
The order in \({{P}_{{{{{\text{O}}}_{2}}}}}\) changes from 1 to 0. The observed activation energy is 68.9 kJ/mol. The kinetic isotope effect is kH/kD = 1, which indicates that the step of transformation of the dimeric ethynyl Cu(II) complex is rate-determining and that the main species of copper in the catalytic cycle is the dimeric ethynyl Cu(II) complex. The ethynyl ligand in this complex can be an η1, η2-ligand, as in complex 1, or a μ-ligand by analogy with the azide anion (see Fig. 1).
New ideas in the theory of the mechanisms of OD of alkynes involving two ethynyl complexes of Cu(II) and Cu(III) appeared as a result of a quantum chemical study of the mechanism of the OD reaction in the CuCl–TMEDA Hay system in acetone using oxygen by the Density Functional Theory (DFT) method [103]. The mechanism of CuCl oxidation by oxygen has long been known [64, 65] and was described above in the discussion of Fedenok et al.’s works [56–63]. Fomina et al. [103] considered the oxidation of ethynyl complexes Cu(C≡CH)L (L-TMEDA) by oxygen. The reaction sequence chosen for analysis in a simplified version (without the participation of protonated forms of the Me2NCH2CH2NMe2H+ ligand) is shown in Scheme 5, which includes a nonelementary step of oxidation of Cu(C≡CH)L to form ethynyl Cu(III) complexes.

Scheme 5 . Mechanism of the OD reaction involving Cu(III) complexes.
Dialkyne is formed at the step with transition state TS1.

Similar transition state TS2 is also accepted for the transformation of the ethynyl Cu(II) complex. A new idea in this work is the participation of Cu(III) complexes in the OD process, although the probable formation of intermediates containing Cu(III) (X5, 7) was also considered earlier [7]. At the same time, the structures of TS1 and TS2 are very doubtful: the convergence of ethynyl cationic (!) complexes in general, and even more so by the proposed method, is very unlikely. More likely, but not for cationic complexes, is the formation of intermediate dimeric ethynyl Cu(III) π-complexes of the type of Bohlmann complexes with structure 1. The use of cationic complexes for theoretical analysis in the presence of chloride anions in solution is not at all clear. Apparently, this theoretical study can only be considered in a conceptual aspect. Many questions arise regarding the quantitative results of the study involving cationic complexes (activation energies, etc.). It is hardly acceptable to use the Poisson–Boltzmann approach to take into account the solvation with LH+ cations and Cl– anions in this system containing cationic complexes of Cu(I), Cu(II), and Cu(III). It was necessary to consider additional intermediates, specifically, several molecules of acetone and water, as well as solvated Cl– ions, i.e., to use ion pairs separated by 3–6 solvent molecules in calculations, and to apply any other model of the polarization continuum. Our experience of theoretical analysis of the mechanism of ethylene oxidation in aqueous acetonitrile solutions of cationic Pd(II) complexes [104] showed the importance of taking into account the solvent in media with proton transfer in the presence of separated ion pairs.
Concluding the analysis of studies on the mechanisms of OD of alkynes in solutions of copper complexes, let us consider the 2013 article of Vilhelmsen et al. [105]. They improved the procedure for integrating the 13C NMR signals of alkynes and diynes to study the kinetics of OD of alkynes in the CuCl–TMEDA Hay system in dichloromethane and to optimize the composition of the catalytic system, including the choice of amines in OD of arylacetylenes. Unfortunately, all experiments were carried out under diffusion control, namely, under the conditions of the dependence of the conversion of alkyne on the speed of a magnetic stirrer. Meanwhile, in the case of very fast reactions, a magnetic stirrer is unsuitable for studying gas (O2)–liquid systems because it poorly mixes these two phases. Therefore, the found “optimal” conditions are such only for the chosen reactor design, the used mixer shape, and the given mixer speed. Nevertheless, Vilhelmsen et al. found a zero order in alkyne and a sharp jump in the reaction rate 50–80 min after the beginning of the reaction at a conversion of 50–80%, regardless of the concentrations of CuCl and TMEDA (1 : 3). The rate of the second slow step increases after the introduction of an additional portion of alkyne, and the inflection of the kinetic curve disappears after the addition of molecular sieves (4 nm) to the system. In Vilhelmsen et al.’s opinion, the decrease in the rate is due to the accumulation of water in the system during the OD reaction. In connection with these results, it would be useful to check the effect of the water concentration on the reaction rate in the CuCl–Py system (V) studied by Fedenok [62]. The zero order in alkyne, which was found by Vilhelmsen et al. [105], is most likely to be due to the fact that the rate-determining step under the studied conditions is the oxygen dissolution and, consequently, the oxidation of CuCl to Cu(II) complexes necessary for the synthesis of dialkyne. Although Vilhelmsen et al. [105] presented no results necessary to put forward hypotheses about the mechanism of the OD reaction, they, based on the literature data and their observations, proposed an original hypothesis of the formation of ethynyl Cu(III) complexes by the redox disproportionation of RC≡CCuCl complexes by the reaction
or
followed by the transformation of the RC≡CCuIII complex into diethynyl complex (RC≡C)2CuII, which decomposes to RC4R with simultaneous or subsequent oxidation of the formed Cu(0) to Cu(I).
1.2. Other Metal Catalysts for OD of Alkynes
Pd(II) complexes became the first copper-free catalysts for OD of alkynes. It is interesting to trace the history of the development of palladium catalysts in the OD reaction, which began with a series of fundamental works of Heck on the oxidative arylation, alkylation, and allylation of unsaturated compounds (olefins, dienes, CO) using organometallic compounds of non-transition metals as a source of aryl and alkyl groups: RHgX, R2Hg, R4Sn, R4Pb, etc. [106–113]. The oxidizing agents in stoichiometric processes were salts and complexes of Pd(II), Ru(III), and Rh(III) [106, 110, 112], which are converted by transmetalation processes into reactive intermediates RMXn; and the oxidizing agents in catalytic processes with Pd(II) were CuCl2, CuBr2 [106, 108–110, 112], and Pb(OAc)4 [111]. In some cases, the catalyst was a polyfunctional catalytic system (PFCS) Li2PdCl4–CuCl2, and the oxidizing agent was oxygen [106]. Below are a few examples:
Heck [113] discussed the mechanisms of the studied reactions and the problems of regio- and stereoselectivity at the steps of insertion of olefins into the R–PdX bond in the formed intermediates and at the step of β-elimination of HPdX.
Note that reactions of electrophilic substitution (or transmetalation) in the chemistry of organometallic compounds have long been known (see [114]). In our Laboratory at the Lomonosov Institute of Fine Chemical Technologies (LIFCF) simultaneously with and independently of Heck’s works [106–113] in 1969–1970 [114], we studied the reactions of PhH-gOAc with NaOAc and in the presence of Cu(II), Pd(II), and Pt(II) in AcOH. In the PhHgOAc–NaOAc system in boiling AcOH, PhOAc was obtained with a yield of 78–84%. In the PhHgOAc–PdCl2 system, diphenyl was synthesized; and in the PhHgOAc–Pd(ОАс)2 system, diphenyl and PhOAc were obtained at 25°C. Numerous examples from the chemistry of acetylene were used [114] to demonstrate the usefulness of using polyfunctional catalytic systems for the design of new catalytic reactions.
Subsequent Heck and colleagues’ works on the oxidative dicarboxylation of alkynes in PdCl2–HgCl2 solutions [115] and the use of Pd(II, 0) complexes in coupling reactions [116, 117], as well as Mizoroki et al.’s work [118] with a heterogeneous palladium catalyst, showed that it is possible to efficiently perform catalytic coupling reactions (XIII) or
which were later named the Heck reactions.
Our Laboratory at LIFCF also took a small part in developing the foundations of this reaction. Using experience in the synthesis of products of oxidative addition of alkyl halides (CHCl3) to PdL4 (L = Ph3P) [119] to catalyze the carbalkoxylation of acetylene to butyl acrylate [120], we carried out the arylation of ethylene to styrene without the use of PhHgX [121]. In a solution of PdL4 in PhBr (25°C), the PhPdBrL2 complex was formed, which was isolated and characterized. A solution of this complex in phenyl bromide was heated in an ethylene atmosphere to 100–120°C, and the formation of styrene (with a small amount of ethylbenzene) was found. Unfortunately, in 1970, we underestimated the importance of this reaction for synthetic chemistry and limited ourselves to a letter to the editors of the journal Kinetika i Kataliz [121]. The formation of ethylbenzene was related to the reduction of the produced olefin by palladium hydride. The Heck reduction reaction [9] was performed using the catalytic system [Pd(L)Cl]2 and CuH with R3SiH as a reducing agent.
The above studies finally led in 1975 to the publication of three articles on the oxidative transformations of alkynes. Cassar [122] proposed a PdL4 catalyst in DMF for arylation of alkynes (cross-coupling):
Heck [123] showed that a reaction similar to reaction (XXXVIII) can be catalyzed by the Pd(II)–Pd(OAc)2L2 complex in Et3N at 100°C. Since Et3N at 100°C does not reduce palladium to a Pd(0) complex, it was assumed that the initial palladium complex is reduced by alkyne to form dialkyne RC4R, followed by the oxidative addition of \({\text{R}' \text{X}}\) to PdL2 to form the intermediate RʹPd(C≡CR)L2 and the product of cross-coupling reaction (XXXVIII). Note that, as early as 1974, Dieck and Heck [117] observed an increase in the yield of dialkyne in the reaction of RC≡CH with \({\text{R}' \text{X}}\) with an increase in the [PhC≡CH]/[\({\text{R}' \text{X}}\)] ratio. Significantly milder conditions with excellent performance were proposed by Sonogashira, Tohda, and Hagihara [124], who used the PdCl2L2–CuI polyfunctional catalytic system in Et2NH (25°C, product yield in the case of PhC≡CH reaches 90–99%) during the formation of the active form of the PdL2 catalyst by the formation of PhC4Ph (!). The reactions of various I- and Br-substituted aromatic compounds and olefins with alkynes are called the Sonogashira reactions (method) and are discussed in chapter 3 of this review.
In further studies, it was found that stoichiometric (in palladium(II)) amounts of disubstituted 1,3-butadiynes are formed in the case of aromatic alkynes, most likely, by the decomposition of the Pd(C≡CR)2L2 intermediate [125]. It turned out that the use of the PdL4–CuI catalytic system in benzene with Et3N and an oxidizing agent (chloroacetone) at 25°C leads to ArC4Ar in the case of arylacetylenes and to RC4R (~50%) in the case of alkylacetylenes, as well as to the product of the addition of two ethynyl groups to diyne RC4R (10):

These studies initiated the use of palladium–copper and palladium catalysts in the synthesis of diynes (OD reaction) with various oxidizing agents.
Let us consider several variants of palladium-containing catalytic systems. Liu and Burton [126] used the L2PdCl2–CuI system in diisopropylamine with I2 as an oxidizing agent at room temperature (L = PPh3). The yield of dialkyl- and diaryldiynes was 84–88% (based on isolated products). Based on the literature data and their observations, Liu and Burton proposed a simplified scheme of the reaction mechanism, in which CuI catalyzes the formation of the key intermediate L2Pd(C≡CR)2:
The Pd(dba)2 complex in the solvent system CH2Cl2–50% NaOH–H2O in the presence of the phase transfer catalyst n-Bu4NBr catalyzes the oxidation of alkynes by allyl bromide [127] with yields of diynes of 72–90% at room temperature:
The proposed [127] mechanism of the participation of allyl bromide in the process is absolutely unrealistic because there is not a single scientifically substantiated step and no products of the reduction of the oxidizing agent allyl bromide were found. It is not clear where 2H+ and 2 electrons are consumed. It is natural to assume that the η1- or η2-allyl complex of Pd(II) can lead to the formation of intermediates BrPd(C≡CR)L2 and L2Pd(C≡CR)2 and С3Н6 (or 1,5-hexadiene) (XLIII). The Pd(PPh3)4 complex with CuI was successfully used for the synthesis of 1,4-bis(3,4-dimethyl-5-formyl-2-pyrryl)butadiine (11) with the oxidizing agent chloroacetone in the benzene–Et3N system at room temperature with a yield of 92% [128].

The L2PdCl2–CuI system with the addition of PPh3 in an Et3N–CH3CN solution at 25°C was studied by Fairlamb et al. in 2003 [129]. The article is called very funny: “Pd-catalysed cross-coupling of terminal alkynes to diynes in the absence of a stoichiometric additive.” In other words, Fairlamb et al. carried out the reaction of not cross-, but homocoupling of alkynes without oxidizing agents. The process was performed in a dried system with a degassed solvent in an argon atmosphere (!) with dialkyne yields of >92%. From the results obtained, it follows that there were neither oxygen, nor products of reduction of alkynes or dialkynes, nor acetonitrile reduction products. For thermodynamic reasons, the hydrogen molecule also cannot be released. Thus, based on the standard thermodynamic characteristics of gaseous participants in the reaction 2C2H2 = C4H2 + H2, we can conclude that this reaction is endoergonic \((\Delta G_{{298\,{\text{K}}}}^{^\circ }\) = 25.6 kJ/mol) and endothermic \((\Delta H_{{298\,{\text{K}}}}^{^\circ }\) = 19.3 kJ/mol), is accompanied by a decrease in entropy \((\Delta S_{{298\,{\text{K}}}}^{^\circ }\) = –21.1 J mol–1 K–1), and has an equilibrium constant of K298K = 3.2310–5. Without balancing the reaction and not finding an oxidizing agent, Fairlamb et al. stated (p. 633): “On mechanistic grounds an oxidant should not be required”! However, in the next Fairlamb’s article with the participation of nine (!) co-authors [130], an important conclusion was made that the oxidative coupling of alkynes to diynes does require an oxidizing agent! It turned out that, without impurities, i.e., without a stoichiometric amount of oxygen, the reaction does not occur. The weak effect of substituents in alkyne on the energy characteristics of the alkyne dehydrocondensation reaction also follows from Fairlamb and colleagues’ quantum chemical calculations [130]. In the presence of oxygen, they found a zero order in alkyne, which, in our opinion, is not explained by the slow step of oxidation of the Pd(0) complex by oxygen, since the this complex (and diyne) is formed by the slow irreversible step of the reductive decomposition of the L2Pd(C≡CR)2 complex. The problem of oxidizing agents was also discussed in interesting McGlacken and Fairlamb’s review of cross-coupling reactions catalyzed by palladium complexes [131].
A very simple catalytic system without amines and phosphines was proposed by Li et al. [132] for OD of aryl- and alkylalkynes. In a solution of PdCl2–CuI and NaOAc in acetonitrile with the oxidizing agent Me3NO at 25°C, diynes were formed with yields of 80–100%. It was shown that both components of the polyfunctional catalytic system play a decisive role in the process. Li et al. considered two routes for the formation of the key intermediate Pd(C≡CR)2 in the reaction mechanism:
Along with palladium(II, 0) complexes, OD reactions are also catalyzed by complexes of other transition metals. In the analysis of the reactions of ethynyl mercury compounds, the participation of Ru(II), Os(II), and Rh(I, III) complexes in the catalysis of Hg(C≡CR)2 transformations with the formation of diynes was already noted [90, 95]. Let us consider some examples of catalysis of OD of alkynes by Ni(II), Rh(I), and Au(I) complexes.
OD reactions with Ni(II), Rh(I), and Au(I) complexes. Lei and colleagues [133] convincingly showed that the addition of NiCl2·6H2O to the Hay catalytic system (CuI–TMEDA–Et3N–THF) in OD of phenylacetylene by oxygen leads to a fourfold increase in the reaction rate and to the formation of diyne with a yield of 93% in 1 h at room temperature. This system also successfully catalyzes the reactions of cross-coupling of phenylacetylene with various derivatives of HC≡CCH2X at a ratio of alkynes of 1 : 5 with yields of unsymmetrical adiynes (11 examples) of 80 to 93%. Interestingly, in a NiCl2 solution without CuI but in the presence of the strong base tBuONa and ZnCl2(!), asymmetric dialkynes are also smoothly synthesized, but at 60°C and in 3 h. Probably, under these conditions, organozinc intermediates ClZnC≡CR are formed, followed by transmetalation with NiCl2, but the formation of an ethynyl zinc compound was not proven. Lei and colleagues assumed that the LNi(C≡CPh)2 intermediate is obtained by the reaction of LNiO2 with two PhC≡CCu molecules, but the fate of O2 was not discussed. In addition, the reductive elimination of diyne from LNi(C≡CPh)2 to form LNi0 without the participation of an oxidizing agent is unlikely, since Ni(II) is not an oxidizing agent. The standard oxidation potential of the Ni2+/Ni pair is –0.23 V (∆F298 > 0). Therefore, the oxidizing agent in this system can be oxygen (step (1)) or the product of the oxidation of CuI by oxygen in the presence of BH+ and Cl–, Cu(II)* (see above):
The oxygenyl nickel complex LNiO2 can participate in the formation of LNi(C≡CPh)2:
In this case, steps (1) and (2) constitute a process catalyzed by the Ni(II) complex:
This stoichiometry is consistent with Lei and colleagues’ data [133]: per 1 mol of the product (PhC4Ph), 1 mol of O2 is consumed, but the possibility of the formation of hydrogen peroxide and its fate remained unclear. If the key intermediate occurs in the reaction of LNiO2 with PhC≡CCu, then the peroxide CuOOCu should be formed, which can also act as an oxidizing agent in the process under discussion. Apparently, the Ni(II)–CuI system with oxygen deserves additional studies.
An interesting mechanism of the reaction of OD of alkynes was discovered by studying the reactivity of dimeric Rh(I) complexes [134]. The dimeric vinylidene Rh(I) complex Rh2(CO)2(dppm)2(C=CHPh) (12) obtained from Rh2(CO)3(dppm)2 and PhC≡CH catalyzes the OD reaction. At 85°C in a solution (benzene) of complex 12, there is a catalytic disproportionation of phenylacetylene to 1,4-diphenylbutadiyne and styrene (90% of the total organic products). Both E- and Z-dimers of phenylacetylene (~10%) are also formed:
This reaction is similar to the well-known Zelinsky disproportionation of cyclohexene:
Thus, in reaction (XLVI), phenylacetylene acts as an oxidizing agent and accepts two protons and two electrons from two PhC≡CH molecules. This original mechanism is shown in Scheme 6. The transformation of complex B (reductive elimination to form a Rh–Rh bond) can lead to butenynes. The transformation of complex D into complex A is similar to the formation of a dinuclear vinylidene Pt(II) complex with an ethynyl ligand by the reaction [135]

Scheme 6 . Mechanism of the reaction of OD of alkynes, catalyzed by dimeric rhodium complexes 12.
Catalysis by Au(I) complexes was first discovered for the Sonogashira cross-coupling reaction [136–140]. It was shown that, in solutions of AuCl(PPh3) complexes, the reaction of ArI with RC2H occurs with a selectivity of ~99%, whereas in solutions of Au(III) complexes with phenolic Schiff bases as ligands at an Au(III) concentration of 20–30 mol %, only the stoichiometric reaction of OD of phenylacetylene (8–10%) is observed. In this reaction, Au(III) acts as an oxidizing agent, rather than a catalyst (see [136])! Corma and colleagues [138] extended the catalysis by Au(I) complexes to other cross-coupling reactions (Suzuki’s and Heck’s). In the course of studying the cascade cyclization of allenoates and oxidative alkynylation of the formed γ-butenolide with phenylacetylene (A) in solutions of the (Ph3P)AuNTf2 complex in the AN–H2O system with the strong oxidizing agent Selectfluor [ClN(CH2CH2)3NF(BF4)2] [138] and the base K3PO4 (B), a catalytic OD reaction was detected with yields of PhC4Ph of 23 to 88%, depending on the substituents in the allenoate R1CH=C=C(Me)(COOR2). The following sequence of steps leading to 1,3-diyne and including the catalytic cycle was assumed:
The 2011 Wegner and Auzias’ review “Gold for C–C coupling reactions: A Swiss-army-knife catalysis” [141] also considered the reactions of OD of alkynes.
Corma and colleagues [142] studied the mechanism of the OD reaction in solutions of the LAuNTf2 complex (L = PPh3) in wet acetonitrile (AN) in the presence of Na2CO3 with the oxidizing agent Selectfluor (Ox) (AuIX + F+ → AuIIIXF+). o-MeC6H4C≡CH and other alkynes were used. The OD reactions involving RC≡CAuL and RC≡CH (R = o-MeC6H4) were compared. It was determined that the conversions of these substrates and the oxidizing agent Selectfluor correspond to the stoichiometry of the reactions
The results of cyclic voltammetry showed the presence of Au(I) and Au(III) complexes in solutions of the intermediate RC≡CAuL and the oxidizing agent. The same forms of gold complexes were also observed in reactions of RC≡CH with the oxidizing agent in LAuNTf2 solutions. It was also observed that, at a conversion of o-MeC6H4C≡CH of >40%, the formation of diyne stopped. Based on the studies performed, it was assumed that the reactions of RC≡CAuL with the oxidizing agent involve both forms of ethynyl gold complexes:
The results obtained also suggested that the intermediate required for the synthesis of diyne is formed by the first of the reactions
Intermediate X7 could also be formed by the disproportionation of two RC≡CAu(III)L molecules, but reaction (L) in this system probably occurs very rapidly. This was also confirmed by experiments in which PhC≡CH was added to a solution of (o‑MeC6H4C≡С)Au(I)L and an oxidizing agent: no product of the cross-coupling of two ethynyl groups R1C4R2 was found. For the initial reaction rates, which were determined very approximately from the yields of diyne by certain times t, the kinetic equation
was obtained, where k and Keq are the observed rate constant and the equilibrium constant of the step of protonation of the base B, respectively.
Corma and colleagues [142] derived kinetic equation (20) based on their proposed four-step mechanism (presented below in their notation):
Equation (20) was derived with small errors. This mechanism together with the material balance of the catalyst suggest that
In the case of a significant binding of the catalyst to alkyne to form a π-complex and ethynyl complex (RC≡C)AuIL, we obtain the equation
which is similar to Eq. (20) with a clear meaning of the observed constants.
One should have not taken the square root during the derivation of Eq. (20) because the reaction rate depends on the concentrations of the complexes (RC≡C)AuIL and (RC≡C)AuIIIL, rather than the stoichiometric number of this step in the final equation of the reaction route.
Shi and colleagues [143] found such LAuIX complexes and reaction conditions that allow efficient cross-coupling of alkynes with high yields of unsymmetrical diynes (80%, ratio R1C4R2 : R1C4R1 = 8–12). Ethynyl derivatives of propargyl alcohols (R2C≡CH), p-fluorophenylacetylene (R1C≡CH), and other ArC≡CH were also used. For example, the (dppm)(AuBr)2 complex in acetonitrile with the addition of the Phen ligand and the oxidizing agent PhI(OAc)2 in a minimum excess of R2С≡CH (1.3) leads to the cross-coupling product R1C4R2 with a yield of 83% at 50°C in 15 min at the total conversion of R1C≡CH and R1C4R2 : R1C4R1 = 12. It was also shown that, already at the step of formation of the ethynyl Au(I) complex, the reactivities of the alkynes R2C≡CH and R1C≡CH were observed to be different. Specifically, the complexes R2C≡CAuL and R1C≡CAuL are formed from LAuOAc into CD3CN in 5 min at room temperature with 100% conversion in a ratio of 3 : 1. Shi and colleagues determined that, in contrast to the results of previous studies, AuCl3 in the presence of Phen and NaOAc stoichiometrically participates in homo- and cross-coupling of the same alkynes with a yield of the product R1C4R2 of about 65% and a product ratio of R1C4R2 : R1C4R1 = 4. In this system, diyne is obtained from the ethynyl complex (R1C≡C)AuIIIL and R2C≡CH (compare with Corma and colleagues’ data [142]). Shi and colleagues also proposed a two-route mechanism for the formation of unsymmetrical alkynes (Scheme 7), which, however, does not take into account the route justified by Shi and colleagues [143] (see above). The catalytic system proposed in that work was tested in the synthesis of 27 asymmetric diynes with yields of 50–93%.
In the 2015 article [144], Corma and colleagues continued to study the mechanism of the OD reaction by the example of the homocoupling of alkynes in solutions of AuLZ complexes (Z = NTf2, Cl).

Scheme 7 . Two-route mechanism for the synthesis of unsymmetrical diynes.
Corma and colleagues synthesized L(Cl)AuIII[C≡ C(CH2)nCH3]2 complexes X7 from alkynes CH3(CH2)nC≡CH (n = 7 and 9) and traced their transformations into diynes by 31Р NMR spectroscopy. It turned out that the rates of reductive elimination of diynes from intermediates X7 are independent of n (yields of diynes were >95% in 1 min at –78°C). At the same time, the oxidation of ethynyl Au(I) complexes LAu[C≡C(CH2)nCH3] with n = 7 and 9 by Selectfluor (SF) under standard conditions (AN, Na2-CO3, room temperature) gives diynes with yields of 95 and 34%, respectively. The catalytic OD of the two alkynes for 24 h showed a unique selectivity related only to the length of the alkyl chain, i.e., to a very remote (peripheral) steric effect. For example, the yield of isolated products at n = 7 was 62%, and at n = 9, it was only 4%! In the Hay catalytic system (CuCl–TMEDA–iPrOH) at 65°C, the yields of diynes at n = 7 and 9 were 63 and 82%, respectively. Those studies led to an important conclusion that, in the case of catalysis by Au(I) complexes, the selectivity of homo- and cross-coupling of different alkynes is affected not only by the different rates of formation of ethynyl Au(I) complexes, but also by the rate of formation of intermediates X7 from (RC≡C)AuIL and (RC≡C)AuIIILZ (Scheme 8). It was assumed that it is at the step of transmetalation with the participation of complex X7 that long-range steric interactions are important. The reductive elimination of diyne from the diethynyl complex occurs in <1 min at –78°C. It is quite probable that this process occurs with the formation of an intermediate complex of the type of dimeric Bohlmann complexes 1. It is also possible that the dimeric complex is a π-complex of only Au(I). It is interesting that the LAuIII[C≡C(CH2)nCH3]Cl2 (L = PPh3) complexes in the temperature range of –30 to –5°C in 15 min after the formation in solution are also converted into diynes without the participation of (RC≡C)AuIL complexes with yields of >95% (n = 7) and 45% (n = 9) and with the formation of free 1‑dodecine (55%). 1-Dodecine is likely to be formed by the disproportionation of the ethynyl complex LAuIII[C≡C(CH2)nCH3]Cl2.

Scheme 8 . Mechanism of the reaction of OD of alkynes with the participation of dimeric ethynyl complex X7 of Au(I) with Au(III).
To increase the selectivity of the intramolecular oxidative cyclization of nonconjugated terminal alkynes in the intermolecular reaction of OD (and oligomerization), Shi and colleagues [145] studied the catalytic properties of complexes containing two Au(I) atoms in order to accelerate the transfer of ethynyl groups between two ethynyl complexes of Au(I) and Au(III) (see [143]). It was found that, in AN solutions in the presence of PhI(OAc)2 (Ox) and Phen (50°C, 24 h) at a dialkyne concentration of 0.003 M (e.g., an o-diphthalate derivative with long alkyl ends, (CH2)nC≡CH)), L(AuCl)2 complexes with L = dppm, dppp, and BINAP are virtually inactive in the formation of a macrocycle with 16 atoms (yield <10%). And only the use of the Au(I) complex tBuXantophos(AuCl)2 led to an increase in the macrocycle yield up to 75%. The study of the structure of this complex showed that one Au(I) atom in the form of an Au+ ion is located between two PtBu2 groups of the diphosphine ligand, and the second gold atom is an AuCl2– anion. These results showed that it is not about the close proximity of two Au(I) atoms. In the catalytic reaction of the macrocycle synthesis with the Xantophos(AuCl)2 complex, in which two AuCl molecules are bound to two PPh2 groups (the Au–Au distance is 2.96 Å), the yield of the product was only 15%.
Further studies demonstrated that the anionic AuCl2– complexes are excellent precursors of active catalysts for the synthesis of macrocycles in comparison with LAuCl complexes, and the optimal precursor is the simple and available [(nBu)4N]+AuCl2– complex, which forms a macrocycle with a yield of 75%. This complex was used to obtain 27 macrocycles containing the –С≡С–С≡С– fragment with 14 to 28 ring atoms and with yields of 20 to 78%, depending on the nature of the dialkyne. In this case, 5 macrocycles were synthesized with higher yields than when using the Collins strategy [51–53]. Shi and colleagues [145] also convincingly showed that the proposed [(nBu)4N]+AuCl2– complex is an excellent catalyst for homo- and cross-coupling of terminal alkynes. They obtained 13 examples of homocoupling of alkynes with yields of 85–95% and 8 examples of cross-coupling with yields of 70–85%, but with lower selectivity than in the case of (dppm)(AuBr)2 complexes [143]. It follows from the results of those works [143, 145] that catalysts based on Au(I) complexes with strong oxidizing agents are much more active and selective than copper-containing catalytic systems (Hay’s and Glazer and Zal’kind’s) and the Eglinton system.
Heterogeneous catalysts for the OD reaction. Another trend in studying the catalysis of the reaction of OD of alkynes appeared during the investigation of the Sonogashira reaction. This is the use of heterogeneous catalysts based on gold nanoparticles or nanoclusters (NC) deposited on various supports. As catalysts for this reaction, Corma and colleagues [136] studied Au/CeO2 and Lambert and colleagues [146] tested Au/La2O3. In the Sonogashira reaction with phenylacetylene (PA) and PhI on a Au/CeO2 catalyst, Corma and colleagues [147, 148] found that PA can be activated on Au+ cation sites at the interface between gold clusters and the CeO2 support. Detailed theoretical (DFT) and experimental studies were carried out in 2014 [148]. A theoretical analysis of the reactivity and structure of the Au3, Au38, AuOx/CeO2, and Au38O16 clusters showed that, on the Au+δ and Au+ sites present on oxygen-containing clusters, the Eact of PA adsorption and the Eact of the OD reaction involving RA are low. The formation of ethynyl fragments on gold NCs Aun involves the breaking of the ≡С–Н bond and the formation of ethynyl hydride clusters. The breaking of this bond is also possible on all gold clusters that have an oxygen atom with basic properties, which is formed by the dissociation of an O2 molecule on the Aun Au/CeO2 surface. Measurements of the initial rates of OD of o-tolylacetylene showed that, on the Au/CeO2, Au/C, and Au/ZnO (1,3-dichlorobenzene, 170°C, 18 h) catalysts, the initial rates pass through maxima with increasing oxygen content in the reaction vessel. The yield of products for 11 alkynes on the Au/C catalyst varied in the range of 60–90%.
Gold nanoclusters 0.8 ± 0.2 nm in size, similar in composition to Au11–Au13 clusters and supported on silicate with immobilized aminopropyl groups (SBA-15), turned out to be active catalysts of various reactions, including OD of alkynes [149]. The yield of dialkynes in the presence of such a catalyst in dichloromethane with the addition of phenanthrene and the oxidizing agent PhI(OAc)2 was 84–98% for 10 aromatic alkynes at room temperature, and the catalyst lost virtually no activity during 5 cycles. The catalyst becomes active after an induction period, during which Aun is partially oxidized. Photoelectron spectroscopy showed the presence of Au0 and Au(III) atoms on its surface, the ratio of which does not change during the reaction. Corma and colleagues suggested that a bis(alkynyl) Au(III) complex stabilized by phenanthrene is formed on the NC surface and is then converted by the reductive elimination into a reaction product and the initial form of a partially oxidized catalyst [Au0 and Au(III)] due to the rapid oxidation of Au(I) to Au(III).
An interesting synergistic effect was detected by Li and colleagues [150] using AuPd alloy NCs (3–4 nm) supported on N-graphitized carbon modified with SiO2 (mpg-C3N4) with phenylacetylene as a catalyst for OD (DMF, 80°C, air, 12 h). The highest activity was shown by Pd2Au clusters with a selectivity of ~100% and a conversion of 60–80%, whereas Pd–mpg-C3N4 and Au–mpg-C3N4 were virtually inactive, and the (Pd + Au)–mpg-C3N4 mixture catalyzed the reaction with a conversion of <10%. In the reactions of various arylalkynes, the conversion ranged from 71 to 99 at 100% selectivity. The role of the mpg-C3N4 support, which has both basic and semiconductor properties and is capable of activating oxygen, is not yet clear. It is assumed that the noticeable synergistic effect is determined by the transfer of charge (electrons) from Pd to Au.
Concluding section 1.2, we note that gold compounds have taken a prominent place in the catalysis of OD of alkynes.
CHAPTER 2. CADIOT–CHODKIEWICZ REACTION
Reaction (XI) was discovered in 1955 and described in detail in Shvartsberg and Fisher’s review in the monograph [5, p. 317], Cadiot and Chodkiewicz’s review in the monograph [151], and in Siemsen et al.’s review [14]. The 1967 review [5] provides references to 145 syntheses of various unsymmetrical 1,4-diynes in the CuCl (CuI)–Et3N systems in organic solvents with the addition of NH2OH·HCl:
Hydroxylamine is necessary for the reduction of Cu(II) formed by the oxidation of Cu(I) by a haloalkyne, since the formation of Cu(II) and the reduction of R2C≡CX can lead to the formation of homodiynes R1C4R1 and R2C4R2, e.g.,
A study of the kinetics of the oxidative chlorination of alkynes RC≡CH (R = H, Me, vinyl) in the CuCl2–CuCl–NH4Cl–H2O system showed [152–154] that the reaction
is indeed kinetically reversible
The reaction kinetics was studied under conditions of high and constant concentrations of free Cl–, i.e., in the case of proportionality of [CuCl2] (or \({{a}_{{{\text{C}}{{{\text{u}}}^{{2 + }}}}}}\)) and [CuCl2]∑ and [CuCl] (or \({{a}_{{{\text{C}}{{{\text{u}}}^{{\text{ + }}}}}}}\)) [CuCl]∑. The study of the kinetics of the synthesis of HC≡CCl (ClC≡CCl and MeC≡CCl) showed that reversible reaction (LII) occurs through the intermediate RC≡CCu2Cl3 (RC≡CCu(II)Cl·CuCl2 or RC≡CCu(III)Cl2⋅CuCl) and comprises the following steps (see also chapter 1 in [70, p. 373]):
The kinetic equation obtained under the assumption that step 2 is the rate-determining step in the forward and reverse directions describes all the experimental results. It follows from the obtained kinetic model that the Cadiot–Chodkiewicz reaction is expediently performed at the lowest concentration of CuX.
The Cadiot–Chodkiewicz reaction has obvious advantages in the syntheses of unsymmetrical 1,4-substituted 1,3-diynes; however, its low rates and often low selectivity, which depend on the nature of the substituents in alkynes, led in the early 1990s to the transition to the Sonogashira catalytic systems: Pd(II), Pd(0)–CuI [124–126]. For example, in 1991, Wityak and Chan found [155] that, in the PdCl2L2–CuI–(iPr)2NH system in THF at room temperature, iodoalkynes react with alkynes at a high rate both in a nitrogen atmosphere and in an oxygen atmosphere, and no OD products were detected. Wityak and Chan “proposed a catalytic version of the Cadiot–Chodkiewicz reaction,” although the classical version of this condensation (without palladium) is also a catalytic process! That system was also used in the synthesis of macrocyclic 1,3-diynes by the Cadiot–Chodkiewicz reaction [156].
Further studies of such bimetallic systems in this reaction revealed new, previously unknown interesting facts about coupling selectivity [157]. In the interaction of R1C≡CH (1) and R2C≡CХ (2) (Х = I, Br) in the same PdCl2L2–CuI–(iPr)2NH system in THF as Wityak and Chan’s [155], Cai and Vasella [157] found that, with some substrates along with heterodiyne R1C4R2 (3), not only homodiynes R1C4R1 (4) and R2C4R2 (5) are formed with comparable yields, but also product R1C≡CX (6) is. In this case, the exchange of ≡С–Н and ≡С–Х groups also occurs without palladium in the system containing only copper. For example, in solutions of CuI and (iPr)2NH in acetonitrile, the ratio of products (6) : (1) reaches 1.1 in 30 min at room temperature. In the same system, Cai and Vasella found that R2C≡CI is converted to homodiyne R2C4R2 to form (iPr)2NH·I2 complex. The reductive dimerization of aliphatic and aromatic iodoalkynes also occurs quite efficiently in solutions of Pd(PPh3)4 in DMF [158] with yields of 75–95%. Cai and Vasella considered the reaction
as the key step in the mechanism of RC4R formation:
In their interesting 2012 work, Jutand and colleagues [159] studied the mechanism of coupling reactions to form compounds with C–N and C–O bonds from ArX and various nucleophiles RZH (CyNH2, H2O, PhOH) that were catalyzed by CuX complexes with 1,3-diketonates. Cyclic voltammetry, H1 NMR spectroscopy, ESI-MS, and DFT were used to study and formulate the reaction mechanism. It includes the formation of intermediate [CuZR], to which PhI is oxidatively added to form PhCu(III)(ZR)L complex. The step of reductive elimination of PhZR is the rate-determining step in the coupling process.
The Pd(OAc)2–CuI (or Pd(dba)2–CuI) system in the presence of tetrabutylammonium bromide (TBAB) and (iPr)2NH as a solvent proved to be a very efficient catalytic system for the Cadiot–Chodkiewicz reaction [160]. In the absence of Pd or CuI, the reaction does not occur even at 70°C. In this case, at a Pd concentration of as low as 0.0001 mol % and an amount of alkyne of 100 mmol in 8 h, the yield of diynes reaches 35% with TON = 350000 and TOF = 43750 h–1. In the case of PhC2H and (CH3)2C(OH)C≡CBr at 0.01 mol % Pd, the yield of diyne is 97–99% without the formation of homodiyne from bromoalkyne. It was shown that the reaction rate versus [Pd] curve plateaus so that the Pd concentration in the range of 2–8 mol % hardly affects the reaction rate. The formation of palladium nanoparticles not participating in the catalytic cycle was assumed. Already in 1995, Amatore et al. [161] found that Cu(I) compounds are not an obligatory component of systems containing palladium complexes. The cross-coupling of haloalkynes with alkynes also occurs in the Pd(OAc)2–L system (where L is sulfonated PPh3) in the acetonitrile–water solvent with yields of 40–65%.
The mechanism of the Cadiot–Chodkiewicz reaction in the case of catalysis by Cu(I) complexes is still under discussion. It has been shown that the reaction with R2C≡CX ethynyl copper complexes involves R1C≡CCu, but the mechanism of this interaction has not yet been determined. The mechanisms under consideration are the following:
(1) the simple nucleophilic substitution of Br– with electrophilic assistance within transition state TS3:

and (2) a more probable mechanism proposed by Cadiot and Chodkiewicz themselves [151], which includes the step of oxidative addition of a haloalkyne to Cu(I) in the π-complex R1C≡CCu with bromoalkyne:

The formation of such an intermediate (see above the mechanism of reduction of RC≡CCl by Cu(I) chloride) also explains the exchange reactions of ethynyl groups in substrates of the Cadiot–Chodkiewicz reaction [157].
In recent years, a number of studies have been made on the Cadiot–Chodkiewicz cross-coupling of ethynyl compounds in which the source of the ethynyl group was not RC≡CХ but ethynyl compounds of hypervalent iodine, ethynylbenziodoxolones (1) and ethynylbenziodoxones (2) [162, 163], and the catalysts were Au(I) complexes. In systems with (Ph3P)AuCl with the addition of Phen, the yield of products reached 87% with 5% homodiyne [162]. The use of aryl azide ligands with the addition of Phen and AgOTs, and ethynylbenziodoxoles as hypervalent iodine compounds increases the yield of cross-coupling products to 90–98% [163].

The successful use of ethynyl compounds of hypervalent iodine in the Cadiot–Chodkiewicz reaction was considered to be due to the use of stronger oxidizing agents than RC≡CI for the oxidation of ethynyl compounds Au(I) by compound (1) or (2) to diethynyl Au(III) complexes with subsequent formation of cross-coupling products:
Comparisons of these data with the results of using the (Ph3P)AuCl complex under standard conditions of the Cadiot–Chodkiewicz reaction failed to be found; therefore, it is difficult to assess the additional effect of hypervalent iodine in this reaction. Note that, although the compositions of the considered catalytic systems [163] are very complex, their high efficiency may attract the attention of synthetic scientists to systems based on Au(I) complexes in the Cadiot–Chodkiewicz reaction. There are reasons to believe that hypervalent iodine compounds are also capable of oxidizing Pd(II) complexes to Pd(IV). Various routes of the studied reactions involving oxidizing agents of type (2), including the roles of Phen and AgOTs, were modeled by DFT [164]. In that work, two versions of the reaction mechanism were considered: a redox mechanism with the formation of LAu(C≡CAr)(C≡CPh)+ and a mechanism with the step of carbometalation of the ethynyl π-complex of Au(I) with iodine compound (2). The pathways of transformations of oxidizing agents of type (2) in the course of the reaction, e.g., to form o-Tol[C(CF3)2O]–Ag(Phen), were also analyzed. Unfortunately, in the considered works, the nature of hypervalent iodine reagents as oxidizing agents was not discussed, and it follows only from the review of the reactions of these compounds with Pd(0, II) complexes and their use in organic synthesis [165] (see also reaction (LV)) that ArI(X,Y) are four-electron oxidizing agents. Let us analyze this issue in more detail.
Frequently used oxidizing agents in RRs are iodine compounds ArI(X,Y), where X and Y are aryl, vinyl, alkynyl, OAc, chloride and iodide; and Ar2I+X–, which are strong oxidizing agents and strong electrophiles, e.g., PhI(OAc)2. When we encounter these unusual oxidizing agents, we try to understand their nature within the framework of the theory of localized electron pairs, all the more so, as the approach to determining the OS of an atom is also based on the localized pair of electrons in the A–X bond. Let us consider the issue of the OS of iodine in this and similar oxidizing agents.
It is known that the simplest method for obtaining ArI(X,Y) compounds is the oxidation of organic iodides by molecular chlorine Cl2 by the reaction
Both chloride anions are replaced by any anions. Hydrolysis of (ArICl)+ gives ArI=O oxide. It was shown in a number of works that this oxide in the presence of AcOH is converted into PhI(OAc)2; i.e., acetate anions replace both Cl– (!) anions.
The assumption about the transfer of two electrons from the negative iodine atom to the Cl2 molecule during the oxidation of ArI is quite plausible. In this case, the Ar+ cation is formally retained in the (ArICl)+Cl– molecule, in which the ArI2+ group can contain two strong electrophiles: the Ar+ group and the I+ iodine cation. The real state of particles in the ArI2+ group in a polar solvent can be represented by a system of electronic mesomers, also including the complete dissociation of the unstable association of two positive cations in ArI2+:
All mesomers are four-electron oxidizing agents. In Ar2I+Cl–, the Ar2I+ group can also contain two cations: [Ar+Ar–I+]+, i.e., two two-electron oxidizers. If both aryl groups have the OS –I, then the OS of iodine is III ([Ar–Ar–I3+]+) and such iodine is also a four-electron oxidizing agent (I3+ → I–). The high probability of the existence of the Ar–I3+(s) mesomer with the Ar– anion is consistent with the exchange reactionFootnote 1
The work under footnote 1 contains a good review of the reactions of oxidation of organic compounds by phenyliododiacetate and other compounds of hypervalent iodine.
Note an example showing that hypervalent iodine compounds are four-electron oxidizing agents. Replacement of one chloride anion in (ArICl)+ by the Ar– anion gives Ar2I+Cl–, which adds oxidatively to Pd0L2:
The Ar+ cation is added to Pd0, and the Ar– anion is oxidized by the iodine atom in the +I oxidation state to form Ar+I–. If the OS of iodine in Ar2I+Cl– is III, then Pd0L2 is first oxidized with the transfer of the aryl anion to palladium(II), and then Ar– is oxidized to form ArI. Thus, hypervalent iodine compounds of this type (and of the type of structures (1) and (2)) oxidize Сu(I) to Cu(III), Pd(0) to Pd(II), Pd(II) to Pd(IV), and Au(I) to Au(III). It was also noted that these compounds have a standard oxidizing potential E0 comparable to that of ArN2+X– salts.
In the scientific literature, there is also a discussion of the assumption that the oxidizing agent in hypervalent iodine compounds is only the iodine atom, which is capable of adding various nucleophilesFootnote 2. Various variants of mechanisms involving iodine compounds in oxidative catalytic processes are considered (see footnote 2). It follows from the discussions that the question of the OS of iodine and Ar in these compounds is still far from being solved. The determination of their structure by X-ray powder diffraction analysis does not clarify the OS of organic substituents at the iodine atom, but at the suggestion of one of the reviewers of this review, we present the structural characteristics of a number of iodine compounds: PhI(C≡CPh)+OSO2Tol (I–Ph, 2.120(30) Å; I–C≡CPh, 1.969(30) Å)2, PhICl2 (ICl2 group is almost linear; C–I, 2.45l Å; C–I, 2.00 Å)Footnote 3, and PhI(OAc)2 (I–C, 2.09 Å; I–O1, 2.136 Å; I–O3, 2.163 Å; I–O2, 3.049 Å; I–O4, 2.93 Å)Footnote 4.
CHAPTER 3. SONOGASHIRA REACTION AND RELATED PROCESSES
C–C coupling reaction (XII) has been the subject of many articles and reviews. The Sonogashira catalytic systems Pd(II), Pd(0)–CuX [123, 124, 166, 167] are also used, as seen above, both in the reactions of OD of alkynes and in the Cadiot–Chodkiewicz reaction. After Chinchilla and Najera’s reviews of 2007 [168] and 2011 [169], and Barbosa and colleagues’ review [170], it is very difficult to characterize the state of the art in this area of catalytic chemistry in a small chapter: just note that the REFERENCES section in the review [168] contains 535 items (916 references)! Nevertheless, in the context of this review, it is useful to discuss the main stages of the design of catalytic systems for the Sonogashira reaction, as well as the results of studies of the mechanisms of this reaction, which is similar to the Cadiot–Chodkiewicz reaction.
Interestingly, as far back as 1968–1969, Shvartsberg and Kotlyarevskii with colleagues used a copper powder as a catalyst for acetylene condensation (later known as the “Sonogashira reaction” (see work under footnote 1)).
3.1. Multifunctional Catalytic Systems Based on Palladium and Copper(I) Complexes
In the Sonogashira reaction named after Kenkichi Sonogashira, one of its discoverers [124]. The organohalides R1X used in this reaction are haloaryls, haloalkenyls, haloalkyls, and halopyrrils (see [149]), and other compounds. In the Sonogashira catalytic system, alkynes efficiently react even with esters of enol compounds, for example, with R1OTf triflates [171]:
The reaction in the presence of L2Pd(OAc)2 in DMF occurs with yields of conjugated enynes of 71–90% at 60°C; and with the addition of CuI and Et2NH, at room temperature.
As catalysts for the Sonogashira reaction, palladium nanoparticles were also studied [172]. One of the first studies of the laws of this reaction, which was carried out by Beletskaya and colleagues [173], showed that the addition of CuI to PdL4 solutions accelerate the cross-coupling process by about an order of magnitude. Later, it was also found that, in most cases, the addition of CuX to systems containing palladium accelerates the condensation process and makes it more selective.
At the same time, it was shown that, depending on the nature of the substrates, CuX can strongly inhibit the process, almost to a complete stop [174]. For example, in an acetonitrile solution of PdCl2(CH3CN)2 in the presence of o-diphenylphosphine ligands and the base Cs2CO3 at 70°C, the reaction
occurs perfectly without CuI; and at [CuI] = 1 mol %, the product yield for 1 h is <10%:
The reaction of aryltosylates with aryl- and alkylacetylenes occurs similarly. Examples of successful syntheses of 19 substances were given.
Since it has long been clear that the main active catalysts for this reaction are the \({\text{PdL}}_{n}^{0},\) complexes necessary for the oxidative addition of R1X,
the attention of researchers was drawn to the ligand environment of the Pd(0) or Pd(II) complexes in the L2PdX2 precursor [168, 169]. At the same time, the Cu(I) complexes were not ignored. For example, additional stabilization of CuI with chelate diphenylphosphines leads to an increase in the selectivity of the Sonogashira reaction [175]. Trisubstituted ferrocenes were used, e.g., [tBuC5H2(PPh2)2]Fe[C5H4(PPh2)](L), the structures of which were studied. In the [Pd(C3H5)Cl]2 (0.2–0.5 mol %)–CuI(L) (0.1–0.4 mol %) system, the reaction of 4-bromoanisole with PhC2H occurs at 120°C with a product yield of 96–99% and with traces of PhC4Ph. Electronically and sterically deactivated aryl bromides successfully react. A 31Р NMR spectroscopy study showed that these phosphine ligands are exchanged between Cu(I) and Pd(II) to form chelate σ-allyl palladium diphosphine and mixed Cu–Pd phosphine complexes. It was found that the condensation in the bimetallic system is inhibited by polyphosphine copper complexes, and that the cross-coupling of iodobenzene with phenylacetylene can occur in the presence of complexes of CuI with other triphosphine-substituted ferrocenes alone. Even the CuX complex with triphenylphosphine (without palladium) slowly but selectively catalyzes the reaction of alkynes with ArI and vinyl iodides to form disubstituted acetylenes and alkenynes. A decrease in the [L] : [Cu] ratio from 2 to 0.5 in a CO atmosphere leads to the appearance of PhCOC≡CPh [176]. The fast cross-coupling reaction also occurs with the participation of CuI alone (Na2CO3, DME–H2O) with the use of RC≡CH with RIPh+ in the reaction [177].
A simple ligand-free system consisting of Na2PdCl4 and CuI in (iPr)2NH with small additions of phosphonium salts (tBu)3PH+BF– and ammonium salts (iPr)2NH2+Br– at 80°C catalyzes the reaction of aryl bromides with various alkynes very efficiently: at [Pd] = 0.005 mol %, the TOF is 3200–10000 h–1 [178].
Probably one of the first mechanisms of action of palladium complexes in the Sonogashira catalytic system was proposed by Kassar [122] in 1975. It was assumed that the intermediate ArPd(C≡CR) leading to the cross-coupling product is formed by the oxidative addition (OA) of ArX to Pd(0) complexes, followed by the interaction with the RC≡C–Na+ anion (in the absence of CuX). The ArC≡CR product is formed by the reductive elimination (RE) from the organopalladium intermediate. The simplest scheme of action of the Pd(0)–CuХ bimetallic system was considered by Gelman and Buchwald [174]. It was assumed that the soluble ethynyl complex Cu(I)RC≡CCuCl–R3NH+ in the case of CuCl as a result of the transmetalation (TM) reaction with the participation of ArPdXL2 is converted into ArPd(C≡CR)L2 intermediate, from which the product of the RE reaction is obtained. Thus, the Sonogashira process occurs by a nonlinear mechanism with two catalytic cycles (Scheme 9), from which the kinetic functions [70] of the two catalysts are clearly visible:
– the kinetic function of CuХ is the catalysis of the reaction of RC≡CH with ArPdXL2 to form I1 and НХ;
– the kinetic function of Pd(0) is the catalysis of the reaction of ArX with CuC≡CR to form the reaction product and CuX.

Scheme 9 . Kinetic functions of components of the Pd(0)–CuX catalytic system.
It follows from Scheme 9 that the Sonogashira reaction can also occur without CuX because the ArPdXL2 intermediate in the presence of bases can give intermediate I1 (see below).
The steps of the process shown in Scheme 9 were studied in great detail, all the more so, as the steps of OA, RE, and TM occur in many C–C and C–X coupling reactions (X is a heteroatom: N, S, O, etc.) [169, 179]: in the Heck, Cadiot–Chodkiewicz, Sonogashira, Stille [181, 182], Suzuki–Miyaura [131, 183], and Negishi [179] reactions and some other processes, as well as the steps of RE and TM (Stille, Suzuki, Negishi, Glazer–Zal’kind reactions) catalyzed by PdLn complexes.
Stille reaction:
Suzuki–Miyaura reaction:
Negishi reaction:
Along with alkyl groups, allyl, aryl, benzyl, and vinyl groups are used as R1 substituents, and Cl, Br, I, OTf, OTs, and OH (LVI) anions are used as X and Y substituents. Mg, Cu, Al, In, and Si are also used in R1МXn reagents [179]. Let us present a number of important works in which the elementary steps characteristic of various processes were studied in detail experimentally and theoretically. Oxidative addition of R2X, mainly to Pd(0) complexes, was studied in numerous works [184–195]. A detailed study of the OD of the RX molecule to PdL2 showed [185] that the trans-PhPdXL2 obtained by Fitton and Rick [186] does not form immediately in the course of the OD reaction; the product of this reaction is cis-PhPdXL2, which rapidly isomerizes to the trans product. The reaction of C6Cl2F3I with Pd[PPh3] was studied in toluene and in THF. In toluene at 60°C, a mixture of cis- and trans-isomeric products of OP is formed, whereas in THF at room temperature, only the cis isomer is obtained in 1 h, which then transforms into the trans isomer. The kinetics of the isomerization in THF was studied by 19F NMR spectroscopy. As a result, an empirical kinetic equation was obtained, which includes the concentrations of THF, PPh3, Pd(0), and the trans-ArPd(I)L2 adduct, with both isomers being autocatalysts of the OP reaction with identical acceleration effects. Unfortunately, when deriving the kinetic equation, the material balance of palladium was not taken into account and the equation for the degree of complexation of Pd (F = [Pd]+/[cis complex of Pd]) was not used. Based on the study, it was concluded that there are at least four routes for the transformation of the cis complex into the trans isomer. One of the routes involves the THF solvent, and the three other involve [Pd]–I complexes. Although the efficiencies of the isomers are almost the same, only the route involving the isomerization product, the trans isomer, is autocatalytic. Interestingly, the ethynyl complex (RC≡C)Pd(Ph)L2 obtained from the trans complex (e.g., from RC≡CCu by the TM reaction) is further isomerized to the cis isomer participating in the step of RE. The role of the amine in the steps of OP was studied by Shekhar et al. [196].
Hartwig studied SE reactions [197], and Osakada et al. [198] and Nova et al. [199] studied TM reactions. Hue and Liu [194] and Jutand et al. [200] studied the role of amines in the steps of the Sonogashira reaction. An der Heiden et al. [195] made a critical theoretical analysis of the steps involving Pd(0, II) complexes and the catalytic cycles of the Heck, Stille, Suzuki, and Negishi reactions; they also discussed the problems of oxidative addition in the Pd(II)/Pd(IV) oxidation cycle.
An der Heiden et al. [195] proposed a “multisubstrate” method for studying the kinetics of complex reactions by the example of the Sonogashira reaction. To determine the first-order rate constants (h–1) or catalyst turnover frequencies (TOFs), measurements were made for 21 aryl bromides in one reactor in the Na2PdCl4–CuI–(iPr)2NH–PR3–HBF4 (80°C) catalytic system using different PR3 (17 ligands). Activation parameters were found for 21 ArX substrates. The rate constants were used to construct the dependences of logki/k0 on the Hammett constants and for other correlations. Note that the study of the kinetics by the proposed method is possible only under the assumption made in that study: in the case of 21 ArX substrates, there is one rate-determining step, and this is the step of OA of ArX to PdLn, and all subsequent steps, including the formation of RC≡CCu, TM, and RE, occur so fast that the active PdLn complex in solution is the only palladium-containing compound (an intermediate of the catalytic cycle). The method of competing reactions with the creation of artificial route-coupling species is typically used to find the details of the mechanism as a result of obtaining more complex kinetic models (see Scheme 3). It is somewhat doubtful that there is only one rate-determining first step in the case of 21 ArX substrates with different X (Cl, Br) and electron-withdrawing and electron-donating substituents. Moreover, PdLn cannot be the only palladium-containing complex, since the OD of the alkyne to PdLn and the formation of anionic complexes LPdX– are possible (see section 3.2).
3.2. Catalytic Systems Based on Pd(0, II) Complexes (Copper-Free Catalysis)
As catalysts for the Sonogashira reaction were studied, catalytic systems containing no Cu(I) began to appear [201–205]. The effect of ligands, amines, solvents, and conditions on process performance was studied. For example, Pd(OAc)2 with water-soluble PR3 (TPPTS) and Et3N in aqueous CH3CN successfully catalyzes the Cadiot–Chodkiewicz reaction of alkenyl iodides with RC≡CH [200] and other cross-coupling reactions (21 reactions) at room temperature. Reactions of alkenyl iodides and aryl iodides with acetylenic alcohols are actively catalyzed by the PdL4 (L = PPh3) systems with piperidine or with pyrrolidine and Pd(OAc)2 with PPh3: the yields of products in the range 90–95% are reached at room temperature in 10–15 min [201]. The effect of 58 potential phosphine, arsine, and carbene (NHC) ligands in the Pd2(dba)3–Et3N–(tBu)3P–THF system at room temperature was studied by Böhm and Herrmann [203]. The TON was 80–200, depending on the nature of the substrates. The use of Pd(OAc)2 complexes with sterically hindered diphosphines [204] makes it possible to introduce ArX (X = I, Br, Cl) with Ph- and Me3Si-substituted terminal alkynes into the Sonogashira reaction at various temperatures and Pd loadings of 0.001 to 1 mol % with high conversion of aryl iodides and RC2H. The TON in this [Pd] range at 80°C ranges from 71000 to 100. It is also recommended to use ionic liquids, e.g., [BMIM][PF6] with PdCl2L2, but with amines (iPr)2NH or PiPy [205]. Such systems facilitate the separation of the catalyst with its subsequent fourfold recycle. It was also proposed [205] to use microflow reactors to increase the efficiency of the process. In the case of PhI and PhC2H at 60°C for 2 h, the product yield in such a reactor is ~95%.
Further improvements in palladium catalytic systems were directed toward the exclusion of amines, ligands, and even solvents. Najera and colleagues [206] used an oxime palladacycle prepared from 4,4-dichlorobenzophenone, hydroxylamine, and PdCl2 in NMP with 1 equiv Bu4NOAc. At 110°C and Pd loadings of 0.1 to 10–3 mol %, cross-coupling of 16 ArX was studied. At [Pd] = 0.1 mol %, the yield of p-chlorophenyl(phenyl)acetylene was >99% (TON = 990). At [Pd] = 0.001 mol %, the yield of the product reaches 72% at TON = 72000. Najera and colleagues [206] determined the optimal conditions for cross-coupling in the presence of an amine, but with neither ligands nor a solvent added. The reaction is completed in 10–20 min in the PdCl2(PPh3)2–PiPy system at 70°C. A system without amines and ligands [208], Pd2(dba)3 or Pd(OAc)2 in DMF was studied by the example of syntheses of 18 disubstituted alkynes at room temperature for 3–6 h with high yields of products of 75–93%. The added salt Bu4NOAc [206–208] acts as a base (see [208]).
Additional ligands and additional activators, e.g., stoichiometric amounts of Ag2O or Bu4NF, for a wide range of aryl iodides and MeC≡CH are used in systems without amines with PdL4 [209]. The role of the fluorine anion is not yet clear, and silver oxide can act as a base and a source of the RC≡CAg intermediate (similar to Cu(I) salts). Salts of carboxylic acids (НСООН) and amines were proposed [210] as low-temperature ionic liquids (IL) (room temperature) for PdCl2, e.g, [NH3+CH2CH2OH][HCOO–] (IL-1) and [C5H10NH2+][HCOO–] (IL-5). These ILs are solvents, bases, and reducing agents of PdCl2 to Pd(0) complexes. In such systems, high yields (up to 100%) were obtained in the homocoupling of ArI and in the reaction of bromotoluene with phenylacetylene. IL-1 was phosphorylated to form IL-OPPh2 (IL-6). This IL is both a solvent and a ligand stabilizing Pd(0). The addition of KOAc to IL-1 and IL-5, as well as of Pr3N to IL-6 lead to disubstituted alkynes with the yield reaching 100% at 100°C. The system allows 10 catalyst recycles with a slight decrease in yields.
The mechanism of the Sonogashira reaction involving palladium complexes has already been partially discussed in section 3.1 [193, 194]. Let us dwell on Jutand and colleagues’ detailed study [193] of the reaction of PhI with acetylenic alcohol HC≡CCH2CH2OH in solutions of Pd(dba)2 and PPh3 or AsPh3 (L) with various amines. Jutand and colleagues studied the kinetics of the substitution of the ligands L by amines and the kinetics of OA of PhI to PdLу complexes. Let us present the main results of that study.
(1) A comparison of the parameters of the processes with two ligands and two amines, piperidine (PiPy) and morpholine (Morph), under the same conditions (3 h) shows that, in the case of PiPy, the product yield for PPh3 is about twice as high as that for AsPh3, and in the case of morpholine, the yields are close and are 56 and 50%, respectively. The transition from piperidine to morpholine is accompanied by a decrease in the yield from 94 to 56%. It was also shown that Pd(dba)2 supplemented with 2L is a more efficient precursor than PdL4 due to the necessary dissociation of the PdL4 complex and the inhibitory effect of excess ligand. Moreover, it follows from the results obtained that the amine takes part in the rate-determining step.
(2) In previous Amatore and Yutand’s works cited by Jutand and colleagues [193], it was already demonstrated that the OA of PhI to PdL4 gives trans-PhPdI(PPh3)2, which reacts with amines, whereas trans-PhPdI(AsPh3)2 in chloroform easily transforms to the dimer Pd2(μ2-I)Ph2L2 (13), which, just as complexes with PPh3, forms complex 14 with amines:

In the case of L = AsPh3, this complex is in equilibrium with the trans-PhPdI(AsPh3) complex and with dimer 13.
(3) The equilibrium constants Keq of the reactions of substitution of L with amines at 25°C were determined by 1H NMR spectroscopy in chloroform.
Amine | PPh3 | AsPh3 |
PiPy | 0.11 | >11 |
Morph | 0.014 | 4.3 |
Thus, the product of substitution of the AsPh3 ligand with amines in the case of PiPy is 100 times more stable than that in the case of PPh3 and is ~300 times more stable than that in the case of morpholine. Note that piperidine (\({\text{p}}{{K}_{a}}_{{\left( {{{{\text{H}}}_{{\text{2}}}}{\text{O}}} \right)}}\) = 11.12) is a stronger base than morpholine (\({\text{p}}{{K}_{a}}_{{\left( {{{{\text{H}}}_{{\text{2}}}}{\text{O}}} \right)}}\) = 8.33).
(4) An important issue of the role of amines in the reaction of OA of PhI to Pd(0) complexes was studied. Shekhar et al. [196] also discussed this issue. The OA kinetics was studied with the PdL4 complex in DMF containing Bu4NBF4 at 20°C by measuring the loss of PdL3 in the reaction by voltammetry with a rotating gold electrode from the current at the PdL3 oxidation potential. The main fact that has been established is that the addition of amine accelerates the OA reaction. After completion of the experiment with PiPy, according to 31P NMR spectroscopy data, the PhPdI(L)(PiPy) complex is formed in the system. The obtained facts point to the participation of complexes with the ligand and amine in the OA reaction. Thus, the amine, replacing L in the PdL2 complex, leads to a less active complex in the reaction of OA of PhI with PdL3 (the inhibitory effect of the amine as a result of the appearance of free ligands in the solution), but, on the other hand, the amine accelerates the OA process due to the formation of a more active Pd intermediate (0)L(amine).
(5) The important role of amines was also found in the steps involving alkynes. If an amine forms a complex with PhPdIL2, then it can compete with alkyne (Ac) for the coordination site at Pd(II). In the case of the PhPdI(AsPh3)2 complex, the reactions with amine and alkyne occur through the dimeric complex. At a stoichiometric ratio of reagents (dimer XIII : amine : alkyne = 1 : 2 : 2), PiPy wins the competition, and the PhPdI(R2NH)L complex (14) is mainly formed, which is more stable than the corresponding π-complex PhPdI(Ас)L. When the concentration of PiPy is doubled, the cross-coupling reaction begins with a product yield of 50%. As a result of the observations made, the scheme of transformations of dimeric palladium complexes can be represented by a sequence of the following reactions:
Since the consumption of Ac (alkyne) in step (2) and the formation of the product (step (4)) obey the same kinetic law, RE step (4) is faster than steps (2) and (3), which, in Jutand and colleagues’ opinion [193], become candidates for the role of the rate-determining step (RDS) of the Sonogashira reaction in the studied systems.
The study of the kinetics of OA reactions and reactions of transformation of dimeric complexes with L = AsPh3 (in the stoichiometric version) allowed Jutand and colleagues [193] to formulate a simplified two-route mechanism of the Sonogashira reaction in a catalytic system with amines (Аm) (Scheme 10). The sequence of transformations of intermediates 1–2–4 corresponds to route A, and the sequence 1–3–5 corresponds to route B.

Scheme 10 . Two-route mechanism of the Sonogashira reaction in a catalytic system with amines.
Thus, in the case of L = РPh3, the substitution of L with an amine in the PhPdХL2 complex does not occur because of the low concentration of the dimer, and, therefore, route A occurs. As was already noted ([193] CONCLUSIONS section, item 5), PdL4 is a less active precursor than Pd(dba)2 + 2L: an excess of L inhibits both the OA of PhI and the substitution of L with alkyne at the next steps. PiPy is more active than morpholine due to its greater basicity at the steps of alkyne deprotonation in the PhPdI(Ac)L π-complex.
In the case of L = AsPh3, there is a substitution of L with amine (PiPy) through dimer 13, which is faster and more efficient than that with alkyne. Therefore, in such a system, route B occurs. The overall result is that (i) amines interfere with the process of OA and accelerate it due to the formation of a more reactive Pd(0) complex; and that (ii) amines replace one of the trans ligands in complex 14, and AsPh3 is substituted more easily than PPh3.
In concluding the analysis of this study, it can be noted that, unfortunately, Jutand and colleagues [193] did not compare the kinetics of OA of PhI to Pd(0) complexes and the kinetics of the transformation of the PhPdХL2 intermediate into the reaction product with one amine and with different ligands under the same conditions. Therefore, it is difficult to draw a conclusion about the rate-determining step of the Sonogashira process in the absence of Cu(I) (see above), although in the case of multiroute reactions, e.g., A and B, the concept of a rate-determining step is generally meaningless. The existence of two routes in systems with amines was also confirmed by Hue and Liu [194] as a result of a detailed kinetic study. At the same time, the detected fact that the Sonogashira reaction mechanism is multistep and multiroute once again confirms our conclusion about the incorrectness of applying the multisubstrate approach [195] based on the assumption that the step of OA is a rate-determining step, regardless of the nature of ArX, to the study of the kinetics of this complex reaction, all the more so in bimetallic catalytic system.
The scientific community is discussing one more mechanism of the Sonogashira reaction involving the step of carbopalladation of alkynes [211–213]. It is assumed that the carbopalladation by the syn addition of Ar–[Pd] in the π-complex ArPd(RC≡CH)L2 to alkyne to form L2PdC(R)=CH(Ar) may be the key step of the cross-coupling reaction. Deprotonation of this intermediate or its isomerization to the anti isomer with HPdL2 elimination can lead to the product RC≡CHAr of the Sonogashira reaction. This mechanism was finally rejected in Ljungdahl et al.’s work [212]: the synthesized intermediate Z-L2Pd[C(Ph)= CH(Ar)]X did not participate in the step of RE to form RC≡CHAr. Ljungdahl et al. [212] also studied the kinetics of the interaction of 4-IC6H2F3 (RI) with electron-donating (ED) and electron-withdrawing (EW) arylalkines in the Pd2(dba)3·CHCl3–AsPh3–Et3N system in chlorobenzene. For ED alkynes, the predominant route is the slow formation of the cationic π-complex by the replacement of the iodine anion by amine (a negative slope of the straight line in the Hammett dependence and an increase in the rate with increasing nucleophilicity of the amine). For EW arylalkines, the key step is the transfer of H+ from the neutral π-complex to the amine (deprotonation) to form an anionic intermediate. In this case, an important role is played by the basicity of the amine, and the slope in the Hammett correlation is positive. A theoretical analysis of the mechanism involving the carbopalladation of alkyne by García-Melchor et al. [213] confirmed the low probability of such a route: the activation energy of the step of deprotonation of Pd–alkenyl intermediates is ~ 40 kcal/mol (the reaction of PhPdI(PH3)2 with PhC≡CH and pyrrolidine as a base was analyzed). The development of the theory of mechanisms of the Sonogashira reaction is currently mainly focused on the deprotonation mechanism involving cationic and anionic intermediates (Scheme 11). In the case of cationic and anionic mechanisms, the activation energy \(\Delta G_{{298\,{\text{K}}}}^{^\circ }\) of all steps is low: for the steps of formation of π-complexes with the PH3 ligand, it is 23 kcal/mol. The total activation energy \(\Delta G_{{298\,{\text{K}}}}^{^\circ }\) of the Sonogashira reaction for the reaction of RC≡CH with ArI (R = NMe2) with the cationic mechanism is 23.7 kcal/mol; and with the anionic mechanism, it is 25.6 kcal/mol.

Scheme 11 . Mechanism of deprotonation steps in the catalytic cycle of the Sonogashira reaction.
Much attention in recent years has been attracted by heterogeneous catalysts for the Sonogashira reaction that are based on palladium nanoparticles (NPs) and nanoclusters (NCs) [168–171].
Three variants of the mechanism of catalysis on these particles are discussed (Scheme 12). The contributions of the three routes depend on the nature of NPs, support, solvent, and ArX reagents. The above reviews provide a huge number of supported and immobilized NPs and NCs. It was also found that the addition of other metals to Pd(0) or the use of alloys with other metals stabilizes active sites of Pd NPs [179]. The 2021 Reina et al.’s review [180] considered the c``atalysis of various reactions, including cross-coupling reactions, by only NPs of palladium and copper and alloys of these metals.

Scheme 12 . Three catalytic cycles of the mechanism of the Sonogashira reaction on Pdn nanoparticles [170]. Het. CC and Hom. CC stand for heterogeneous and homogeneous catalytic cycles, respectively.
3.3. Catalytic Systems Containing Other Metals
A short Plenio’s review of the Sonogashira reaction [137] compared compounds of various metals (Cu, Au, Ag, Pd, Ru, Fe, In) as catalysts for this reaction and also binary (polyfunctional) catalytic systems (Pd–Cu, Pd–Au, Pd–Ag). It was noted that palladium catalysts with Cu(I) and without copper compounds have the highest activity.
Gold compounds in the Sonogashira reaction. Among the studied systems based on other metals, catalysts containing gold compounds are most actively studied. It follows from section 1.1 that, in solutions of Au(I) complexes and on the surface of heterogeneous catalysts (AuX/support, Au/support), the reaction of OD of alkynes with oxygen and with stronger oxidizing agents efficiently occurs.
The problems of catalysis of the Sonogashira reaction by gold compounds turned out to be more complex and controversial. Corma and colleagues [136, 138] described the catalysis of the reaction of R1C6H4I with alkynes (xylene, K3PO4 at 130°C) by complexes of AuCl(PPh3) and Au(I) with Schiff bases and oxidizing agents (Selectfluor, hypervalent iodine compounds).
A comparison of soluble complexes of Au(I) and Pd(OАc)2−L under the same conditions showed their similar activity in the synthesis of diphenylacetylenes from PhI and PhC2H. The heterogeneous Au/СеО2 catalyst [139] was also used for the synthesis of Ph2C2 in DMF at 150°C with Na2CO3 as a base. The PhI conversion in 24 h was complete with the following yields: Ph2C2, 89%; Ph2, 11%; and PhC4Ph, 30% (due to excess of PhC2H). A study of the reaction in xylene detected no passing of gold into solution. It was concluded that Au/СеО2 can be a catalyst for the Sonogashira reaction, but is inactive. With colloidal gold (~5 nm), results close to those for a heterogeneous catalyst were obtained.
Reactions of synthesis of diarylacetylenes were also studied with AuI on CeO2 in 11 solvents with various ligands [140]. The best components of the catalytic system for the reaction of iodoanisole with phenylacetylene turned out to be the following: the solvent, toluene (at 130°C); the ligand, dppf (1,1-bis(diphenylphosphine)ferrocene); the gold compound, AuI (1 or 2 mol %); and the base, K2CO3; high yields were also observed in solutions of Au(III) with dppf, with AuCl, and even with colloidal gold. The products were 23 disubstituted acetylenes with yields of 35–99% (24 h). In the case of o-iodoaniline and various alkynes, in the same systems, cross-coupling with intramolecular cyclization to substituted indoles occurs with yields of 41–99%. The main conclusions made in that work [140] were that (a) the activity of the catalyst is determined by the ability of nanocrystalline CeO2 to stabilize a small amount of gold in the form of Au(I), although the dominant gold species are Au(0) and Au(III); and that (b) gold-based catalytic systems are unlikely to be widely used in the Sonogashira reaction.
Heterogeneous catalysts were studied in more detail by De Souza et al. [214]. Gold salts were deposited on SiO2, Ce2O3, and Nb2O3 and, after reduction, were studied in a DMF medium with K2CO3 under microwave irradiation. The best result was obtained for the Au/SiO2 catalyst: 87% yield in 1 h. However, it was concluded that a heterogeneous catalyst is active if it is a source of soluble active gold particles(!). The issue of soluble active gold NPs was also studied by Lambert and colleagues [215], who showed that the AuCl(PPh3) complex in DMF at 145°C is inert in the reaction of PhI with PhC2H for ~100 h and the PhI conversion of about 13% is achieved after 160 h. During the reaction, this complex is reduced to gold NPs, on which X-ray photoelectron spectroscopy (XPS) detects the signal of an iodine atom adsorbed on a gold atom. Since the mechanism of catalysis of the cross-coupling reaction adopted for palladium(0) complexes requires a change in the oxidation state of Au(I) to Au(III), Lambert and colleagues [215] questioned such a mechanism. For a gold atom to pass into solution from the surface of nanoclusters, it should first be oxidized to Au(I) and only after that it can participate in the steps of transformation of Au(I) to Au(III), which, in Lambert and colleagues’ opinion [215], is unlikely. Therefore, they came to the fundamental conclusion that gold compounds on the surface of the formed NPs are involved in heterogeneous catalysis. A pronounced increase in the activity of the catalysts and the selectivity of the process with increasing NP size was also detected. It was noted that a noticeable passing of gold atoms into solution from the NP surface takes place, but the emerging solution demonstrates an immeasurably low activity. It was determined that the cross-coupling reaction stops when NPs disappear, and the activity of NPs is an order of magnitude higher than the activity of the formed solution. The results of the work [215] contradict the conclusions made by De Souza et al. [214]. In subsequent works, the problem of choosing between heterogeneous and homogeneous types of catalysis by gold compounds was actively studied.
The question of whether or not there are thermal activation and dissociation of PhI on the Au(0) surface was answered by the methods of temperature-programmed adsorption–desorption and IR reflection–absorption spectroscopy during the adsorption of PhI on the (111) surface of Au NCs [216]. The formation of adsorbed Ph and Ph2 was determined. A monolayer coating with phenyl iodide was observed at 90 K and is 0.16 (mole fraction) of multilayer adsorption. Ph2 was reversibly adsorbed in a planar molecule conformation oriented parallel to the Au(111) plane. Three desorption peaks were found for PhI(ads) layers at 290–308, 211, and 185 K. PhI dissociated in the range of 200–250 K to form an I(ads) atom and Ph2 without accumulation of Au–Ph groups. In Lambert and colleagues’ opinion [215], it is natural to expect that, in the presence of phenylacetylene, the Au–Ph surface group can interact with alkyne to form a cross-coupling product. At this stage, Lambert and colleagues conducted two more studies, which confirmed their opinion on the heterogeneous nature of gold catalysis. In one of the studies [217], they investigated the reaction of PhI with PhС2Н on the surface of an Au(111) single crystal (gold content 99.999%) in a vacuum. Using the temperature-programmed reaction (TPR) method together with scanning tunneling microscopy (STM), it was found that these molecules react on a smooth Au(111) surface to form Ph2, PhC4Ph, and Ph2C2. The temperature threshold for the Sonogashira reaction was found, which is determined by the conditions for breaking the C–I bond on the metal surface. The main conclusion is that active sites are located at the boundary of islands of adsorbed reagents. The formation of two intermediates, AuPh and AuI, on two active sites on the Au(111) surface was considered. Thus, that study proved for the first time that heterogeneous catalysis is an inherent property of metallic gold. It follows from the works [215, 217] that Au(0) NPs on the support surface are the active form of the Au/SiO2 and Au/СеО2 catalysts.
To resolve the contradictions between the results obtained by Corma and colleagues [138, 142, 144, 147, 149] and Lambert’s group [146], one more study was performed to refine the nature of active sites in the heterogeneous catalytic reaction and increase the selectivity of the process using La and Ce oxides as supports. It was shown that immobilized Au(0) NPs in all the systems studied are actual catalysts for this reaction, whereas immobilized PdL2 compounds are inactive. Goguet et al. in 2009 [218] studied the Au/La2O3 system by various physical methods and concluded that, on this catalyst at a small amount of gold, only ionic species Au+ and Au3+ are present in the form of monoatomic (molecular) particles in the complete absence of Au(0). Checking the results of that work, Lambert and colleagues [146] prepared three samples, A, B, and C:
– A (0.5 wt % Au on La2O3) demonstrated the ionic species Au+ and Au3+ in Au/La2O3 according to Goguet et al. [218];
– B (10 wt % Au on La2O3) contained Au(0) NPs 20 nm in size;
– C (10 wt % Au on SiO2) contained the same Au(0) NPs 20 nm in size.
These catalysts were studied in the reaction of PhI with PhC2H at 145°C in DMF for 160 h. On samples A, B, and C, DPA was obtained in amounts of about 0, 0.4, and 0.16 mmol, respectively. The TON (mole of DPA per mole of Au atoms on the catalyst surface) was 10, 275, and 158, respectively. In the gas phase, H2 and I2 were found. The selectivity in DPA is 0, 0.82, and 38%, respectively. It was assumed that the spillover effect of H2(H) with Au(0) on La2O3 or Ce2O3 accelerates the synthesis of DPA and DPDA.
Since most studies of the Sonogashira reaction with gold compounds were carried out under different conditions (solvent, ligands, homogeneous and heterogeneous systems, different amounts of gold on supports and colloidal gold, temperature), questions naturally arise about the nature of the active forms in gold catalysis and about the mechanism (or mechanisms) of this reaction.
(1) Is catalysis by gold compounds homogeneous catalysis, metal complex catalysis, or heterogeneous catalysis by gold nanoparticles formed by the reduction of LAuX complexes in solution or on the surface?
(2) What is the mechanism of RX activation by gold complexes or nanoclusters?
(3) Are gold compounds [Au(0), Au(I)] indeed catalysts for this reaction?
Already the first acquaintance with the above works causes the reader to associate the studied gold catalysts with catalytically active Pd(0) complexes, which in the PdL2–PdL4 complexes have the d10 electronic configuration, the same as Au(I) in the LAuX complexes. This immediately implies the idea of using the mechanism of the Sonogashira reaction in solutions of palladium complexes for catalysis by Au(I) complexes, i.e., of considering the formation of reaction products as a result of the steps of OA of R1X or LAu(C≡CR2) to form, in both cases, the key intermediate R1Au(C≡CR2)ХL, which transforms into a product.
The analysis of this problem was very thoroughly approached by Echavarren and colleagues [219]. They reported that they failed to find any indication of the possibility of OA of R1X to LAuX complexes and that, for complexes with the LAu–C bond, a slow step of OA of methyl halides only to MeAu(PR3) is known, the reactivity of which is characteristic of SN2 reactions (MeI > EtI > iPrI). The interaction of Au(C≡CC6H4Me)L with RC6H4I for 24 h at 130°C was studied, but no reaction product was obtained even with RC6H4I as a solvent, although AuR1R2X compounds have long been known to participate in the step of RE in statu nascendi. This property of organometallic Au(III) compounds was also confirmed. The addition of even a small amount of Pd(II) compounds to a system containing LAuX complexes leads to the formation of Sonogashira reaction products. At a PdCl2L2 concentration of 1.4 mol %, the yield of products for 16 h was 100%. Naturally, such a system is the classical Sonogashira catalytic system, in which the functions of CuX are performed by Au(I) complexes. The gold used in this work contained 0.3%, or 3000 ppm, palladium. Since the results obtained were fundamentally different from the data of Corma’s [136, 138] and Li’s [139] groups, Echavarren and colleagues [219] repeated the synthesis of phenyltolylacetylene under conditions close to those in Corma’s works and showed that, in the AuI–dppe (1,2-bis(diphenylphosphino)ethane) in toluene at 130°C in the presence of K2CO3 for 16 h, the yield of products was <2%; i.e., the Au(I) complex with a palladium content of 3.1 μg/g (3.1 ppm) in AuI is virtually inactive. The categorical conclusions of Echavarren and colleagues were the following [219]:
(1) the observed facts about catalysis by gold compounds are related to the presence of palladium impurities in the gold samples used;
(2) there are no mechanisms explaining the catalytic properties of Au(I) compounds.
Immediately after the appearance of that article, Corma et al. published the results of their experiments [220], which disproved Echavarren and colleagues’ conclusions [219]. It was noted that supported gold NPs are not individual gold atoms or Au(I) molecular complexes. Corma et al. studied the kinetics of the reaction of phenylacetylene with PhI on a Au/СеО2 catalyst (with a Pd content in the original gold of 1.1 ppm) with Pd additives and obtained the dependence of the initial velocity r0 = b + a[Pd] with a correlation coefficient of 0.99. The b value is 5.8 ± 0.4 h–1. The r0 value is ~90% of the r0 achieved on a Au/СеО2 catalyst with a gold purity of 99.9%. The r0 value for gold with a purity of 99.999% is 7.8 h–1.
These results were confirmed by Lambert and colleagues’ works [146, 215, 217]. A theoretical analysis of mechanism steps (DFT) suggested [220] that the reaction of OA of PhI to the Au(I) complex is a difficult process with Еact = 31.6 kcal/mol and ∆Н0 = 11.1 kcal/mol. In the case of Au38 NPs (~1 nm), the OA of PhI occurs at different sites to form adsorbed Ph and I with Eact = 11.1 kcal/mol.
Meanwhile, in a 2011 short communication [221], Echavarren and colleagues summarized the results of previous studies of the catalysis of the Sonogashira reaction by gold compounds. They acknowledged that gold NPs and NCs on the surface of supports can be catalysts for this reaction, but they categorically opposed the possibility of catalysis by homogeneous Au(I) complexes, still believing that the observed catalysis is due to palladium impurities. That article was followed by works by Corma and colleagues [147, 221, 222] and other authors [223–225], in which additional information was obtained on the reactivity of gold NPs in various transformations of alkynes and RI.
Alkynes activated by electron-withdrawing groups, e.g., EtO2C≡CH, are converted on the surface of Au NPs (2–10 nm) on TiO2 [222] by the cyclomerization reaction into benzene isomers (1, 3, 4 and 1, 3, 5) at 120°C in 1,2-dichlorobenzene for 30 min with a yield of 85% and TOF = 30500 h–1. The reaction rate is described by an equation of the first order in alkyne and the first order in [Au]∑. It was noted that gold NCs <1 nm in size are virtually inactive in the catalysis of trimerization and alkynes because they are quasi-molecular particles with a tendency to stabilize gold cations (Au+). When an Au/TiO2 sample is treated with methyl iodide, only small clusters (<0.75 nm), which are inactive in the EtO2C≡CH trimerization reaction, remain on the TiO2 surface. The maximum activity was found for NPs ~3 nm in size. In the presence of O2, on the studied catalyst, a catalytic reaction of oxidative alkynylation of arenes occurs.
Corma and colleagues [223] made a theoretical analysis (DFT) of PhX adsorption processes (X = I, Br, Cl) and steps of breaking of the Ph–X bond on the surface of gold clusters. It was found that the enthalpy of ArI adsorption increases in the order Au(111) < Au38 < Au13 < Au3 (from –6.8 to –30.5 kcal/mol), whereas the activation energy of breaking of the C–I bond passes through a minimum for Au13 NCs (8.8 kcal/mol), where Au38 and Au13 are three-dimensional NCs, and Au7, Au6, and Au3 are planar NCs. It was found that the electronic properties of NCs determine both Eads and Eact of the reaction.
At the same time (2012–2014), an efficient new catalyst based on an Au cluster with Au25(SR)18 thiolate ligands supported on CeO2, TiO2, MgO, and SiO2 was proposed [224]. The reaction of phenylacetylene with o-iodoanisole occurs with a conversion of 96% and a selectivity of 88% (DMF, 160°C, Au/СeO2, K2CO3). It follows from the results obtained by the DFT method that, on the open faces of the cluster, both reagents are adsorbed in a configuration convenient for cross-coupling. As a result, three new intermediates, [Au]25Ar2, [Au]25(C≡CPh)2, and [Au]25(C≡CPh)Ar, are formed, ready for reductive elimination (RE) with the predominant formation of ArC≡CPh.
The reactivity of mononuclear cationic complexes [Au]\(({\text{P}}{{{\text{R}}}_{3}})_{n}^{ + }\) (R = Me, Ph; n = 1, 2) and Au3L clusters (L = Ph2P(CH2)nPPh2, n = 3–6) in reactions with PhI in the gas phase was studied [225] by mass spectrometry, DFT, and electrospray ionization (ESI) in combination with multistage mass spectrometry. The initial reagents for ESI were (PR3)AuCl and large gold clusters synthesized in the condensed phase. Rapid fragmentation of particles and products to be studied was carried out collision-induced dissociation (CID). It was determined that (PR3)2Au+ cations do not interact with PhI, whereas (PR3)Au+ cations interact with it to form (PR3)Au(PhI)+ adduct and two products, AuI and \({\text{PHPR}}_{3}^{ + }.\) PhI addition product is also obtained from Au3L+. Transformations of the (PR3)Au(PhI)+ complex (using CID) give Au3L+ + PhI and LAu2I+ + PhAu. Thus, it was proven that PhI oxidizes Au(I) complexes and small gold clusters. The results of the DFT analysis showed that the formation of the products of the oxidative addition of PhI to (PR3)Au+ ((Me3P)Au(Ph)I+, Me3PPh+, and AuI) is an exothermic processes, whereas the formation of OP products in the case of (Me3P)2Au+ is an endothermic process (5.9 kcal/mol) with an activation energy of 19.1 kcal/mol. These results are consistent with Corma et al.’s data [220]. In the case of the most reactive Au3L+ cluster with n = 6 in the (PPh3)2(CH2)n(PPh3)2 ligand, the enthalpy of formation of the complex with PhI is –17.7 kcal/mol and Eact = 1.48 kcal/mol.
A similar theoretical DFT study [226] of the reactions of Aun clusters and \({\text{Au}}_{n}^{ + }\) with RI (R = Et, Ph, vinyl) included the reactions of coordination of RI and OA of RI to gold clusters. It was found that cationic clusters coordinate more strongly, e.g., PhI (with higher exothermicity in comparison with neutral clusters), but the activation energy of bond breaking in the OA reaction is higher for cationic complexes. The exothermicity of the overall process is higher for neutral clusters. The enthalpy of binding RI decreases with increasing n in cationic and neutral clusters in the order 3 > 4 > 14 > 20, and the Eact values of the OP reaction depend weakly on n for cationic and neutral clusters, except for the reactions of C2H5I with Au3 and Au3+. After the above studies, over the past 8 years, several more works have appeared aimed at studying the laws of the cross-coupling reaction on the faces of Au0 nanocrystals, as well as the processes of ArX activation by Au(I) complexes.
Li et al. [227] in continuation of Li and Jiang’s work [224] studied the catalytic activity of gold nanorods with the Au(111) and Au(100) faces in the reaction of PhC2H with 4-iodoanisole to PhC≡CC6H4OMe, diphenyldiacetylene (DPDA), and Ar2. It was shown that short rods (~33 nm) with a large fraction of Au(111) faces catalyze the cross-coupling reaction much more selectively (90% at a conversion of 57%). The selectivity of cross-coupling in the presence of longer nanorods (42 and 50 nm) with a larger fraction of Au(100) faces, as well as shapeless NPs (from 2 to 20 nm), turned out to be below 59%. A theoretical (DFT) analysis of the reaction mechanism for the formation of three products on the Au(111) and Au(100) faces showed that the adsorption of PhI, its dissociation, surface diffusion of reagents, and the step of formation of the Ph2 product occur more efficiently on the Au(111) face. PhI dissociation on the Au(100) face occurs with the participation of three surface gold atoms to form Au2I and AuPh; and on the Au(111) face, with the participation of four gold atoms to form Au3I and AuPh. The activation energy of the step of PhI dissociation on the Au(111) face is 16.1 kcal/mol, and on the Au(100) face, it is 20.3 kcal/mol. Eact of the reaction of cross-coupling on the Au(100) face is 8.3 kcal/mol higher than that on the Au(111) face.
The reaction of PhC2H with PhX (X = Cl, Br, I) as the most common model of the Sonogashira reaction was studied under ultrahigh vacuum conditions and at normal pressure on the surface of the Au(111) face by X-ray photoelectron spectroscopy (XPS) [228]. It was found that PhCl and PhI participate in the reaction of DPA formation, whereas PhBr does not react with PhC2H under these conditions. At normal pressure, in the case of PhBr, the surface of the Au(111) face was found to be poisoned by carbon particles. A study of the adsorption of PhC2H and PhX by X-ray adsorption spectroscopy showed that the adsorption of undissociated molecules occurs at –195°C, and the dissociation of PhCl and PhI occurs in the range of –80 to –15°C. The dissociation of PhBr is observed at –123°C with the formation of Ph2 and Br2.
When studying the arylation of aromatic compounds (ArX + ArH) [229] in solutions of Au(I) complexes, the problem of accelerating the step of OA of ArX to Au(I) to form Au(III) intermediates was solved. The use of the phosphine–nitrogen ligands o‑С6H4(PR2)NR2 (L1) and o-С6H4(PAd2)NR2 (L2) (Ad is adamantyl) allows the OA reaction to be performed in the range of –80°C to room temperature even with cationic Au(I) complexes. In the case of the L2 ligand, the L2AuCl complex adds PhI in the presence of AgX to form the AuIII(Ph)I+ complex in less than 1 min at room temperature with a yield of 99%! p-ArX molecules with X = Me, OMe, NO2, F, etc., are also readily attached. The DFT method in the B97D/SDM variant with taking into account the effect of the solvent (dichloroethane) and the presence of cations and anions (ion pairs) was used to analyze the reaction of OA of PhI to the L2Au+[SbF6]– complex. The energy barrier \(\Delta {{G}^{ \ne }}\) of the step of OA is 11.2 kcal/mol, and ΔG° = –8.9 kcal/mol. Thus, Echavarren and colleagues’ assertion [219, 221] about the impossibility of the reaction of OA of ArX to Au(I) complexes is erroneous. The article also considered interesting details of the process of arylation of 1,3,5-methoxybenzene with phenyl iodide using the synthesized complexes L2AuCl and AgSbF6.
Bimetallic systems Pd–Au, Pd–Ag, Pd–Ni, Co–Cu, and other options. A study of bimetallic systems in the Sonogashira reaction showed that the addition of Au(I) complexes to PdCl2(PPh3)2 instead of CuI gives rise to a very active catalytic system [230]. For example, the AuCl(THT), AuC6F5(THT), and (iPr)2NH complexes in THF make it possible to achieve a 100% yield of diarylacetylene with ArX (X = Br, I) at room temperature or under reflux conditions for 14 h. It was found that the catalyst with CuI under the same conditions is 12–20 times more active than systems with gold complexes. At the same time, under the conditions of a two-phase system with the PdCl2(tppts) complex in CH2Cl2–H2O, catalytic systems with Au complexes are close in activity to systems with CuI and are even more active in the case of the AuCl(tppts) complex in the reaction of alkynes with PhI. The same polyfunctional catalytic systems with Au–Pd were studied by Panda and Sarkar [231, 232]. Some of the results of Jones et al. [230] and Echavarren and colleagues [219] were confirmed. It was found [231] that, in the PdCl2(PPh3)2–AuCl(PPh3)2 system, the alkyne conversion reaches 96% with a product yield of 54%, rather than 1% as obtained by Jones et al. [230]. In the reactions of PhBr with various alkynes and in various solvents, the yield of products at 70°C ranged from 33 to 96%, depending on the process time (3–12 h). An increase in the concentration of the Pd(II) and Au(I) complexes from 1 to 2% in a DMF–Et3N solution at 60–80°C decreases the reaction time to 0.5 h and increases the product yield to 80–98%. Cross-coupling with the ethynyl complex of Au(I) and PdCl2(PPh3)2 in DMF at 70°C for 1 h with a yield of 79% was also performed by the reaction
A similar study of catalysis by various [Au]–R complexes with ArI in the presence of PdCl2(dppf)2 was performed by Hashmi et al. [233] in an acetonitrile solution. Seventeen different products were obtained, including the Sonogashira reaction product R=(C≡CPh) with PhI. It was noted that the transmetalation in the [Au]–R reaction with ArPdХL2 does not require less probable transformations of intermediates with Au(I) to intermediates with Au(III). In that work, an interesting variant of using the AuX–PdII system for the synthesis of arylalkenyl products was proposed, in which the [Au]–R intermediate is formed from an alkyne and a Y–H nucleophile:
As a result of the reaction of TM with ArPdХL2, the alkenyl intermediate is converted into the product of the Heck reaction: ArCH=C(R)Y. This product can also be considered as a result of the addition of the Y–H molecule to the product of the Sonogashira reaction.
An interesting variant of the cross-coupling reaction was proposed by Panda and Sarkar [232]. They used the aryldiazonium salt ArN2BF4 instead of R1X in the reaction with phenylacetylene in the AuCl–PdCl2–2,6-lutidine system in THF at room temperature. The reaction product was obtained with a yield of 54%. The yield of the product of homocoupling of alkyne was 8%. Various aryldiazonium salts were studied with the 2,6-di-tert-butyl-4-methylpyridine ligand in 15 syntheses with product yields of 50–80%.
The Pd–Au system turned out to be also very active in the form of nanoparticles. Pd–Au or Pd–Au–Ag alloys in the form of nanowires perfectly catalyze the activation of RX. The history of the use of reactions involving the activation of R–X bonds by palladium and gold alloys in catalysis was described in a large work by Chinese researchers [234]. They studied the factors affecting the breaking of the R–X bond on these NPs by systematically varying the coordination numbers of the Pd–Pd pair, the occupation of Pd d‑orbitals, the structures of the surrounding of the palladium active site by gold atoms, and the catalytic properties of various NPs of the Pd–Au alloy in the hydrodehalogenation of RX. Among palladium nanoclusters on the gold surface, three types of sites that are active in reactions with various X were found:
(1) 5–7 close Pd atoms (Х = Cl);
(2) 3–4 adjacent Pd atoms (Х = Br);
(3) isolated Pd atoms (X = I).
It was also found that the addition of other metals and alloys of other metals to a Pd/support catalyst stabilizes Pd(0) active sites [234]. For example, in the case of a Pd/С catalyst, Au(0) additives in the form of a Pd–Au alloy manifest themselves [235]. Pd–Cu alloys were described in the review [236], and Pd–Ag alloys were used in a reaction close to the Sonogashira reaction with the participation of terminal alkynes and aryl iodonium salts [237]. The use of Pd–Au–Ag ternary alloys [238] in the Sonogashira reaction is also known.
The use of silver catalysts was described in Pale and colleagues’ review [239]. Li and Wang [240] found that AgI in the presence of PPh3 and K2CO3 in DMF catalyzes the reaction of cross-coupling of various ArX with various alkynes (Ph, Tol, PhF, hexyne) with high yields (85–99%) but at a high temperature of 100°C. Other silver salts were also active in the presence of phosphines. As intermediates when using silver compounds, AgC≡CR was found to be formed [241]. In the case of silver, the AgX–PdII binary systems [239], which also catalyze the Heck, Stille, and Suzuki reactions [242], were the most active. In these systems, the role of AgX is similar to that of CuX in the classical Sonogashira system: it is the formation of LAgC≡CR, followed by transmetalation and the appearance of the intermediate R1Pd(C≡CR)L2 [241]. However, an additional function of silver salts (nitrates, perchlorates, acetates, and triflates) in coupling processes to form a C–C bond is also important: this is the ability to abstract a halide anion from PdX2L2 complexes or from R1PdXL2 and convert these compounds into more reactive cationic complexes [239]. Pale and colleagues’ review [239] described many examples of addition reactions of strong nucleophiles to alkynes, catalyzed by Ag(I) complexes, in which the nucleophile is added to LAgC≡CR. Both NPs and NCs of Ag(0) are used in the Sonogashira processes [242–245].
The possibility of RX activation on Ag(0) NCs attracted the attention of researchers as early as 1999. The reactions of CH3I with CH2I2 during their surface adsorption on Ag(111) were studied by Wu et al. [243]. The following steps were detected by TPR and mass spectrometry:
– 110 K: CH3I and CH2I2 are adsorbed on the silver surface;
– <200 K: CH3Iads + CH2I2ads → CH3ads + CH2ads + 3Iads;
– ~225 K: CH3ads + CH2ads → CH3CH2ads (insertion of CH2ads into the CH3–Ag bond);
– 2CH3CH2ads + CH3ads → CH3CH2CH3r (cross-coupling);
– 2CH3CH2ads → CH3(CH2)2CH3h (homocoupling).
The adsorbed iodine atom is converted into I2r at 230 K. When CD2I2 is used, the corresponding deuterium derivatives are obtained, and in the case of adsorption of CH2I2 alone, ethylene is formed, which is desorbed at 260 K. The mechanism of NC formation and growth of Ag(0) particles was considered by Han et al. [244]. The synthesized NCs with sizes of 6–9 and 11.5 nm were used to catalyze the reaction of ArI with R1C≡CH. In a system containing silver NCs, CuI, PPh3, and K2CO3 in NMP at 80°C for various AuI, AuBr, and alkynes, the yield of products was in the range of 74–94%. The best results were detected on NCs 6 nm in size. Ag(0) NCs were characterized by various physical methods (X-ray powder diffraction analysis (XRD), transmission electron microscopy (TEM), high resolution transmission electron microscopy (HR TEM), field emission scanning electron microscopy (FE SEM), and others). It was determined that the catalyst components do not change during the reaction, only the size and shape of NCs change. AgI formation was not detected. A comparison of Ag(0) and Pd(0) NCs of similar sizes in the reaction of p-tolyliodide with PhC≡CH [244] showed that the activity of the catalyst (mmol product per gram of catalyst (!) per hour) is somewhat higher for silver: a 50% conversion is achieved in 54 min in the case of Ag(0) and in 64 min in the case of Pd(0). Han et al. believed that silver NCs are an ideal candidate for replacing palladium in the catalytic Sonogashira reaction.
The adsorption of PhC≡CH and PhCl on the Ag(100) surface of single crystals was studied [244] by scanning tunneling microscopy (STM (TEM)), temperature-programmed reaction (TPR), XRD, and DFT. The adsorbed molecules have a planar configuration, for which the experimental and calculated values of Eads are close. The Eads values indicated the decisive role of dispersion forces in the adsorption act. The study also showed that, on the Ag(100) surface of NCs, the Sonogashira reaction with usually low-reactive PhCl occurs. The reaction occurs at the contact boundaries of islands of adsorbed reagents and gives Ph2, DPA, and also DPDA. According to the TPR data, the maximum product yields were in the temperature range of 184–188 K and were in the order Ph2 ≈ DPA > DPDA. It was found that the activity of the catalyst depends on the size of the islands of adsorbed reagents: the smaller the island, the more active the Ag NCs. In Han et al.’s opinion [244], for practically interesting silver catalysts, it is necessary to inhibit the growth of reagent islands on the silver surface. According to Wu et al.’s results [243], the size of the islands is determined by the size of NCs: higher activity is characteristic of smaller silver NCs.
Interesting results in the catalysis of the cross-coupling reaction were also obtained with catalytic systems containing other transition and non-transition metals. As far back as 1986, Bumagin et al. [246] showed that NiCl2 catalyzes the reaction of PhC≡CCu with RX to form PhC≡CR and CuX in the presence of 1 mol % PhPdI(PPh3)2 and Bu4NI in рexamethylphosphoramide (RX is 4-nitrophenyl iodide). A more detailed study of the catalytic properties of Ni(II) complexes in the Sonogashira reaction was made by Beletskaya et al. [247]. They studied the effect of the solvent and the Ni(II) ligand environment and finally opted for the Ni(PPh3)2Cl2–CuI (10 mol %)–K2CO3 system in the dioxane–H2O (3 : 1) solvent. Under reflux conditions for 2 h for various PhC≡CH and ArI, the yield of isolated products was 86–100%. An interesting system based on pincer complexes of Ni(II) and CuI (dioxane, Cs2CO3, 100°C) was proposed for the reactions of alkyl bromides and alkyl chlorides with alkynes [248]. In the case of chlorides, Bu4NI was added to the system, and the process was carried out at 140°C for 16 h. The yield of products in the reactions of 14 alkyl halides with various alkynes was 57–89%. The structure of the pincer complex is represented by structure 15:

The synthesis of Ni(0)–Pd(0) NCs with a particle size of 1.5–4.2 nm was performed from Pd(OAc)2 and Ni(OAc)2 in trioctylphosphine [249]. The solution was loaded into oleylamine and heated to 205–235°C for 0.5 h. The synthesized NCs have a core–shell structure, where the core and the shell are dominated by nickel and palladium, respectively. The yield of products from various ArBr and phenylacetylene in the presence of Ni(0)–Pd(0) NCs and CuI in diisopropylamine at 80°C was 90–95%. At the same Pd content, the activity of Ni–Pd NCs is about three times higher than that of Pd NCs.
Hollow spherical Co NPs (~50 nm) in the presence of CuI and PPh3 (K2CO3, N-methyl-2-pyrrolidone (NMP)) also proved to be good catalysts for the Sonogashira reaction for ArBr and ArI [250]. The product yield for 20 h at 120°C was 82–90%. It was assumed that intermediates R1CoL4X and R1CoLn(C≡CR2) are formed. Co(0) NPs as a catalyst were believed [250] to have a number of advantages over palladium catalysts: they are much cheaper, are easier to synthesize, and can be used repeatedly.
Lambert and colleagues [251] studied the heterogeneous catalysis of the Sonogashira reaction on Rh(0) NCs deposited on various supports. Rh(0)/γ-Al2O3 catalysts were used in the reaction of PhI with PhC≡CH. The turnover number of the catalyst for particles ~2 nm in size in the presence of tetrabutylammonium acetate as a base turned out to be 562 with a selectivity in DPA of 57%. In the case of 8-nm particles, the TON value in the presence of the same base was 2745 with a selectivity of 76% and a DPA yield of 41%. In the absence of a base, the TON value and the selectivity of the process do not depend on the NC size. A TEM study showed that rhodium NCs are mainly cuboctahedrons demonstrating the Rh(100) and Rh(111) faces. Reuse of the rhodium catalyst with NPs showed much better results for large NCs. The loss of rhodium in these experiments was 0.1 ppm. An increase in the activity of the catalyst with an increase in the NC size according to the data of that article contradicts the results of studying NCs of other metals, e.g., silver NCs [245].
Fe and In compounds. Along with the interest in the use of transition (Ni, Pd, Pt, Rh) and post-transition (Cu, Ag, Au) metals in homogeneous catalysis, a great number of publications also consider catalysis by Fe(III) complexes [252–254]. Correa et al.’s review [253] contained 1551 literature citations. Many works also focused on the catalysis of the Sonogashira reaction by various Fe(III) compounds. Bolm and colleagues [255] studied the reactions of various aryl halides and alkynes in solutions of iron salts and complexes. The choice of iron complexes was based on the studies of reactions of ArX with various nucleophiles, i.e., nucleophilic substitution reactions: the formation of C–N [256], C–O [257], and C–S [258] bonds, catalyzed by FeCl3 complexes. The effect of bases, anions, and ligands DMED, TMED, Phen, etc., and also the process conditions on the product yield and reaction time was studied [259]. Optimal conditions: FeCl3, DMED, Cs2CO3, solvent PhMe, temperature 135°C, and time 72 h. Under these conditions, 19 products of the Sonogashira reaction were synthesized, 10 of which were obtained with high yields, reaching 100%. It was assumed that the Lewis acidity of Fe(III) salts plays a major role in the activation of reagents. A similar inexpensive, but not very active, catalyst was also proposed for the synthesis of enynes from R1CH = CHI and R2CH≡CH [260].
In a catalytic system comprising 15 mol % FeCl3, 30 mol % Phen, and Cs2CO3 in toluene at 110°C, 11 enynes were synthesized with yields of 67–98% in 36–60 h. Diazotic ligands (dipyridyl (dipy), substituted phenanthrolines, and diquinoline) were also studied under typical conditions of the Sonogashira reaction with various bases and iron complexes (chloride, acac, acrylate) [260]. The best results were obtained with Fe(acac)3, dipy, and Cs2CO3 for 42 h at 135°C in toluene. The products of 22 syntheses were obtained with yields of isolated products of 80–96%.
Cross-coupling of ArI with RC6H4C≡CC(Me)2OH with simultaneous elimination of acetone and formation of RC6H4C≡CAr readily occurs in an aqueous solution of FeCl3∙6H2O, cationic 2,2-dipyridyl (Br–Me3N+-dipy-N+Me3Br–), and KOH in the presence of O2 and zinc powder [261]. The product yield at 140°C using FeCl3 containing 99.99% iron is 99% in 48 h. The cationic ligand allows dipyridyl Fe(III) complexes to be dissolved in water, which makes it possible to use a very convenient protocol (H2O, air). Without such a ligand, cross-coupling is not observed. The protocol also ensures that there is no homocoupling of alkynes. Zn(0) powder increases the probability of formation of iron complexes with oxidation states 0, I, and II. Cross-coupling of PhC≡CH with o‑iodophenol easily occurs in solutions of FeCl3 with Phen in xylene (N2, 120–145°C, 36 h) [262]. The cross-coupling product under these conditions is almost quantitatively converted into phenylbenzofuran XVI 16 with a yield of 82%. Seven different arylbenzofurans were synthesized with yields of 50–82%. Note that arylbenzopyrrole was obtained on a Pd/C palladium catalyst with o-iodoaniline [263].

It was shown [264] that 4-iodotoluene reacts with phenylacetylene in the Fe(acac)3–DMF–Cs2CO3 system at 140°C with a cross-coupling product yield of 93% in 36 h. In the NMP solvent, the yield was 95% in 20 h. The use of a microwave reactor significantly increases the reaction rate: a yield of 94% is achieved in 2.5 h. Various alkynes with aryl and alkyl substituents in reactions with various ArX are converted into products with yields of 79–96%. For the same ligand-free protocol of the Sonogashira reaction, a system containing FeX3 and CuI was proposed [265]. For example, 4-iodoanisole does not react with phenylacetylene in solutions of FeCl3 and Fe(acac)3 in DMF in the presence of K2CO3. The addition of CuI (10%) to a solution of Fe(acac)3 in DMSO at 140°C in 24 h leads to almost a quantitative ArX yield. The same results were obtained with FeCl3. However, the use of only CuI leads to a yield of 42%. The presence of FeCl3 in the system suppresses the homocoupling of both ArX and PhC≡CH. Quantitative yields of products in a series of solvents are achieved only in DMSO. Twenty syntheses were carried out with various ArI and PhC≡CH in the Fe(acac)3 (20 mol %)–CuI (20 mol %) system containing K3PO4 in DMSO at 140°C in argon, mainly with very high yields. The synthesis of arylbenzofuran also occurs well [262].
Interestingly, in the case of Fe(III) complexes, just as in the case of catalysis by palladium complexes [219], the question arose about the role of impurities in the manifestation of catalytic activity [266–269]. Buchwald and Bolm [266] drew attention to the fact that the FeCl3 manufacturer and impurities in iron play an important role in the catalytic cross-coupling of ArI with various aromatic nucleophiles. For example, it was assumed that the admixture of CuX in FeCl3 in the reaction of pyrazole arylation (toluene, DMDA, K3PO4, 135°C, 24 h) increases the activity of the iron catalyst: the product yields when using FeCl3 with a content of iron atoms >99.99% are lower than those at iron content >98%. The product yield without FeCl3, but in the presence of Cu2O (3 ppm) is 77%, and in the presence of FeCl3 with a purity of 99.99% and Cu2O (5 ppm), it is 78%. Similar results were obtained in the arylation of amides, phenol, and thiophenol.
Bolm and colleagues [267] also used copper compounds with concentrations at the ppm level to catalyze the reaction of cross-coupling of PhI with nucleophiles. It can be said that they used a “homeopathic catalyst” (or homeopathic amounts of catalyst). This definition for such catalysts was first introduced in Beletskaya and Cheprakov’s review [189]. In the CuO–DMDA system in toluene at 135°C and a reaction time of 24 h, sufficient concentrations of CuO turned out to be 0.001–0.1 mol % (0.001 mol % corresponds to 3 ppm Cu in the system with ArI). A similar situation with impurities was also observed in the case of arylation of phenylboronic acid with aryl bromide (Suzuki–Miyaura reaction) [268]. The synthesized Fe(Py)4Cl2, Fe(Py)3Cl2, and FeCl3(Py)3 complexes turned out to be inactive in this synthesis. It was assumed that, in cases where FeCl3 catalyzes this reaction, the effect of palladium impurity in iron is exhibited. Palladium compounds are known to catalyze the Suzuki–Miyaura reaction at palladium concentrations at the ppb level, i.e., 10–3 ppm.
The opposite conclusion about the role of impurities in the catalysis of the Sonogashira reaction was made by Savant et al. [269]. They studied the reaction of PhI with PhC≡CH in solutions of complexes of FeCl3 (>95% pure) with PPh3 and diphosphines, which was quite successful: the yield in 48 h was 98% at 135°C and a FeCl3 content of 0.15 mol % and a content of ligands of 0.3 mol %, i.e., at very low catalyst loading. Studies of the effect of the purity of commercial FeCl3 showed that the transition from FeCl3 with Fe > 95% (S.D. Fine Chem.) to Fe > 99.99% (Aldrich) leads to an increase in the DPA yield from 96 to 99%. Eighteen syntheses were performed for various reagents with high to very high yields. These results, in Savant et al.’s opinion, indicated that the actual catalyst of the reaction is FeCl3, rather than CuI impurities, as was assumed by Bolm and colleagues [266, 267].
The mechanism of catalysis of the Sonogashira reaction by Fe(III) complexes has been virtually unstudied, but a lot of useful results have been obtained by studying the reactions of cross-coupling of ArX with RMgX compounds, in particular, with RC≡CMgBr [270–273]. The results obtained made it possible to put forward hypotheses about the mechanisms of the Sonogashira reaction catalyzed by Fe(III) compounds. Nakamura and colleagues studied the synthesis of enyne compounds by the reaction R1C≡CMgBr with XCH = CHR (X = Br) in FeCl3–LiBr solutions in THF in the range 0–50°C [268]. The product yields reached 99% in 24 h. Ethynyl Mg compounds are easily obtained by the reaction of MeMgBr with alkynes. Interestingly, the Bolm catalytic system [255] does not catalyze this reaction. The addition of LiBr increases the yield of enynes from 12 to 85%. In the reaction of C6H13C≡CMgBr with BrCH = CHPh with the addition of tetramethylethylenediamine (TMEDA) to FeCl3, the yield of the product is 60%. Different 14 products were synthesized. Berben and Long [271] detected the strong stabilization of the Fe–C bond in the presence of ethynyl groups and assumed that this stabilizes iron ferrate complexes and stops (slows down) the homocoupling reaction of alkynyl substrates. Berben and Long [271] described the formation of stable ferrate complexes Li4[Fe(C≡CSiMe3)6]. Nakamura and colleagues [270] based on Berben and Long’s data [271] and their own experimental results proposed the first mechanism of the C–C coupling reaction involving iron complexes:
Thus, the catalytic cycle in this scheme is determined by the Fe0 complex (A). A very informative effect of accelerating the reaction by adding LiBr was found in that work. It was also noted that a partial loss of selectivity with respect to the geometry of the initial E- and Z-alkenyl bromides indicates the involvement of the electron transfer process at the step of reductive elimination. A convenient and simple protocol for the synthesis of enynes was also proposed.
Quite significant data on the mechanism of cross-coupling of R1X with R2MgX were obtained by Fürstner et al. [272], who described the synthesis of organoiron complexes not stabilized by ligands and their reactivity in the cross-coupling reaction. They informed that a partial consensus has been reached on the formal OS of iron. Transitions of complexes to Fe(II)/Fe(0), Fe(0)/Fe(II), or Fe(I)/Fe(III) were reported. The differences in the points of view are due to the lack of serious studies of the structure and reactivity of organoiron derivatives devoid of stabilizing ligands. Interestingly, under the same conditions in the presence of FeCl3–LiBr, MeMgBr does not react with 4-ClC6H4COOMe, but EtMgBr reacts with this halide to form cross-coupling products with yields of >95%. This difference is explained by the fact that EtMgBr is capable of β-Н-elimination with the release of ethylene from the [FeС2Н5(Br)] fragment. As a result, the reaction gives ethylene, ethane, and the Fe(–II)–Fe(MgBr)2 complex. In the reactions of alkyl halides, [Fe(C2H4)4][Li(TMEDA)]2 and [Fe(cod)2][Li(DME)]2 complexes are also formed. Moreover, it was shown that the reactions of MeMgBr and PhMgBr with FeCl2 give Fe(II) ferrates [Me4Fe2–Li(OEt2)22+(MeLi)] and Fe(0) ferrates [Ph4Fe4–Li(OEt2)44+]. The Fe(II) ferrate complex catalyzes the cross-coupling of MeMgBr with various aryl bromides and aryl triflates. Fürstner et al. [272] presented many structures of iron complexes with the OS –II, 0, +I, and +II and proposed mechanisms of the cross-coupling reaction. As a result, Fürstner et al. formulated three variants of routes of Ar–R formation from ArX and RMgX (Scheme 13):
– a route in which reductive elimination from L2Fe(Ar)(R) occurs in one step;
– a route in which RE occurs with electron transfer and the formation of Ar2FeIII(R)L and ArR + ArFeIL2;
– a route that involves Fe0 complexes and Fe–II(MgBr)2 complexes, and also the ArFe0L2(MgX) and ArFe0(R)(MgX)2 complexes.

Scheme 13 . Probable routes of Ar–R synthesis [272].
Interesting products and intermediates were obtained with CpFeX and Cp*FeX complexes, namely, CpFe(CH2Ph)L2 and Cp*Fe(C3H5)(C2H4), and the mechanisms of the corresponding steps of the cross-coupling reaction were also discussed.
An experimental and theoretical study of the effect of N-heterocyclic saturated and unsaturated carbene ligands (NHC) and fluoride ions on the cross-coupling reaction using Fe, Co, and Ni fluoride salts was published in a long Nakamura and colleagues’ article [273]. These systems proved to be excellent catalysts for the cross-coupling of Ar1MgBr with Ar2Cl to produce unsymmetrical bis-aryls (without palladium complexes and phosphine ligands!). The role of fluoride ions is in the formation of stable [Ar1FeIIF2]MgBr metallates, which are key intermediates in putative catalytic cycles. In the steps of the mechanism, an important role is played by the strong coordination of F– with Fe(II), which inhibits the reduction of iron and promotes the formation of ferrates in high OSs. A mechanism for the studied reaction involving M0Ln/MII and Ar1FeII/Ar1Ar2FeIV was proposed. Calculated (DFT) mechanisms involving bimetallic intermediates 17 with fluoride bridges were also considered:

Complex 17 with the saturated NHC ligand dimethylimidazoline reacts with PhCl. In general, three products are obtained: Ar1–Ar2, Ar1–Ar1, and Ar2–Ar2. By choosing the conditions and combinations of ligands, it is possible to reduce the homocoupling to 5%. The NHC ligands were mesitylene and isopropyl derivatives of imidazole and imidazoline. Experimental studies and theoretical (DFT) calculations confirmed that the formation of fluoride ferrates with Fe(II) determines the high selectivity with respect to the cross-coupling reaction. Numerous cross-coupling products were synthesized, and the conditions for five syntheses were optimized.
In the case of Fe(II) complexes, high catalyst activity and process selectivity were achieved with aryl chlorides; in the case of Co(II), with heteroaromatic aryls; and in the case of Ni(II), in reactions of aryl bromides with sterically hindered substrates. The 2009 Nakamura and colleagues’ article [273] discussed above is an excellent review of the literature (137 citations).
If one returns to the mechanisms of the Sonogashira reaction catalyzed by FeCl3 complexes, it becomes obvious that the results of the above studies of cross-coupling reactions involving RMgX make it possible to propose its main steps involving Fe(II), Fe(I), and Fe(0) complexes after replacing of RC≡CMgX by RC≡CH in the presence of bases. Scheme 14 presents a variant of a three-route mechanism of the Sonogashira reaction (the number of routes is determined by the difference between the number of steps and the number of linearly independent intermediates).

Scheme 14 . Proposed three-route mechanism of the Sonogashira reaction in solutions of Fe(II), Fe(I), and Fe(0) complexes.
In conclusion of the section describing iron compounds, let us note a study of a heterogeneous “iron” catalyst, paramagnetic magnetite Fe3O4 [274]. The magnetic properties of Fe3O4 NPs have long attracted researchers by the possibility of easy isolation of the catalyst, including a supported catalyst, from the reaction medium (see review in [274]). The reaction was carried out with NPs (<30 nm) obtained from iron samples with a Fe content of 18 to 99.99% (different sources) in ethylene glycol with K2CO3 at 125°C.
The presence of Pd, Ni, Cu, and Cr impurities was determined by ICP emission spectrometry. For example, in NP samples obtained in reactions with FeCl2 and FeCl3 with an Fe content of 99% in both NP samples (Merck), the contents of impurities were the following (ppb): Рd, 850; Cu, 3; Ni, 65; and Co, 563. The presence of impurities in ethylene glycol and K2CO3 was also studied. The product yield in the reaction of iodotoluene with phenylacetylene was 75–88%. Interestingly, the addition of Pd and Cu salts in amounts of 200–1000 ppb in the absence of Fe3O4 lead to product yields of about 40 and 10%, respectively, and the presence of 5 mol % Fe3O4 NP increases the yield up to 88%.
Numerous experiments determined that the best of the solvents (DMF, toluene, ethylene glycol, NMP) is ethylene glycol (EG). The protocol used allowed the synthesis of 19 products of cross-coupling of various ArI, ArBr, and heteroaromatic ArX with various aryl- and alkynylalkynes. The convenience of complete catalyst extraction using external magnets was demonstrated, and fivefold reproducibility of the results with yields of 89–92% was shown.
Heterogeneous catalysts for the cross-coupling reaction from groups 8, 9, and 10 of the periodic table (Co, Fe, Pd, Rh) have already been discussed in this review. Note also Ru/Al2O3 [275]. Dodecylamine-stabilized Ru NPs on Al2O3 catalyze the Heck and Suzuki reactions [276]. It was noted that colloidal NPs 2–3 nm in size satisfactorily similar in structure and properties to NPs extracted from catalytic systems for the Heck synthesis under conditions of homogeneous catalysis by [RuCl2(p-cumene)2] complexes in the presence of NaOAc NPs. Park et al.’s study of Ru NPs on various supports [275] showed that, at a ruthenium content on the support of 2.2–8.5 wt %, NPs are uniformly distributed on the surface of the support with a support particle diameter of 2.3–10 nm. The most active supports in the reaction of 4-AcC6H4I with PhC2H (5 mol % Ru/support and 10 mol % CuCl2 in a solution of Et3N and CH3CN) at 90°C for 12 h were activated carbons and Al2O3. The yield of the cross-coupling reaction product was 90%. In the absence of CuCl2, this yield was 70%. In Park et al.’s opinion, the above catalytic system is the first example of the use of a heterogeneous ruthenium catalyst in the Sonogashira reaction. The optimal catalyst without CuCl2 was tested in the synthesis of 14 products with various aryl iodides and alkynes and showed good results: the product yield was 66–98%. In two cycles of use, its catalytic activity remained constant.
Concluding section 3.3, we note the surprising fact that the Sonogashira cross-coupling reaction was catalyzed by chloride of the non-transition metal In (InCl3) [277], which turned out to be a very efficient catalyst. For example, dry InCl3 in dry benzene at 80°C (reflux) catalyzes the synthesis of the product from RC6H4I and PhC2H with a yield of 80% in 3.5 h, rather than in 20 and 48 h! Nine more ArX (Cl, Br, I) are converted in 3.5–4 h with yields of 75–83%. The properties of InCl3 as a Lewis acid are known; therefore, the formation of the In(C≡CR)nCl3–n intermediate is quite probable, just as is the synthesis of organoindium compounds in general in the absence of bases. The known indium compounds in the OSs II and I are very unstable, and the OSs of the intermediates in this synthesis are not yet clear. The further fate of the InCl3 complexes is not clear, especially in connection with the weak oxidizing ability of In(III), and then only in an alkaline medium. The question of the presence of transition metal impurities in In(III) was not discussed in that work.
CHAPTER 4. MAIN INTERMEDIATES IN THE REACTIONS OF OXIDATIVE DIMERIZATION OF ALKYNES AND THE CADIOT–CHODKIEWICZ REACTION, THE SONOGASHIRA REACTION, AND RELATED CATALYTIC PROCESSES
In conclusion, let us consider, with minor comments, intermediates with М–С bonds and other intermediate complexes occurring in the reactions considered in the review, as well as in reactions of alkynes with similar mechanisms (oxidation, oxidative halogenation and carbonylation reactions, dimerization and oligomerization of alkynes, and addition reactions of HX molecules to alkynes). This summarization was made using articles cited in the review and monographs [3, 7, 8, 70, 167, 278–295].
4.1. Cu(I, II, III) Compounds
In the introduction to this section, we note the fundamental studies of Meyerstein and colleagues, which were performed over the last 40 years [278–289] and focused on unstable and highly reactive compounds RCuI, RCuII, and RCuIII formed from various free radicals R• and copper salts. The kinetics of radical formation under conditions of radiolysis of various media in situ and the kinetics and mechanism of radical transformations were investigated. The formation and transformation reactions of RCuII and RCuIII compounds, including radicals derived from •R1R2(OH) alcohols, with the formation of CuIICH3+, CuIIICH2Ph2+, and LCuIIICR1R2(OH)2+ were studied [278–281, 283]. The β-elimination of CuIII–Н from the last intermediate leads to R1R2С=O, whereas the β-elimination of CuIII–OH leads to olefins [282]. It was shown that the compound CuIIIC6H10(OH)2+ (from the 2-hydroxycyclohexyl radical) isomerizes to cyclopentylaldehyde C5H9CHO [286]. Thus, in the presence of a strong oxidizing agent, the following catalytic reaction can occur:
From the radical •CH2CH(NH2)COOH and Cu(I). one can obtain CH2=CHCOOH by the β-elimination of CuII–NH2 or CH3CHO by the β-elimination of CuII–COOH. The effect of various ligands on the mechanism and kinetics of the transformation of RCuII and RCuIII compounds was studied [279, 280, 286, 288, 289]. A detailed study of the kinetics of transformation reactions of σ-organometallic compounds of copper(II) showed [285, 288] that RCu(II) compounds participate in the reactions of homolysis, heterolysis (H2O), β -elimination of CuII–X (X = H, OH, OR, NH2), insertion into the R–CuII bond,
and isomerization. Meyerstein and colleagues [289] also studied in detail the mechanism of the formation of C2H6 from MIIC\({\text{H}}_{3}^{ + }\), where M = Cu(II), Ni(II), Fe(II), and Mn(II). It was shown that the fate of RMn+1 depends on the nature of L and the composition of the medium. A systematic study of the interaction of Cu(II) complexes with alkyl radicals by stationary and pulsed femtosecond photolysis, EPR spectroscopy, and quantum chemical methods was undertaken by Zubanova [296].
In the case of alkynes, in view of the special stability of the Cu–C≡ bond, a variety of σ-organometallic copper intermediates are formed in various reactions (Schemes 15–17). The intermediates CuCH=CHX (Scheme 15) participate in the reactions of oxidative halogenation of alkynes (steps 4–7, 11, 12) and addition (steps 8–10). The intermediate CuC≡CH (CuC≡CR) is also involved in the reactions of oxidative halogenation (steps 17–20, 22), OD (21), dimerization (20), and oligomerization (22). The intermediates R1CuX2 and R1Cu(C≡CH)X are participants in the Sonogashira process.
Scheme 16 presents the intermediates formed with the participation of Cu(II) and possibly also from Cu(II) π-complexes. The intermediate R1CuX is converted into R1X (5) and the product of Sonogashira reaction (6, 7), whereas the compound RC≡CCuIIX (2) is converted into the products of oxidative halogenation (8) and oxidative dehydrocondensation (9). The vinyl intermediate XCuCH=CHX (3) is involved in the oxidative halogenation reaction (10). The intermediate CuCH = CHC≡CH (4) is oxidatively halogenated (11–13) to form 2-chlorovinylacetylene.

Scheme 15 . Intermediates formed with the participation of CuCl.

Scheme 16 . Intermediates formed with the participation of Cu(II).
Copper compounds RCuIII are also involved in the mechanisms of catalytic reactions (Scheme 17). Using CuX2, through steps (4, 5), bicyclic intermediate (16) is obtained, which is most likely to participate in OD reaction (16). Compounds RCuIIIX(I) (1) are intermediates in Sonogashira reactions (2, 3), and R=C≡CR1 also participate in oxidative cross-coupling of alkynes to form R1C≡C–C≡CR2 (7–9). Stable Cu(III) complexes with macrocyclic ligands were described by Fedenok [62] and Temkin et al. [7].

Scheme 17 . Formation of intermediates RCu(III) and R2Cu(III) with the participation of CuX and CuX2 and CuIII(X) with the participation of CuX.
In O2-involving oxidative processes catalyzed by Cu(I) complexes, an important role is played by oxygen complexes formed by reactions (10–12). It is important to note the role of polynuclear Cu(I) complexes in the catalytic transformations of alkynes, including oxidative processes. These complexes were described in our works [7, 70, 71].
Ethynyl polynuclear complexes have a very diverse structure (Fig. 2) [1, 71]. The structure of acetylide complexes obtained by dissolving Cu2C2 in concentrated CuCl–MCl solutions is also very interesting [290].

Structures of ethynyl complexes occurring in polynuclear complexes of Cu(I) and other metals.
4.2. Au(I, III) Compounds
Gold complexes in catalysis were considered in numerous works, e.g., [291–295]. A high electrophilicity (increased Lewis acidity) and an increase in oxidizing properties in the order Cu < Ag <Au were noted [292, 295]. The backbonding properties of LAu+ are less pronounced than those of Cu(I), but are quite noticeable in the carbene and carbine complexes Au=CHR+ and Au≡CR+. This gives rise to stable cationic intermediates [295–297]. In Zang’s study [298] of the catalysis of alkyne oxidation reactions by Au(I) complexes, the formation of an α-oxocarbene complex of the [LAu=CR–C(O)–R1]+ type was detected.
The action of the strong oxidizing agent OZ+ on Au(I) π-complexes leads to the [Au]–CR1=CR2OZ+ intermediate, which detaches Z (Z are various organic groups) to form a carbene intermediate (Scheme 18, steps 6, 14, 15). Interestingly, Au(I) carbene complexes are more stable than the corresponding gold carbenium ions. Another version of the synthesis of the gold carbene complex is shown in Scheme 18. The Au(I) complexes, just as the Cu(I) complexes, play an important role in oxidative reactions. The LAuX complexes react with O2 to form L(X)OOAu(X)L [299]. The hydride complexes LAuH with the ligand L = Ar2NHC also easily interact with O2 (at a pressure of 4–9 bar and temperatures of 40–60°C) and are converted into LAuOOH. The reaction is described by a second-order kinetic equation, and the kinetic isotope effect is kH/kD = 2.4. Note that entire volume 47(3) of Account of Chemical Research in 2014 was devoted to reactions involving gold.
Scheme 18 shows the intermediates formed from LAuX3 and the products of their transformations in the processes of С–С coupling (steps 1–5), as well as the intermediates formed from LAuX (6, 5) and the products of their transformations in the Sonogashira reactions (9–11), cross-coupling reactions of R1X and RZH to form a С–Z bond (9, 12, 1 3), OD reactions (1, 3, 5) and oxidative cross-coupling of alkynes with vinyl compounds (1, 2, 9). Gold complexes are used in many coupling reactions.
4.3. Pd(0, I, II) Compounds
Palladium complexes in various oxidation states are quite widely represented in homogeneous metal complex catalysis and were studied by Moiseev, Henry, Maitlis, Heck, Tsuji, Negishi, and many other researchers (see [58]). The same applies to the reactions of OD, Sonogashira reaction, and other reactions of formation of C–C and C–heteroatom bonds. Schemes 19–23 show the intermediates formed from complexes formed from Pd(0), Pd(I), and Pd(II) and their transformations in the processes of oxidation, C–C coupling, oxidative halogenation and carbonylation, and addition of HX molecules. Scheme 19 presents the reactions of ethynyl complexes obtained from C2H2 and terminal alkynes, RC≡CX, and R1PdX. These are oxidative carbonylation of alkynes (1, 6–9), dimerization of alkynes (1, 10–13), reactions involving vinylidene carbene intermediates (1–5), and nonoxidative carbonylation RC≡CX (14–16).
Pd(0) complexes efficiently interact with various R1X (14, 15), in particular, with alkyl and aryl halides, in catalytic reactions with H2O, C2H2 to give R1COOH (17–19), and vinyl ketones (17, 18, 20, 21), as well as in the synthesis of unsaturated keto acids (17, 18, 20, 22). The mechanism of the Sonogashira synthesis in Pd(0) solutions is represented by steps (17, 26, 27).

Scheme 18 . Participation of LAuX and LAuIIIX3 complexes in the synthesis of gold-containing intermediates.
Scheme 20 shows the intermediates formed from Pd(I) complexes and participating mainly in the reactions of oxidative and nonoxidative carbonylation to α-, β-unsaturated and saturated acids and esters and the corresponding anhydrides. The catalysis of the Sonogashira reactions and other C–C coupling processes by Pd(I) complexes, to the best of my knowledge, has not been studied. On σ-organopalladic compounds Pd(II), there is a huge amount of information that cannot be fully reflected in this review. Scheme 21 shows the intermediates and steps of the formation and transformations of ethynyl Pd(II) complexes obtained from Pd(I) complexes: these are carbonylation reactions (4–7), OD (1, 8, 9), oxidative amination of alkynes (1, 3), and syntheses involving Pd(IV) complexes (10–12).
Scheme 22 characterizes the vinyl pathway of alkyne transformations. These are oxidative reactions of halogencarbonylation (1, 4–6), halogenation (1, 12–14), hydration (1, 7), and synthesis of unsaturated aldehydes (1, 8, 9) and η3-allyl complexes (1, 8, 10). Heating of η3-allyl complexes gives rise to acrolein and formaldehyde [7, 8]. Scheme 23 shows intermediates (1–3) formed from CO and H2O (1), CO and ROH (2), and CO and R2NH (3). These intermediates participate in the reactions of oxidative and additive carbonylation of alkynes (1, 5, 7, 8), (1, 5, 6), and (2, 13, 15, 16) and also in catalytic reactions with olefins, dienes, and cycloolefins [58]. The hydride complex of palladium (17) is an active site in the carbonylation reactions of alkynes (18, 19) and olefins (20, 21). Scheme 23 shows two routes for the synthesis of acrylates: (2, 13, 15) and (1, 4, 18, 19), as well as two routes for the oxidative synthesis of α-, β-unsaturated acids and anhydrides (1, 5, 7, 8) and esters (2, 13, 16, 17). The diversity of palladium intermediates makes it possible to generate hypotheses of mechanisms that can be discriminated experimentally and theoretically [58].

Scheme 19 . Mechanisms and intermediates of reactions involving ethynyl complexes formed from Pd(0) complexes.

Scheme 20 . Intermediates and steps of the participation of ethynyl Pd(II) complexes obtained from the initial Pd(I) complexes.

Scheme 21 . Ethynyl intermediates formed directly from alkynes and Pd(II).

Scheme 22 . Vinyl, alkenyl, and other organopalladium intermediates obtained with the participation of Pd(II) complexes.

Scheme 23 . Intermediates and steps involving CO and Pd(II) complexes.
4.4. Fe(0, I, II, III) Compounds
Section 1.3 presents a lot of mechanisms and intermediates containing iron atoms; therefore, Scheme 24 shows only one mechanism for the Sonogashira reaction with the Fe(I) complex as a catalyst (1–3). A very large amount of material on catalysis by iron compounds was collected in Bauer and Knölker’s review [254]. Scheme 24 shows organometallic compounds of iron in various OSs. As can be seen, various ethynyl complexes occur in the case of Fe(0), Fe(II), and Fe(III). Therefore, there are no restrictions on the choice of routes of mechanisms in various catalytic systems containing iron.

Scheme 24 . Intermediates containing Fe in the oxidation states 0, I, II, and III.
CONCLUSIONS
The review considered three reactions of alkynes: oxidative dehydrocondensation (OD) and Cadiot–Chodkiewicz and Sonogashira syntheses. In all cases, the kinetic laws and the nature of the intermediates were critically analyzed in detail. In view of the theoretical and practical importance of the considered processes, numerous experimental protocols of syntheses were briefly presented and discussed in the review. Examples of use in the chemical and pharmacological industries were given in monographs and in a number of reviews mentioned in the text [7, 15, 70, 168, 169, 254, 291, 293, 294, 297].
Notes
Merkushev, E.B. and Shvartsberg, M.S., Iodine Organic Compounds and Syntheses Based on Them: A Tutorial, Tomsk: Tomsk. Gos. Pedagog. Inst. im. Leninskogo Komsomola, 1978.
Stang, P.J., Surber, B.W., Chen, Z.-C., Roberts, K.A., and Anderson A.G., Preparation and mechanism of formation of alkynyl tosylates and mesylates via tricoordinate iodonium species, J. Am. Chem. Soc., 1987, vol. 109, p. 228.
Archer, E.M. and Van Schalkwyk, T.G.D., The crystal structure of benzene iododichloride, Acta Cryst., 1953, vol. 6, p. 88.
Alcock, N.W., Countryman, R.M., Esperas, S., and Sawyer, J.F., Secondary bonding. Part 5.l. The crystal and molecular structures of phenyliodine(III) diacetate and bis(dichloroacetate), JCS Dalton Trans., 1979, p. 854.
REFERENCES
Temkin, O.N., “Golden Age” of homogeneous catalysis chemistry of alkynes: Dimerization and oligomerization of alkynes, Kinet. Catal., 2019, vol. 60, p. 689.
Temkin, O.N. and Bruk, L.G., The use of the concept of “oxidation state of an atom” and electronic balances in redox processes in organic and organoelement chemistry, Ross. Khim. Zh. (Zh. Ros. Khim. o-va im. D.I. Mendeleeva), 2014, vol. 58, nos. 5–6, p. 90.
Modern Alkyne Chemistry. Catalytic and Atom-Economic Transformations, Trost, B.M. and Li, Ch.-J., Eds., Weinheim: Wiley-VCH, 2015.
Stefanj, H.A., Guarezemini, A.S., and Cella, R., Tetrahedron, 2010, p. 7871.
Kotlyarovskii, I.L., Shvartsberg, M.S., and Fisher, L.B., in Reaktsii atsetilenovykh soedinenii (Reactions of acetylenic compounds), Novosibirsk: Nauka, 1967, p. 173.
Temkin, O.N. and Flid, R.M., Kataliticheskie prevrashcheniya atsetilenovykh soedinenii v rastvorakh kompleksov metallov (Catalytic Transformations of Acetylenic Compounds in Solutions of Metal Complexes), Moscow: Nauka, 1968.
Temkin, O.N., Shestakov, G.K., and Treger, Yu.A., Atsetilen. Khimiya. Mekhanizmy reaktsii. Tekhnologiya (Acetylene. Chemistry. Reaction Mechanisms. Technology), Moscow: Khimiya, 1991.
Lu, W. and Zhou, L., Oxidation of C–H Bonds, Wiley and Sons, 2017, ch. 8, p. 209.
Friis, S.D., Pirnot, M.T., Dupuis, L.N., and Buchwald, S.L., Angew. Chem., Int. Ed., 2017, vol. 56, p. 7242.
Kudryavtsev, Yu.P., Sladkov, A.M., Aseev, Yu.G., Nedoshivin, Yu.N., Kasatochkin, V.I., and Korshak, V.V., Dokl. Akad. Nauk SSSR, 1964, vol. 158, p. 389.
Eglinton, G. and Mc Grae, W., Advances in Organic Chemistry. Methods and Results, 1963, vol. 4, p. 225.
Sondheimer, F., Pure Appl. Chem., 1963, vol. 7, p. 363.
Bohlmann, F., in Chemistry of Acetylenes, Viehe, H.G., Ed., New York: M. Dekker, 1969, ch. 14, p. 977.
Siemsen, P., Livingston, R.C., and Diederich, F., Angew. Chem., Int. Ed., 2000, vol. 19, p. 2632.
Acetylene Chemistry: Chemistry, Biology, and Material Science, Diederich, F., Stang, P.J., and Tykwinski, R.R., Eds., Weinheim: Wiley-VCH, 2005.
Allen, S.E., Walvood, R.R., Padilla-Salinas, R., and Kozlowcki, M., Chem. Rev., 2013, vol. 113, p. 6234.
Zal’kind, Yu.S. and Fundyler, B.M., Zh. Org. Khim., 1936, vol. 6, p. 530.
Zal’kind, Yu.S. and Aizikovich, M.A., Zh. Org. Khim., 1937, vol. 7, p. 227.
Zal’kind, Yu.S. and Fundyler, B.M., Zh. Org. Khim., 1939, vol. 9, p. 1725.
Straus, F., Justus Liebigs Annalen der Chemie, 1905, vol. 342, no. 5, p. 190.
Akhtar, F., Richards, T.A., and Weedon, B.C.L., J. Chem. Soc., 1959, p. 933.
Balcioglu, N., Uraz, I., Bozkunt, C., and Sevin, F., Polyhedron, 1997, vol. 16, no. 2, p. 327.
Salkind, J.S. and Fundyler, B.M., Chem. Berichte, 1936, vol. 69, p. 128.
Zal’kind, Yu.S. and Gverdtsiteli, I.M., Zh. Org. Khim., 1939, vol. 9, p. 971.
Bowden, K., Heilbron, I., Jones, E.R.H., and Sargent, K.H., J. Chem. Soc., 1947, p. 1579.
Heilbron, I., Jones, E.R.H., and Sondheimer, F., J. Chem. Soc., 1947, p. 1586.
Zal’kind, Yu.S. and Kolyaskina, Z.N., Zh. Org. Khim., 1952, vol. 22, p. 2148.
Mkryan, G.M. and Papazyan, M.A., Dokl. Akad. Nauk Arm. SSR, 1953, vol. 16, p. 17.
Bohlmann, F. and Viehe, H.G., Chem. Berichte, 1954, vol. 87, p. 712.
Klebanskii, A.L., Grachev, I.V., and Kuznetsova, O.M., Zh. Org. Khim., 1957, vol. 27, p. 2977.
Bennett, E.G., Org. Chem., 1957, vol. 22, p. 557.
Copenhaver, J.W. and Bigelow, M.H., Acetylene and Carbon Monoxide Chemistry, New York: Reinhold, 1949, p. 121.
Reppe, W., Justus Liebigs Annalen der Chemie, 1955, vol. 596, p. 51.
Franke, W. and Meister, H., US Patent 2796442, 1957.
Hay, A.S., Org. Chem., 1960, vol. 25, p. 1275.
Hay, A.S., Org. Chem., 1962, vol. 27, p. 3320.
Eglinton, G. and Galbraith, A.R., Chem. Ind., 1956, p. 737.
Eglinton, G. and Galbraith, A.R., J. Chem. Soc., 1959, p. 889.
Sondheimer, F. and Amiel, Y.J., J. Am. Chem. Soc., 1956, vol. 78, p. 4178.
Sondheimer, F. and Amiel, Y., J. Am. Chem. Soc., 1957, vol. 79, p. 5817.
Sondheimer, F., Amiel, Y.J., and Wolovsky, R.J., J. Am. Chem. Soc., 1957, vol. 79, p. 6263.
Sondheimer, F., Wolovsky, R., and Ben-Efraim, D.A., J. Am. Chem. Soc., 1961, vol. 83, p. 1686.
Sondheimer, F. and Wolovsky, R.J., J. Am. Chem. Soc., 1962, vol. 84, p. 260.
Yadav, J.S., Reddy, B.V.S., Reddy, K.B., Uma, K., and Prasad, A.R., Tetrahedron Lett., 2003, vol. 44, p. 6493.
Li, Y.-N., Wang, J.-L., and He, L.-N., Tetrahedron Lett., 2011, vol. 52, p. 3485.
Wang, D., Li, J., Gao, T., Hou, S., and Chen, B., Green Chem., 2010, vol. 12, p. 45.
Adimurthy, S., Chandi, C., Malakar, C.C., and Beifuss, U., Org. Chem., 2009, vol. 74, p. 5648.
Zhu, M., Jin, J.C., and Tong, J.Y., J. Chem. Res., 2008, no. 4, p. 218.
Alcaide, B., Almendros, P., Carrascosa, R., and Rodriguez-Acehes, R., Eur. J. Org. Chem., 2008, p. 1375.
Balaraman, K. and Kesavan, V., Synthesis, 2010, no. 20, p. 3461.
Bedard, A.-C. and Collins, S.K., J. Am. Chem. Soc., 2011, vol. 133, p. 19976.
Bedard, A.-C. and Collins, S.K., Chem. Commun., 2012, vol. 48, p. 6420.
Godin, E., Bedard, A.-C., Raymond, M., and Collins, S.K., Org. Chem., 2017, vol. 82, p. 7576.
Clifford, O.A. and Waters, W., J. Chem. Soc., 1963, p. 3056.
Bohlmann, F., Schonowsky, H., Inhoffen, E., and Grau, G., Chem. Berichte, 1964, vol. 97, p. 94.
Fedenok, L.G., Berdnikov, V.M., and Shvartsberg, M.S., Zh. Org. Khim., 1973, vol. 9, p. 1781.
Fedenok, L.G., Berdnikov, V.M., and Shvartsberg, M.S., Zh. Org. Khim., 1975, vol. 11, p. 2492.
Fedenok, L.G., Berdnikov, V.M., and Shvartsberg, M.S., Zh. Org. Khim., 1974, vol. 10, p. 922.
Fedenok, L.G., Berdnikov, V.M., and Shvartsberg, M.S., Zh. Org. Khim., 1976, vol. 12, p. 1395.
Fedenok, L.G., Berdnikov, V.M., and Shvartsberg, M.S., Zh. Org. Khim., 1978, vol. 14, p. 1425.
Fedenok, L.G., Berdnikov, V.M., and Shvartsberg, M.S., Zh. Org. Khim., 1978, vol. 14, p. 1429.
Fedenok, L.G., Mechanism and synthetic possibilities of some reactions of acetylenic compounds, Doctor Sci. (Chem.) Dissertation, Novosibirsk: Inst. Khim. Kinet. Goreniya SO RAN, 2008.
Fedenok, L.G. and Shvartsberg, M.S., Tetrahedron Lett., vol. 52, p. 3776.
Tolman, W.B., Acc. Chem. Res., 1997, vol. 30, p. 227.
Holland, P. and Tolman, W.B., Coord. Chem. Rev., 1999, vols. 190–192, p. 855.
Khoan, Kh.M., Physical and chemical foundations of the catalytic synthesis of dialkynes, Cand. Sci. (Chem.) Dissertation, Moscow: Mosk. Inst. Tonkoi Khim. Teknol., 1990.
Khoan, Kh.M., Brailovskii, S.M., and Temkin, O.N., Kinet. Katal., 1994, vol. 35, no. 2, p. 266.
Khoan, Kh.M., Brailovskii, S.M., and Temkin, O.N., Kinet. Katal., 1994, vol. 35, no. 3, p. 367.
Sladkov, A.M., Gol’ding, I.R., Usp. Khim., 1979, vol. 10, no. 9, p. 1625.
Temkin, O.N., Homogeneous Catalysis with Metal Complexes. Kinetic Aspects and Mechanisms, Wiley, 2012.
Mykhalichko, B.M., Temkin, O.N., and Mys’kiv, M.G., Usp. Khim., 2000, vol. 69, no. 11, p. 1042.
Mc Connell, H.M. and Davidson, H., J. Am. Chem. Soc., 1950, vol. 72, no. 7, p. 3168.
Mc Connell, H.M. and Weaver, H.E., J. Chem. Phys., 1956, vol. 25, no. 2, p. 304.
Wilson, E.E., Oliver, A.G., Hughes, R.P., and Ashfeld, B.L., Organometallics, 2011, vol. 30, p. 5214.
Secutowski, D.G. and Stucky, G.D., J. Am. Chem. Soc., 1976, vol. 98, p. 1376.
Erker, G., Angew. Chem., 1986, vol. 98, p. 456.
Rosenthal, U. and Görls, H., J. Organomet. Chem., 1992, vol. 439, p. C36.
Rosenthal, U., Ohff, A., Tillack, A., and Baumann, W., J. Organomet. Chem., 1994, vol. 468, p. C4.
Pellny, P.-M., Burlakov, V.V., Peulecke, N., Spannenberg, A., Kempe, R., and Rosenthal, U., J. Organomet. Chem., 1999, vol. 578, p. 135.
Burlakov, V.V., Acetylene complexes of metallocenes of group IV B (4), Doctor Sci. (Chem.) Dissertation, Moscow: INEOS im. A.N. Nesmeyanova, 2012.
Wendlandt, A.E., Suess, A.M., and Stahl, S.S., Angew. Chem., Int. Ed., 2011, vol. 50, p. 11062.
Rosenthal, U., Ohff, A., Baumann, W., Kempe, R., Tillack, A., and Burlakov, V.V., Organometallics, 1994, vol. 13, p. 2903.
Rosenthal, U., Pubst, S., Arndt, P., Ohff, A., Tillack, A., Baumann, W., Kempe, R., and Burlakov, V.V., Organometallics, 1995, vol. 14, p. 2961.
Pubst, S., Arndt, P., Heller, B., Baumann, W., Kempe, R., and Rosenthal, U., Angew. Chem., Int. Ed., 1996, vol. 35, no. 10, p. 2454.
Heeres, H.J., Nijhoff, J., and Teuben, J.H., Organometallics, 1993, vol. 12, p. 2609.
Choukroun, R. and Cassoux, P., Acc. Chem. Res., 1999, vol. 32, p. 494.
Zhao, J., Zhang, S., Zhang, W.-X., and Xi, Z., Coord. Chem. Rev., 2014, vols. 270–271, p. 2.
Lttenauer, M.S., Mobian, P., and Barloy, L., Coord. Chem. Rev., 2022, vol. 459, p. 214.
Evans, W.J., Keyer, P.A., and Ziller, J.W., Organometallics, 1993, vol. 12, no. 7, p. 2618.
Khoan, Kh.M., Brailovskii, S.M., and Temkin, O.N., Kinet. Katal., 1994, vol. 35, no. 6, p. 889.
Coates, G.E. and Parkin, C., J. Inorg. Nucl. Chem., 1961, vol. 22, p. 59.
Corfield, P.W.R. and Shearer, H.M.M., Abst. Am. Cryst. Assoc. Meeting. Bozeman. Mont., 1964, p. 96.
Blake, D., Calvin, G., and Coates, G.E., Proc. Chem. Soc., 1959, p. 396.
Organocopper Compounds, Gmelin Handbook of Inorganic Chemistry, 8th ed., Berlin: Springer, 1986, vol. 60, p. 3.
Bedford, R.R., Hill, A.F., Thompsett, A.R., White, A.J.P., and Williams, D.J., Chem. Commun., 1996, p. 1059.
Collman, J.P. and Kang, J.W., J. Am. Chem. Soc., 1967, vol. 89, p. 844.
Nast, R. and Pfab, W., Chem. Berichte, 1956, vol. 89, p. 415.
Negishi, E., J. Am. Chem. Soc., 1991, vol. 113, p. 1440.
Kamata, K., Yamaguchi, S., Kotani, H., Yamaguchi, K., and Mizuno, N., Angew. Chem., Int. Ed., 2008, vol. 47, p. 2407.
Yamaguchi, K., Kamata, K., Yamaguchi, S., Kotani, H., and Mizuno, N.J., J. Catal., 2008, vol. 258, p. 121.
Milane, P., Duboc, C., Marrot, J., Riviere, E., Dolberg, A., and Secheresse, E., Chem. Eur. J., 2006, vol. 12, p. 1950.
Chaudhuri, P. and Wieghardt, K., Prog. Inorg. Chem., 1987, vol. 35, p. 330.
Fomina, L., Vazquez, B., Tkatchouk, E., and Fomine, S., Tetrahedron, 2002, vol. 58, p. 6741.
Efremov, G.E., Bovyrina, E.A., Katsman, E.A., Shamsiev, R.S., and Temkin, O.N., Izv. Akad. Nauk, Ser. Khim., 2019, no. 7, p. 1366.
Vilhelmsen, M.H., Jensen, J., Tortzen, C.G., and Nilsen, M.B., Eur. J. Org. Chem., 2013, p. 701.
Heck, R.F., J. Am. Chem. Soc., 1968, vol. 90, p. 5518.
Heck, R.F., J. Am. Chem. Soc., 1968, vol. 90, p. 5526.
Heck, R.F., J. Am. Chem. Soc., 1968, vol. 90, p. 5531.
Heck, R.F., J. Am. Chem. Soc., 1968, vol. 90, p. 5535.
Heck, R.F., J. Am. Chem. Soc., 1968, vol. 90, p. 5538.
Heck, R.F., J. Am. Chem. Soc., 1968, vol. 90, p. 5542.
Heck, R.F., J. Am. Chem. Soc., 1968, vol. 90, p. 5546.
Heck, R.F., J. Am. Chem. Soc., 1969, vol. 91, p. 6707.
Temkin, O.N., Kaliya, O.L., Shestakov, G.K., and Flid, R.M., Dokl. Akad. Nauk SSSR, 1970, vol. 190, p. 398.
Heck, R.F., J. Am. Chem. Soc., 1972, vol. 94, p. 2712.
Heck, R.F. and Nolley, J.P., Org. Chem., 1972, vol. 37, p. 2320.
Dieck, H.A. and Heck, R.F., J. Am. Chem. Soc., 1974, vol. 96, p. 1133.
Mizoroki, T., Mori, K., and Ozaki, A., Bull. Chem. Soc. Jpn., 1971, vol. 44, p. 581.
Kaliya, O.L., Temkin, O.N., Kirchenkova, G.S., Smirnova, E.M., Kimel’fel’d, L.M., and Flid, R.M., Izv. Akad. Nauk SSSR, Ser. Khim., 1969, p. 2854.
Kaliya, O.L., Kirchenkova, G.S., and Temkin, O.N., Kinet. Katal., 1969, vol. 10, p. 1186.
Temkin, O.N., Kaliya, O.L., Shestakov, G.K., Brailovskii, S.M., Flid, R.M., and Aseeva, A.P., Kinet. Katal., 1970, vol. 11, p. 1592.
Cassar, L., J. Organomet. Chem., 1975, vol. 93, p. 253.
Diek, H.A. and Heck, R.F., J. Organomet. Chem., 1975, vol. 93, p. 259.
Sonogashira, K., Tohda, Y., and Hagihara, N., Tetrahedron Lett., 1975, no. 50, p. 4467.
Rossi, R., Carpita, A., Quirici, M.G., and Gandenzi, M.L., Tetrahedron, 1982, vol. 38, p. 631.
Liu, Q. and Burton, D.J., Tetrahedron Lett., 1997, vol. 38, no. 25, p. 4371.
Vlassa, M., Ciocan-Tarta, I., Margineanu, F., and Oprean, I., Tetrahedron, 1996, vol. 54, no. 4, p. 1337.
Cho, D.H., Lee, J.H., and Kim, B.H., Org. Chem., 1999, vol. 64, p. 8048.
Fairlamb, I.J.S., Bäuerlein, P.S., Marrison, L.R., and Dickinson, J.M., Chem. Commun., 2003, p. 632.
Batsanov, A.S., Collings, J.C., Fairlamb, I.J.S., Holland, J.P., Howard, A.K., Liu, Z., Mard, T.R., Parsons, A.C., Ward, R.M., and Zhu, J., Org. Chem., 2005, vol. 70, p. 703.
Mc Glacken, G.P. and Fairlamb, I.J.S., Eur. J. Org. Chem., 2009, p. 4011.
Li, J.-H., Liang, Y., and Zhang, X.-D., Tetrahedron, 2005, vol. 61, p. 1903.
Yin, W., He, C., Chen, M., Zhang, H., and Lei, A., Org. Lett., 2009, vol. 11, no. 3, p. 709.
Berry, D.H. and Eisenberg, R., Organometallics, 1987, vol. 6, p. 1796.
Afzal, D., Lenhert, P.G., and Lukenhart, C.M., J. Am. Chem. Soc., 1984, vol. 106, p. 3050.
Gonzalez-Arellano, C., Abad, A., Corma, A., Garsia, H., Iglesias, M., and Sanchez, F., Angew. Chem., Int. Ed., 2007, vol. 46, p. 1536.
Plenio, H., Angew. Chem., Int. Ed., 2008, vol. 47, p. 6954.
Gonzalez-Arellano, C., Corma, A., Iglesias, M., and Sanchez, F., Eur. J. Inorg. Chem., 2008, p. 1107.
Li, P., Wang, L., Wang, M., and You, F., Eur. J. Org. Chem., 2008, p. 5946.
Hopkinson, M.N., Ross, J.E., Gluffredi, G.T., Gee, A.D., and Gouverneur, V., Org. Lett., 2010, vol. 12, no. 21, p. 4904.
Wegner, H.A. and Auzias, M., Angew. Chem., Int. Ed., 2011, vol. 50, p. 8236.
Leyva-Perez, A., Domenech, A., Al-Resayes, S.I., and Corma, A., ACS Catal., 2012, vol. 3, p. 121.
Peng, H., Xi, Y., Ronagi, N., Dong, B., Akhmedov, N.G., and Shi, X., J. Am. Chem. Soc., 2014, vol. 136, p. 13174.
Leyva-Perez, A., Domenech-Carbo, A., and Corma, A., Nat. Commun., 2015, vol. 6, p. 6703.
Ye, X., Peng, H., Wei, C., Yuan, T., Wojtas, L., and Shi, X., Chemistry, 2018, vol. 1983.
Beaumont, S.K., Kyriakou, G., and Lambert, R.M., J. Am. Chem. Soc., 2010, vol. 132, no. 35, p. 12246.
Boronat, M., Combita, D., Concepcion, P., Corma, A., Hermenegildo Garcia, H., Juarez, R., Laursen, S., and De Dios Lopez-Castro, J., J. Phys. Chem. C, 2012, vol. 116, p. 24855.
Boronat, M., Laursen, S., Leyva-Perez, A., Oliver-Meseguer, J., Combita, D., and Corma, A., J. Catal., 2014, vol. 315, p. 6.
Vulhanova, B., Vaclavik, J., Artiglia, L., Ranocchiari, M., Togni, A., and Van Bokhoven, J.A., ACS Catal., 2017, no. 7, p. 3414.
Chen, Z., Shen, R., Chen, C., Li, J., and Li, Y., Chem. Commun., 2018, vol. 54, p. 13155.
Cadio, P. and Chodkiewicz, W., in Chemistry of Acetylenes, Viehe, H.G., Ed., New York: M. Dekker, 1969, p. 597.
Bruk, L.G., Brailovskii, S.M., Temkin, O.N., Flid, R.M., and Kostyushin, A.S., Zh. Org. Khim., 1974, vol. 10, no. 11, p. 2262.
Shchel’tsyn, L.V., Brailovskii, S.M., Murugova, U.Yu., and Temkin, O.N., Kinet. Katal., 1988, vol. 29, no. 5, p. 1044.
Shchel’tsyn, L.V., Brailovskii, S.M., and Temkin, O.N., Kinet. Katal., 1990, vol. 31, no. 6, p. 1361.
Wityak, J. and Chan, J.B., Synth. Commun., 1991, vol. 21, p. 977.
Elbaum, D., Nguyen, T.B., Jorgensen, W.L., and Schreiber, S.L., Tetrahedron, 1994, vol. 50, p. 1503.
Cai, C. and Vasella, A., Helv. Chim. Acta, 1995, vol. 78, p. 2053.
Damie, S.V., Seomoon, D., and Lee, P.H., Org. Chem., 2003, vol. 68, p. 7085.
Lefevre, G., Franc, G., Tlili, A., Adamo, G., Taillefer, M., Ciofini, I., and Jutand, A., Organometallics, 2012, vol. 31, no. 22, p. 7694.
Weng, Y., Cheng, B., He, C., and Lei, A., Angew. Chem., Int. Ed., 2012, vol. 51, no. 38, p. 9547.
Amatore, K., Blart, E., Genet, J.P., Jutand, A., Lenaire-Audoire, S., and Savignac, M., Org. Chem., 1995, vol. 60, p. P. 6829.
Banerjee, S. and Patil, N.T., Chem. Commun., 2017, vol. 53, p. 7937.
Li, Y., Xie, X., Sun, N., and Liu, Y., Angew. Chem., Int. Ed., 2017, vol. 56, p. 6994.
Liu, Y., Yang, Y., Zhu, R., Liu, C., and Zhang, D., Catal. Sci. Technol., 2019, vol. 9, no. 15, p. 4091.
Deprez, N.R. and Sanford, M.S., Inorg. Chem., 2007, vol. 48, p. 1924.
Sonogashira, K., in Metal-Catalyzed Cross-coupling Reactions, Diederich, F. and Stang, P.J., Eds., Weinheim: Wiley-VCH, 1998, p. 203.
Sonogashira, K., Alkynes synthesis, in Handbook of Organopalladium Chemistry for Organic Synthesis, Negishi E.-I., Ed., New York: Wiley, 2002, vol. 1, p. 493.
Chinchilla, R. and Najera, C., Chem. Rev., 2007, vol. 107, p. 874.
Chinchilla, R. and Najera, C., Chem. Soc. Rev., 2011, vol. 40, p. 5084.
Karak, M., Barbosa, L., and Hargaden, G.C., RSC Adv., 2014, vol. 4, p. 53442.
Cacchi, S., Synthesis, 1986, p. 320.
Balanta, A., Godard, C., and Claver, C., Chem. Soc. Rev., 2011, vol. 40, p. 4973.
Bumagin, N.A., Ponomarev, A.B., and Beletskaya, I.P., Izv. Akad. Nauk SSSR, Ser. Khim., 1984, no. 7, p. 1561.
Gelman, D. and Buchwald, S.L., Angew. Chem., Int. Ed., 2003, vol. 2, p. 5991.
Beauperin, M., Job, A., Cattey, H., Royer, S., Meunier, P., and Hierso, J.-C., Organometallics, 2010, vol. 29, p. 2815.
Okuro, K. and Furuune, M., Enna, M., Miura, M., and Nomura, M., Org. Chem., 1993, vol. 58, p. 4716.
Kang, S.K., Yoon, S.-K., and Kim, Y.-M., Org. Lett., 2001, vol. 3, p. 2697.
Kollhofer, A. and Plenio, H., Adv. Synth. Catal., 2005, vol. 347, p. 1295.
Negishi, E. and Anastasia, L., Chem. Rev., 2003, vol. 103, p. 1979.
Reina, A., Dang-Bao, T., Guerrero-Rios, I., and Gómez, M., Nanomaterials, 2021, vol. 11, p. 1891.
Farina, V., Krishnamurthy, V., and Scott, W.J., The Stille Reaction, New York: J. Wiley and Sons, 1998.
Milstein, D. and Stille, J.K., J. Am. Chem. Soc., 2007, vol. 129, p. 11340.
Miyaura, N. and Suzuki, A., Chem. Commun., 1979, p. 866.
Portnoy, M. and Milstein, D., Organometallics, 1994, vol. 13, p. 3465.
Casado, A.L. and Espinet, P., Organometallics, 1998, vol. 17, no. 5, p. 954.
Fitton, P. and Rick, E.A., J. Organomet. Chem., 1971, vol. 28, p. 287.
Amatore, C. and Jutand, A., J. Organomet. Chem., 1999, vol. 576, p. 254.
Amatore, C. and Jutand, A., Acc. Chem. Res., 2000, vol. 33, p. 314.
Beletskaya, I.P. and Cheprakov, A.V., Chem. Rev., 2000, vol. 100, p. 3009.
Kozuch, S. and Jutand, A., Organometallics, 2005, vol. 24, p. 2319.
Barrios-Landeros, F. and Hartvig, J.F., J. Am. Chem. Soc., 2005, vol. 127, p. 6944.
Kozuch, S. and Shai, S., J. Am. Chem. Soc., 2006, vol. 128, p. 3355.
Tougerti, S. and Jutand, A., Chem. Eur. J., 2007, vol. 13, p. 666.
Hue, L. and Liu, Z., Chem. Soc. Rev., 2010, vol. 39, p. 1692.
Heiden, M.R., Plenio, H., Immel, S., Burello, E., Ruthenberg, G., and Hoefsloot, H.C., Chem. Eur. J., 2008, vol. 14, p. 2857.
Shekhar, S., Riberg, P., Hartwig, J.F., Mathiew, J.S., Blackmond, D.G., Strieter, E.R., and Buchwald, S.L., J. Am. Chem. Soc., 2006, vol. 128, p. 3584.
Hartwig, J.F., Inorg. Chem., 2007, vol. 46, p. 1936.
Osakada, K., Takisawa, T., and Yamamoto, T., Organometallics, 1995, vol. 14, p. 3531.
Nova, A., Ujaque, G., Maseros, F., Ledos, A., and Espinet, P., J. Am. Chem. Soc., 2006, vol. 128, p. 14571.
Jutand, A., Negri, S., and Principaud, A., Eur. J. Inorg. Chem., 2005, p. 631.
Genet, J.P., Blart, E., and Savignac, M., Synlett, 1992, p. 715.
Alami, M., Ferri, F., and Linstrumelle, G., Tetrahedron Lett., 1993, vol. 34, no. 40, p. 6403.
Böhm, V.P.W. and Herrmann, W.A., Eur. J. Org. Chem., 2000, p. 3679.
Mery, D., Heuze, K., and Astruc, D., Chem. Commun., 2003, p. 1934.
Fukuyama, T., Shinmen, M., Nishitani, S., Sato, M., and Ryu, L., Org. Lett., 2002, vol. 4, no. 10, p. 1691.
Alonso, D.A., Najera, C., and Pacheco, C., Tetrahedron Lett., 2002, vol. 43, p. 9365.
Leadbeater, N.E. and Tominack, B.J., Tetrahedron Lett., 2003, vol. 44, p. 8653.
Urgaonkar, S. and Verkade, J.G., Org. Chem., 2004, vol. 69, p. 5752.
Mori, A., Kawoshima, J., Shimada, T., Suguro, M., Hirabayashi, K., and Nishihara, Y., Org. Lett., 2000, vol. 2, no. 19, p. 2935.
Iranpoor, N., Firouzabadi, H., and Ahmadi, Y., Eur. J. Org. Chem., 2012, p. 305.
Amatore, C., Bensalem, S., GHam, S., and Jutand, A., J. Organometall. Chem., 2004, vol. 689, p. 4642.
Ljungdahl, T., Bennur, T., Dallas, A., Emtenas, H., and Matensson, J., Organometallics, 2008, vol. 27, p. 2490.
Gacia-Meldhor, M., Pacheco, M.C., Najera, C., Ledos, A., and Ujaque, G., ACS Catal., 2012, vol. 2, p. 135.
De Souza, R.O.M.A., Bittar, M.S., Mendes, L.V.P., and Da Silva, C.M.F., Synlett, 2008, no. 12, p. 1777.
Kuriakou, G., Beaumont, S.K., Humphrey, S.M., Antonetti, C., and Lambert, R.M., ChemCatChem, 2010, vol. 2, p. 1444.
Suomin, D. and Koel, B.E., Surf. Sci., 2001, vol. 490, p. 265.
Kanuru, V., Kuriakow, G., Beaumont, S.K., Papageorgiu, A.C., Watson, D.J., and Lambert, R.M., J. Am. Chem. Soc., 2010, vol. 132, p. 8081.
Goguet, A., Ace, M., Saih, Y., Sa, J., Kavanagh, J., and Hardacre, C., Chem. Commun., 2009, p. 4889.
Lauterbach, T., Livendahl, M., Rosellon, A., Espinet, P., and Echavarren, A.M., Org. Lett., 2010, vol. 12, no. 13, p. 3006.
Corma, A., Juarez, R., Boronat, M., Sanchez, F., Iglesias, M., and Garsia, H., Chem. Commun., 2011, vol. 47, p. 1446.
Espinet, P. and Echavarren, A.M., Platinum Metals Rev., 2011, vol. 55, no. 3, p. 212.
Leuva-Perez, A., Oliver-Meseguer, J., Cabrero-Antonio, J.P., Rubio-Marques, R., Serna, P., Al-Resayes, S.I., and Corma, A., ACS Catal., 2013, vol. 3, no. 8, p. 1865.
Boronat, M., Lopes-Ausens, T., and Corma, A., J. Phys. Chem., 2014, vol. 118, no. 17, p. 9018.
Li, G. and Jiang, D., J. Catal., 2013, vol. 306, p. 177.
Robinson, P.S.D. and Khairallah, G.K., Da Silva, G., Lioe, H., and O’Hair, R.A.J., Angew. Chem., Int. Ed., 2012, vol. 51, p. 3812.
Nijamudheen, A. and Datta, A., J. Phys. Chem., 2013, vol. 117, p. 21433.
Lin, J., Abroshan, H., Liu, C., Zhu, M., Li, G., and Haruta, M., J. Catal., 2015, vol. 330, p. 354.
Johansson, N., Sisodiyas, S., Shyesteh, P., Chaudhary, S., Andersen, J.N., Knudsen, J., Wendt, O.F., and Schnadt, J., J. Phys. Condens. Matter, 2017, vol. 29, p. 444005.
Zeineddine, A., Estevez, L., Malet-Ladeira, S., Miquen, K., Amgoune, L., and Bourosson, D., Nat. Commun., 2017, vol. 8, no. 1, p. 565.
Jones, L.A., Sanz, S., and Laguna, M., Catal. Today, 2007, vol. 122, p. 403.
Panda, B. and Sarkar, T.K., Tetrahedron Lett., 2010, vol. 51, p. 301.
Panda, B. and Sarkar, T.K., Chem. Commun., 2010, vol. 46, p. 3131.
Hashmi, A.S.K., Lothschutz, C., Dopp, R., Rudolph, M., Ramamurthi, T.D., and Rominger, F., Angew. Chem., 2009, vol. 48, no. 44, p. 8243.
Liu, R., Chen, H., Fang, L., Xu, C., He, Z., Lai, Y., Zhao, H., Bekana, D., and Liu, J.-F., Environ. Sci. Technol., 2018, vol. 52, p. 4244.
Rossy, J., Majimel, J., Fouqeuet, E., Delacote, C., Boujita, M., Labrugere, C., Treguer-Dlapierre, M., and Felkin, F.-X., Chem. Eur. J., 2013, vol. 19, p. 14024.
Reina, A., Dang-Bao, T., Guerrero-Rios, I., and Gomez, M., Nanomaterials, 2021, vol. 11, p. 1891.
Zhu, M., Zhou, Z., and Chen, R., Synthesis, 2018, no. 17, p. 2680.
Venkatesan, P. and Santhanalakshmi, J., Langmuir, 2010, vol. 26, no. 14, p. 12225.
Weibel, J.-M., Blanc, A., and Pale, P., Chem. Rev., 2008, vol. 108, p. 3149.
Li, P.H. and Wang, L., Synlett, 2006, p. 2261.
Halbes-Letinois, H., Pale, P., and Berger, S., Org. Chem., 2005, vol. 70, p. 9185.
Jeffery, T., JCS Chem. Commun., 1991, p. 324.
Wu, H.-J., Hsu, H.-K., and Chiang, C.-M., J. Am. Chem. Soc., 1999, vol. 121, no. 18, p. 4433.
Han, M., Liu, S., Nie, X., Yuan, D., Sun, P., Dai, Z., and Bao, J., RSC Adv., 2012, vol. 2, p. 6061.
Sanches-Sanches, C., Orozco, N., Holgado, J.P., Beaumont, S.K., Kuriakou, G., Watson, D.J., Gonzalez-Elipe, A.R., Feria, L., Sanz, J.F., and Lambert, R.M., J. Am. Chem. Soc., 2015, vol. 137, p. 940.
Bumagin, N.A., Kalinovskii, I.O., and Ponamorev, A.B., Izv. Akad. Nauk SSSR, Ser. Khim., 1986, no. 12, p. 2836.
Beletskaya, I.P., Latyshev, G.V., Tsvetkov, A.V., and Lukashev, N.V., Tetrahedron Lett., 2003, vol. 44, p. 5011.
Vechorkin, O., Barmaz, D., Pronst, V., and Hu, X., J. Am. Chem. Soc., 2009, vol. 131, p. 12078.
Son, S.U., Jang, Y., Park, J., Bin, N.H., Park, H.M., Yun, H.J., Lee, J., and Hueon, T., J. Am. Chem. Soc., 2004, vol. 126, p. 5026.
Feng, L., Liu, F., Sun, P., and Bao, J., Synlett, 2008, no. 9, p. 1415.
Kanuru, V.K., Humphrey, S.M., Kyffi, J.M.W., Jefferson, D.A., Burton, J.W., Armbruster, M., and Lambert, R.M., Dalton Trans., 2009, p. 7602.
Enthaler, S., Junge, K., and Beller, M., Angew. Chem., Int. Ed., 2008, vol. 47, p. 3317.
Correa, A., Mancheno, O.G., and Bolm, C., Chem. Soc. Rev., 2008, vol. 37, p. 1108.
Bauer, I. and Knolker, H.J., Chem. Rev., 2015, vol. 115, pp. 3170–3387.
Carril, M., Correa, A., and Bolm, K., Angew. Chem., Int. Ed., 2008, vol. 47, p. 4862.
Correa, A., Elmore, S., and Bolm, K., Chem. Eur. J., 2008, vol. 14, p. 3527.
Bistri, O., Correa, A., and Bolm, K., Angew. Chem., Int. Ed., 2008, vol. 47, no. 3, p. 596.
Correa, A., Corril, M., and Bolm, K., Angew. Chem., Int. Ed., 2008, vol. 47, p. 2880.
Xie, X., Xu, X., Li, H., Xu, Y., Yang, J., and Li, Y., Adv. Synth. Catal., 2009, vol. 351, p. 1263.
Pan, C., Luo, F., Wang, W., Ye, Z., and Liu, M., J. Chem. Res., 2009, p. 478.
Hung, T.-T., Huang, C.-M., and Tsai, F.-Y., ChemCatChem, 2012, vol. 4, p. 540.
Yang, J., Shen, G., and Chen, D., Synth. Commun., 2013, vol. 43, p. 837.
Gruber, M., Chouzier, S., Koehler, K., and Djakovitch, L., Appl. Catal. A: Chem., 2004, vol. 265, p. 161.
Rao Volla, C.M. and Vogel, P., Tetrahedron Lett., 2008, vol. 49, p. 5961.
Mao, J., Xie, G., Wu, M., Guo, J., and Li, S., Adv. Synth. Catal., vol. 350, p. 2477.
Buchwald, S. and Bolm, C., Angew. Chem., Int. Ed., 2009, vol. 48, p. 5586.
Larsson, P.-F., Correa, A., Carril, M., Norrby, P.-O., and Bolm, C., Angew. Chem., Int. Ed., 2009, vol. 48, p. 5691.
Bedford, R.B., Nakamura, M., Gower, J., Haddow, M.F., Hall, M.A., Huwe, M., Hashimoto, T., and Okopie, R.A., Tetrahedron Lett., 2009, vol. 50, p. 6110.
Savant, D.N., Tambade, J., Wagh, S., and Bhanage, B.M., Tetrahedron Lett., 2010, vol. 51, p. 2758.
Hatakeyama, T., Yoshimoto, Y., Gabriel, T., and Nakamura, M., J. Org. Lett., 2008, vol. 10, no. 23, p. 5341.
Berben, L.A. and Long, J.R., Inorg. Chem., 2005, vol. 44, no. 23, p. 8459.
Furstner, A., Nartin, R., Krause, H., Seidel, G., Goddard, R., and Lehman, C.W., J. Am. Chem. Soc., 2008, vol. 130, p. 8773.
Hatakeyama, T., Hashimoto, S., Ishizuka, K., and Nakamura, M., J. Am. Chem. Soc., 2009, vol. 131, p. 11949.
Firouzabadi, H., Iranpoor, N., Gholinejad, M., and Hoseini, J., Adv. Synth. Catal., 2011, vol. 353, p. 125.
Park, S., Kim, M., Koo, D.H., and Cheng, S., Adv. Synth. Catal., 2004, vol. 346, p. 1638.
Na, Y., Park, S., Han, S.B., Han, H., Ko, S., and Chang, S., J. Am. Chem. Soc., 2004, vol. 126, no. 1, p. 250.
Borah, H.N., Prajapati, D., and Boruah, R.C., Synlett, 2005, no. 18, p. 2823.
Freiberg, M., Mulac, W.A., Schmidt, K.H., and Meyerstein, D., JCS Faraday I, 1980, vol. 76, p. 1838.
Freiberg, M., Meyerstein, D., and Yamamoto, Y., JCS. Dalton Trans., 1982, p. 1137.
Cohen, H. and Meyerstein, D., Inorg. Chem., 1986, vol. 25, p. 1505.
Masarawa, M., Cohen, H., and Meyerstein, D., Inorg. Chem., 1986, vol. 25, p. 4897.
Cohen, H. and Meyerstein, D., JCS Faraday I, 1988, vol. 84, no. 11, p. 4157.
Masarawa, M., Cohen, H., Glaser, R., and Meyerstein, D., Inorg. Chem., 1990, vol. 24, p. 5031.
Goldstein, S., Czapski, G., Cohen, H., and Meyerstein, D., Inorg. Chim. Acta, 1992, vol. 192, p. 87.
Navon, X., Golub, G., Cohen, H., and Meyerstein, D., Organometallics, 1995, vol. 14, p. 5670.
Szulc, A., Meyerstein, D., and Cohen, H., Inorg. Chim. Acta, 1998, vol. 270, p. 440.
Epstein, D.M., Cohen, H., Masarawa, A., and Meyerstein, D., Inorg. Chim. Acta, 2002, vol. 339, p. 281.
Burg, A. and Meyerstein, D., in Inorganic/Bioinorganic Reaction Mechanisms, 2012, vol. 64, p. 220.
Yardent, G., Meyerstein, D., Kats, L., Cohen, H., Zilberman, I., and Maimon, E., J. Coord. Chem., 2019, vol. 72, nos. 22–24, p. 3445.
Mykhalichko, I.M., Mys’kiv, M.G., and Davydov, V.N., Zh. Neorg. Khim., 1999, vol. 44, no. 1, p. 46.
Catalysis by Gold. Catalytic Science, Bond, G.C., Louis C., and Thompson, D.T., Eds., London: Imp. College Press, 2006, vol. 6.
Gorin, D.J., Toste, F.D., and Reina, A., Nature, 2007, vol. 446, p. 395.
Modern Gold Supramolecular Chemistry. Gold–Metal Interaction and Application, Laguna, A., Ed., Weinheim: Wiley-VCH, 2009.
Gold Chemistry: Applications and Future Directions in the Life Sciences, Mohr, F., Ed., Weinheim: Wiley-VCH, 2009, p. 408.
Soriano, E. and Marco-Conteeles, J., Top Curr. Chem., 2011, vol. 302, p. 1.
Zubanova, E.M., Reaction mechanisms of copper complexes with alkyl radicals, Extended Abstract of Cand. Sci. (Chem.) Dissertation, Moscow: Mosk. Gos. Univ. im. M.V. Lomonosova, 2015.
Modern Gold Catalyzed Synthesis, Hashmi, A.S.K. and Toste, F.D., Eds., Weinheim: Wiley-VCH, 2012.
Zang, L., Acc. Chem. Res., 2014, vol. 47, no. 3, p. 877.
Rosca, D.-A., Fernandez-Cestau, J., Huges, D.-L., and Bohmann, M., Organometallics, 2015, vol. 34, no. 11, p. 2098.
ACKNOWLEDGMENTS
I thank K. Egiazaryan, graduate student, Chair of Physical Chemistry, MIREA—Russian Technological University, Moscow, Russia, for his invaluable help in preparing this review; Dr. L.G. Fedenok for very useful discussions concerning the mechanisms of the OD reaction; Professor O.L. Kalija and Professor L.G. Bruk for valuable advice on the content and structure of the review.
Funding
No support was received for the preparation of this review.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The author declares that he has no conflicts of interest.
Additional information
Translated by V. Glyanchenko
Abbreviations and notation: EDA, ethylenediamine; TMEDA, tetramethylethylenediamine; THF, tetrahydrofuran; MA, methylacetylene; DMDA, dimethyldiacetylene; CMA, chloromethylacetylene; DA, diacetylene; MDA, methyldiacetylene; DCE, dichloroethylene; PEG, polyethylene glycol; BMA, bromomethylacetylene; CMA, copper methylacetylide; TBA, tetrabutylamine; DMF, dimethylformamide; EG, ethylene glycol; Py, pyridine; PiPy, piperidine; DiPy, dipyridyl; AN, acetonitrile; Phen, phenanthrene; BMIM, butylmethylimidazole; dba, dibenzylideneacetone; hal, halides; dppm, 1,2-bis(diphenylphosphino)methane; dppp, 1,2-bis(diphenylphosphino)propane; dppe, 1,2-bis(diphenylphosphino)ethane; dppf, 1,1-bis(diphenylphosphino)ferrocene; BINAP, bis(naphthyldiphenylphosphine); Xantophos, 9,9-dimethyl-4,5-bis(diphenylphosphino)xanthene; TEMPO, (2,2,6,6-tetramethylpiperidin-1-yl)oxyl; TMEDA, tetramethylethylenediamine; TMED, tetramethylethylenediamine; DME, dimethyl ether; DMED, dimethylethylenediamine; NMP, N-methyl-2-pyrrolidone; dipy, dipyridyl; NHC, N-heterocyclic carbene; HX, acids; D, optical density; POCC, polyfunctional oxygen-containing compounds; NMR, nuclear magnetic resonance; OS, oxidation state; ORR, redox reactions; OD, oxidative dehydrocondensation of alkynes (coupling); B, base; NC, nanocluster; NP, nanoparticle; OA, oxidative addition; TM, transmetalation; RE, reductive elimination; rt, room temperature; TOF, turnover frequency of catalyst; TON, turnover number of catalyst; EDG, electron donating group; EWG, electron withdrawing group; KIE, kinetic isotope effect; IL, ionic liquids; RDS, rate-determining step; TPR, temperature-programmed reaction; CSI-MS, cold-spray ionization mass spectrometry; STM, scanning tunneling microscopy; ESI-MS, electrospray ionization mass spectrometry; CID, collision-induced dissociation; XPS, X-ray photoelectron spectroscopy; HR TEM, high-resolution transmission electron microscopy, FE SEM, field emission scanning electron microscopy; STM, scanning tunneling microscopy; DFT, density functional theory; XRD, X-ray powder diffraction analysis; and ICP-ES, inductively coupled plasma emission spectroscopy.
Rights and permissions
About this article

Cite this article
Temkin, O.N. “Golden Age” of Homogeneous Catalytic Chemistry of Alkynes: Some Oxidative Transformations of Alkynes (A Review). Kinet Catal 64, 521–577 (2023). https://doi.org/10.1134/S0023158423050117
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0023158423050117