
Abstract
The results of studying the reaction kinetics of the catalytic oxidation of methyl oleate with hydrogen peroxide in a two-phase system (aqueous phase-organic phase) in the presence of the bifunctional catalyst [(Octn)3NMe]3{PO4[WO(O2)2]4} are presented. For the selected reaction conditions, the first orders of reaction with respect to the catalyst, substrate, and oxidizing agent were established. The activation energy was 47 ± 3 kJ/mol in a temperature range of 313–353 K, and the preexponential factor was (6.0 ± 0.3) × 107 L2 mol–2 min–1.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Fatty acids (FAs) are among the most valuable components of natural biomass, and the area of their applications is constantly expanding [1]. In this regard, they were the objects of study of many research teams around the world [2]. Thus, FA esterification with methanol in the presence of cation-exchange resins [3] or with phosphotungstic acid in the presence of potassium oxide and silica [4] makes it possible to synthesize FA methyl esters, components of biodiesel fuel [5]. Epoxides obtained by the oxidation of fatty acids and/or their methyl esters are in demand as polymer stabilizers and plasticizers [6]. At the same time, despite the solutions known from the literature for the oxidation of the alkenyl groups of unsaturated fatty acids to corresponding epoxides, a search for new more efficient methods and a study of the reaction kinetic of the oxidation remain of considerable current interest [7]. In 1944, Swern et al. [8] published one of the first studies of these reactions where oleic acid (OA) and perbenzoic acid acted as a substrate and an oxidizing agent, respectively. Subsequently, both peroxo acids and hydrogen peroxide were used to oxidize OA to epoxides in the presence of homogeneous [9] or heterogeneous catalysts [10]. The disadvantages of these latter include the occurrence of a decomposition reaction of hydrogen peroxide along with the main reaction, but it can be eliminated almost completely by using liquid-phase systems. Among them, tungsten-containing systems based on tungsten peroxo complexes can be considered as the most convenient ones for the epoxidation of FAs. In this case, an elegant solution is the use of bifunctional metal complex catalysts Q3{PO4[WO(O2)2]4} containing an organic cation (Q) and combining the functions of phase-transfer and homogeneous catalysis [11].
Khlebnikova and coauthors [12, 13] carried out primary studies and established basic ratios between the components of a reaction mixture for the oxidation of both oleic acid (OA) and methyl oleate (MO) with hydrogen peroxide under the conditions of phase-transfer catalysis. In particular, using the oxidation of MO with hydrogen peroxide as an example, they found that the epoxide yield can reach about 85% at a complete conversion of the substrate in the presence of the catalyst [(Octn)3NMe]3{PO4[WO(O2)2]4}. Although the kinetics of this reaction was not studied in detail, these published data [12, 13] were crucial for scaling up a catalytic epoxidation process in order to develop an industrial technology. Therefore, the purpose of this work was to study in detail the reaction kinetics of the catalytic oxidation of oleic acid methyl ester with hydrogen peroxide to the corresponding epoxide, which proceeds without the use of additional solvents.
EXPERIEMNTAL
Reagents. Oleic acid methyl ester (methyl oleate) (96%) from Alfa Aesar and hydrogen peroxide (special purity grade 8–4, 35%) from OOO Leta were used in this study. The catalysts were synthesized using the heteropoly acid H3[PW12O40]·2H2O from Panreac and the ammonium salt [(Octn)3NMe]3Cl (Aliquat® 336) from Alfa Aesar. Solvents were chemically pure dichloromethane and ethyl acetate.
IR spectroscopy. The IR spectrum of the complex [(Octn)3NMe]3{PO4[WO(O2)2]4} was recorded on an IRAffinity-1 FT-IR spectrometer (Shimadzu, Japan) in a range 400–4000 cm–1 between Zn–Se glasses.
1H and 31P NMR spectroscopy. The 1Н and 31P NMR spectra of the complex [(Octn)3NMe]3-{PO4[WO(O2)2]4} were recorded on an AV-300 spectrometer (Bruker, Germany) in C6D6 (operating frequencies of 300.13 MHz for 1Н and 121.49 MHz for 31P) for solutions of substances. The proton signals (0 ppm) of tetramethylsilane (TMS) were used as an external standard for the 1Н NMR spectra. The 31P NMR spectra were recorded with suppression of heteronuclear resonance relative to H3PO4 (0 ppm) as an external standard.
Synthesis of the complex [(Octn)3NMe]3-{PO4[WO(O2)2]4}. The synthesis was carried out in accordance with a published procedure [11], and the structure of the complex was confirmed by Fourier transform IR (FTIR) spectroscopy and NMR spectroscopy.
FTIR spectrum, ν max (cm–1): 3523.9, 2961.7, 2860.2, 1726.4, 1565.9, 1483.6, 1467.1, 1378.0, 1091.5, 1058.5, 974.0, 888.9, 855.9, 846.2, 723.3, 650.9, 628.6, 590.9, 576.0, 549.3, 523.5.
1Н NMR spectrum, δ (ppm): 4.29 (s, 1H), 3.20 (m, 5H), 2.94 (m, 10H), 1.39 (m, 78H), 1.03 (m, 20H), 0.29 (s, 2H); 31P NMR spectrum, δ (ppm): 4.41 (m).
MO oxidation procedure. The study of catalytic activity was carried out in a 15-mL reactor equipped with a thermostatically controlled jacket, a reflux condenser, and a magnetic stirring activator. The rotation frequency of the activator was chosen so that it did not affect the reaction rate of the oxidation (Fig. 1a).

(a) Dependence of the rate of MOE formation on the stirrer rotation frequency (rpm); (b) kinetic curve of MOE formation versus time. Reaction conditions: Т = 333 K; substrate amount, 10.12 mmol; oxidizing agent, 35% aqueous solution of H2O2; [Cat]/[MO]/[H2O2] = 1/1000/2000.
An LT-205a thermostat (Loip, Russia) was used for temperature control, and the heat carrier was water. The reaction mixture was stirred using an RCT basic magnetic stirrer (IKA, Germany). The oxidation reaction was carried out in accordance with a previously described procedure [12]. The initial reaction rate (W) of the formation of methyl oleate epoxide (MOE) was determined from the slope of the initial section of a kinetic curve for the time dependence of MOE concentration (Fig. 1b).
Quantitative analysis. The quantitative analysis of reaction mixtures was carried out using a GKh-1000 chromatograph (Khromos, Russia) equipped with a flame ionization detector. The chromatographing conditions were the following: Tcolumn = 200°C (5 min); then, heating to 250°C at a rate of 2 K/min; Tdetector = 280°C and Tinjector = 330°C; and carrier gas, He (р = 1 kg/cm2). The retention times of MO and MOE were 6.6 and 14.9 min, respectively. A DB-Wax column of 30 m × 0.25 mm × 0.25 µm (Agilent Technologies, the United States) was used. GC–MS analysis performed on a Shimadzu GCMS-QP2010 gas chromatograph mass spectrometer (Japan) was used to identify MOE. The chromatographing conditions were the following: GsBP1-MS column, 30 m × 0.32 mm; Tinjector = 300°C; Tcolumn = 50°C (3 min); then, heating at a rate of 20 K/min to 300°C (10 min) (total time, 25.5 min); carrier gas, He; flow rate, 50 cm/s; split ratio, 1 : 50; Tdetector = 300°C; ion source temperature Tion source = 290°C; scan interval, 0.2 s; and determined m/z, 35–500.
RESULTS AND DISCUSSION
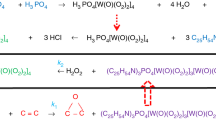
Previous studies have shown that catalysts based on tungsten peroxo complexes in combination with quaternary ammonium cations are effective in the oxidation of various substrates, namely, cycloalkenes, alcohols to carboxylic acids, unsaturated fatty acids to epoxides, and tertiary amines to corresponding N-oxides [11]. As noted above, the aim of this work was to study in detail the main reaction kinetics of the of catalytic (Cat) oxidation of MO (1) with hydrogen peroxide to the corresponding epoxide MOE (2) (Scheme 1).

Scheme 1 . Catalytic oxidation of oleic acid methyl ester with hydrogen peroxide.
Initially, it was found that the stirring mode of a reaction mixture at a rotation frequency of >900 rpm did not affect the reaction rate of epoxidation—the curve reached a plateau (Fig. 1a). Therefore, all further experiments were carried out at a stirring rate of 1000 rpm. In this case, the expression for the rate of MOE formation has the form
where W is the reaction rate of MOE formation, mol/L; A is the preexponential factor, L2 mol–2 min–1; l, m, and p are the orders of reaction with respect to the catalyst, oxidizing agent, and substrate, respectively; Ea is the apparent activation energy, kJ/mol; R is the gas constant, 8.314 J mol–1 K–1; and T is the temperature, K.
Dependence of the Reaction Rate of MOE Formation on the Catalyst Concentration
To determine an optimal catalyst concentration, we obtained the kinetic function W = f[Cat], where W is the rate of reaction and [Cat] is the concentration of the catalyst [(Octn)3NMe]3{PO4[WO(O2)2]4. The catalyst concentration was varied in the range (1.5–5.8) × 10–3 M ([MO]/[Cat] ≈ 550–2100).
The order of reaction (l) found from the slope of a logarithmic anamorphosis for catalyst concentrations in the range (1.5–5.8) × 10–3 M was 1, and the observed reaction rate of MOE formation increased with the catalyst concentration in the reaction mixture (Fig. 2).

Anamorphoses of the concentration dependences (1) ln W–ln [Cat] and (2) ln W–ln [H2O2]. Conditions: T = 333 K; substrate amount, 10.12 mmol; oxidizing agent, 35% aqueous solution of H2O2; (1) f[W]–[Cat], [MO]/[H2O2] = 1/2, and [Cat] = (1.5–5.8) × 10–3 M; (2) f[W]–[H2O2], [MO]/[Cat] = 1000, and [H2O2] = 2–12 M.
Dependence of the Rate of MOE Formation on the Concentration of Hydrogen Peroxide
In order to determine the order of reaction with respect to the oxidizing agent in the presence of the catalyst [(Octn)3NMe]3{PO4[WO(O2)2]4, we studied the kinetic function W = f[H2O2] of the reaction rate of MOE formation on the concentration of hydrogen peroxide. From the experimental data (Fig. 2), it follows that the order of the reaction with respect to hydrogen peroxide (m) was 1 for a hydrogen peroxide concentration range of 2–12 M. It was also shown that the reaction rate increased with the concentration of the oxidizing agent, and the reaction with the formation of MOE did not occur in the absence of hydrogen peroxide [13].
Order of the Reaction of MOE Formation with Respect to the Substrate
The order of the reaction of MOE formation with respect to the substrate was determined as follows: In accordance with Eq. (1), the reaction rate of MOE formation depends on the catalyst concentrations, the preexponential factor, and the activation energy. In the case of constant catalyst concentration and reaction temperature, expression (1) can be represented in the form
where kobs = A[Cat]\({{{\text{e}}}^{{\frac{{ - {{E}_{{\text{a}}}}}}{{RT}}}}}\) (L mol–1 min–1) is a constant value at fixed catalyst concentration and temperature. Considering the ratio [MO]/[H2O2] = 1/2 in the course of the reaction, we can represent expression (2) in the form
where [MO]0 is the initial concentration of methyl oleate. The analytical solution of this equation shows that the linear dependences of
\(\frac{{\ln \left( {\frac{{{{{[{\text{MO}}]}}_{0}}}}{{[{\text{MOE}}] - {{{[{\text{MO}}]}}_{0}}}} - 1} \right)}}{{{{{[{\text{MO}}]}}_{0}}}}\)
on time t are indicative of the first order with respect to the substrate (inset in Fig. 3), and the slope of the plot of ln kobs vs. 1000/T (Fig. 3) corresponds to the reaction rate constant kobs = A[Cat]\({\text{exp(}}{{ - {{E}_{{\text{a}}}}} \mathord{\left/ {\vphantom {{ - {{E}_{{\text{a}}}}} {RT}}} \right. \kern-0em} {RT}}{\text{)}}\) (L mol–1 min–1) of MOE formation.

Arrhenius relationship ln kobs–1000/T for MOE formation upon MO oxidation with hydrogen peroxide. Inset: dependence of MOE formation on time t at different temperatures. Reaction conditions: Т = 313–353 K; substrate amount, 10.12 mmol; oxidizing agent, 35% aqueous solution of H2O2; [Cat]/[MO]/[H2O2] = 1/1000/2000.
Thus, an analysis of the graphic dependences confirmed the first orders (l, m, and p) with respect to the catalyst, oxidizing agent, and substrate for the catalytic oxidation of MO with hydrogen peroxide in a temperature range of 313–353 K (Fig. 3) and an increase in the rate constant of the formation of MOE with temperature (Table 1).
The apparent activation energy Ea found from the temperature dependence of the reaction rate constants using an anamorphosis (Fig. 3), where Ea = –R(Δln kobs/Δ(T–1)), was 47 ± 3 kJ/mol, and the preexponential factor determined from the intercept on the ordinate axis was A[Cat] = (1.8 ± 0.2) × 105 L mol–1 min–1 (Fig. 3).
Taking into account that the catalyst concentration in the expression A[Cat] is constant, we can determine the preexponential factor as A = (6.0 ± 0.3) × 107 L2 mol–2 min–1; therefore, the expression for the rate of formation of MOE in the range of [MO]/[Cat] ≈ 550–2100 at [MO]/[H2O2] = 2 takes the following form:
The values of activation energy known from the literature for this reaction are 51–53 kJ/mol (T = 35–50°С) in the case of oxidation in the presence of the heteropoly acid H3PW12O40 in combination with various phosphonium cations [9] and 85 kJ/mol (T = 15–30°C) in the presence of the catalyst [(Octn)3NMe]3{PO4[WO(O2)2]4} [14]. In the latter case, the reaction was carried out in a steel autoclave at overpressure (40 psi) in an inert (nitrogen) atmosphere. It is likely that differences in the activation energies observed by Maiti et al. [14] and in this work are due to different reaction mechanisms of epoxidation in the temperature ranges of 15–30°С (85 kJ/mol [14]) and 40–80°С (47 kJ/mol, this work).
A comparison of the found value of Ea = 47 kJ/mol with the activation energy of the N-oxide formation reaction (37 kJ/mol), which was determined in the catalytic oxidation of N-(phosphonomethyl)iminodiacetic acid with hydrogen peroxide in the presence of the catalyst [(Octn)3NMe]3{PO4[WO(O2)2]4} [15], demonstrated their comparability. Therefore, it can be assumed that the active forms of the catalyst are identical in the oxidation reactions of different substrates (tertiary amines and alkenes). The found first orders of the reaction with respect to the substrate, catalyst, and oxidizing agent allowed us to assume that the oxidation of MO also proceeds in accordance with the reaction schemes proposed earlier [9, 11, 15].
Thus, because W(VI) is a relatively weak oxidizing agent, the oxidation reaction of MO in the presence of the catalyst proceeds according to a peroxometalate mechanism, when the oxidation state of the metal remains unchanged and stoichiometric oxidation does not occur in the absence of H2O2, in contrast to an oxometalate mechanism [16].
The reaction proceeds through the following main steps:
— complex formation in a organic phase upon the interaction of the substrate with a catalyst containing active peroxo groups;
— formation of the target product MOE and the inactive oxo form (W=O) of the catalyst;
— catalyst regeneration by the interaction of the oxo form with H2O2 at the (aqueous/organic) phase boundary.
The proposed mechanism of oxidation is consistent with the observed first order of reaction with respect to hydrogen peroxide, and it indicates the presence of only one stage at which the oxidizing agent is consumed: the stage of catalyst regeneration.
CONCLUSIONS
The catalytic oxidation of methyl oleate with hydrogen peroxide in a two-phase liquid system in the presence of the metal complex catalyst [(Octn)3-NMe]3{PO4[WO(O2)2]4} to the corresponding epoxide can be studied without the use of additional organic solvents. At the same time, the study of the formal kinetics for the selected boundary parameters (the ratios [MO]/[Cat] ≈ 550–2100 and [MO]/[H2O2] ≈ 0.5–3.8 in a temperature range from 313 to 353 K) showed that the reaction is of the first order with respect to the catalyst, substrate, and oxidizing agent. The preexponential factor and activation energy determined for these conditions are 6 × 107 L2 mol–2 min–1 and 47 ± 3 kJ/mol, respectively.
The results obtained indicate the possibility of successfully expanding the scope of the catalyst [(Octn)3NMe]3{PO4[WO(O2)2]4} and its application to the oxidation of various polyfunctional substrates.
REFERENCES
Cerone, M. and Smith, T.K., Front. Nutr., 2021, vol. 8, p. 1.
Biermann, U., Bornscheuer, U.T., Feussner, I., Meier, M.A.R., and Metzger, J.O., Angew. Chem. Int. Ed., 2021, vol. 60, no. 37, p. 20144.
Otopkova, K.V., Esipovich, A.L., Kanakov, E.A., Charykova, T.A., Baidachenko, V.E., and Ryabova, T.A., Kinet. Katal., 2022, vol. 63, no. 6.
Singh, H. and Ali, A., Kinet. Katal., 2022, vol. 63, no. 6.
Belousov, A.S., Esipovich, A.L., Kanakov, E.A., and Otopkova, K.V., Sust. Energy Fuels, 2021, vol. 5, no. 18, p. 4512.
Wypych, G., PVC Degradation and Stabilization, ChemTec Publishing, 2015, p. 287.
Meng, Y., Taddeo, F., Aguilera, A.F., Cai, X., Russo, V., Tolvanen, P., and Leveneur, S., Catalysts, 2021, vol. 11, no. 7, p. 765.
Swern, D., Findley, T.W., and Scanlan, J.T., J. Am. Chem. Soc., 1944, vol. 66, no. 11, p. 1925.
Musik, M., Janus, E., Pelech, R., and Salaciński, L., Catalysts, 2021, vol. 11, no. 9, p. 1058.
Esipovich, A.L., Belousov, A.S., Kanakov, E.A., Mironova, V.Yu., Rogozhin, A.E., Danov, S.M., Vorotyntsev, A.V., and Makarov, D.A., Kinet. Catal., 2019, vol. 60, no. 1, p. 62.
Pai, Z.P., Chesalov, Y.A., Berdnikova, P.V., Uslamin, E.A., Yushchenko, D.Y., Uchenova, Y.V., Khlebnikova, T.B., Baltakhinov, V.P., Kochubey, D.I., and Bukhtiyarov, V.I., Appl. Catal. A: Gen., 2020, vol. 604, p. 117786.
Khlebnikova, T.B., Pai, Z.P., Fedoseeva, L.A., and Mattsat, Y.V., React. Kinet. Catal. Lett., 2009, vol. 98, no. 1, p. 9.
Pai, Z.P., Khlebnikova, T.B., Mattsat, Y.V., and Parmon, V.N., React. Kinet. Catal. Lett., 2009, vol. 98, no. 1, p. 1.
Maiti, S.K., Snavely, W.K., Venkitasubramanian, P., Hagberg, E.C., Busch, D.H., and Subramaniam, B., Ind. Eng. Chem. Res., 2019, vol. 58, no. 7, p. 2514.
Yushchenko, D.Y., Pai, Z.P., and Khlebnikova, T.B., Catal. Lett., 2022, vol. 152, no. 7, p. 2025.
Sheldon, R.A., Arends, I.W.C.E., and Dijksman, A., Catal. Today, 2000, vol. 57, nos. 1–2, p. 157.
Funding
This work was carried out within the framework of a state contract of the Boreskov Institute of Catalysis, Siberian Branch, Russian Academy of Sciences (project no. AAAA-A21-121011390007-7).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of interest.
Additional information
Translated by V. Makhlyarchuk
Abbreviations and notation: MO, methyl oleate; Cat, (N-methyl-N,N,N-trioctyl)-tetrakisoxodiperoxotungstophosphate [(Octn)3-NMe]3{PO4[WO(O2)2]4}; concentration, mol/L; T, temperature, K; t, reaction time, min; W, rate of reaction, mol L–1 min–1; rpm, revolutions per minute; A, preexponential factor, L2 mol–2 min–1; l, order of reaction with respect to the catalyst [(Octn)3NMe]3{PO4[WO(O2)2]4}; m, order of reaction with respect to the oxidizing agent H2O2; p, order of reaction with respect to the substrate, MO; Ea, apparent activation energy, kJ/mol; kobs, reaction rate constant of the formation of methyl oleate epoxide, L mol–1 min–1; FA, fatty acid; MOE, methyl oleate epoxide
Rights and permissions
About this article

Cite this article
Yushchenko, D.Y., Pai, Z.P., Uchenova, Y.V. et al. Kinetics of the Catalytic Oxidation of Methyl Oleate. Kinet Catal 64, 270–275 (2023). https://doi.org/10.1134/S0023158423030102
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0023158423030102