
Abstract
Peculiar coordination compound Cu(Hbpt)(CH3CN)2PF6 (1) derived from 3,5-bis(4-pyridyl)-1H-1,2,4-triazole (Hbpt) is synthesized by the layer diffusion method and structurally characterized by single crystal and powder X-ray diffraction, elemental analysis, and infrared spectroscopy. Compound 1 exhibits a structure formed by cationic \([\text{Cu}(\text{Hbpt}){{(\text{C}{{\text{H}}_{\text{3}}}\text{CN})}_{\text{2}}}]_{n}^{n+}\) chains and \(\text{PF}_{6}^{-}\) anions with abundant supramolecular interactions. Solid-state photoluminescence experiments show that compound 1 exhibits relatively strong red emission with a peak around 656 nm, and the origin of the emission band may be ascribed to the metal-to-ligand charge transfer according to the calculation of the density of states.

Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
The design and synthesis of coordination compounds with strong luminescence and useful photochemical properties have received considerable interest in coordination chemistry and crystal engineering [1-3]. Among the reported various luminescent coordination compounds, d10 metal complexes containing conjugated organic bridging ligands are attractive candidates since most of them possess strong and long-lived photoluminescence with emission energies spanning a wide range in the visible spectrum. Other [4-6] and our [7-12] studies have revealed that the fascinating luminescence of these compounds was mainly associated with the d10 electronic configurations of metal ions [13-15], as well as the extended π-electron systems of conjugated organic ligands. The conjugated ligands commonly have rigid linear or planar structures which may enhance photoelectron transfer. For an example, 3,5-bis(4-pyridyl)-1H-1,2,4-triazole (Hbpt) is a typical dipyridyl ligand with a slightly bent backbone, and shows many advantages in the construction of luminescent coordination compounds with diverse structures when it coordinates to various transition metal ions such as Cu+, Cd2+, Zn2+, etc. [10-12, 16-19]. Accordingly, the combination of inorganic d10 metal ions and conjugated organic ligands may lead to coordination compounds with special structural architectures and excellent photoluminescence properties. In the present paper, copper(I) salt and a Hbpt ligand were selected to assemble a coordination compound with the composition Cu(Hbpt)(CH3CN)2PF6 (1). Based on a single crystal X-ray diffraction study, compound 1 exhibits a peculiar structure formed by cationic \([\text{Cu}(\text{Hbpt}){{(\text{C}{{\text{H}}_{\text{3}}}\text{CN})}_{\text{2}}}]_{n}^{n+}\) chains and \(\text{PF}_{6}^{-}\) anions with abundant supramolecular interactions. The photoluminescence properties and the relationship between the structure and photoluminescence of 1 were also investigated.
EXPERIMENTAL
Materials and instrumentation. The Hbpt ligand was prepared according to our previous reported method [12]. Other chemicals were purchased from commercial sources and used without further purification. The FTIR spectra were recorded from KBr pellets on a FTIR 8400S (CE) instrument in the range of 4000-400 cm–1. C, H, and N elemental analyses were carried out on a Vario EL III elemental analyzer. Powder X-ray diffraction data were recorded with CuKα radiation (λ = 1.5406 Å) on an XRD-6100 apparatus with a scan speed of 2 deg/min. Photoluminescence analyses were carried out on a PerkinElmer LS55 fluorescence spectrometer.
Synthesis. A solution of Hbpt (10.4 mg, 0.04 mmol) in ethanol (3 mL) was carefully layered onto a mixed acetonitrile/water solution (5 mL, volume ratio 1:1) of Cu(CH3CN)4PF6 (14.9 mg, 0.04 mmol) in a straight glass tube. After about 7 days light yellow rod-like single crystals were obtained. Yield: 12 mg (60% based on the Hbpt ligand). Anal. calc. for C16H15CuF6N7P (%): C 37.40, H 2.94, N 19.08; found (%): C 37.86, H 3.02, N 18.97. FTIR (KBr, 4000-400 cm–1): 3549 s, 3379 s, 2258 w, 1636 m, 1583 m, 1543 w, 1489 w, 1420 m, 1366 m, 1314 w, 1217 w, 1059 w, 982 w, 843 s, 754 m, 721 m, 559 m, 503 w; ν(C≡N) vibration of coordinated CH3CN: 2258 w.
Single crystal X-ray diffraction determination. A suitable single crystal of 1 was carefully selected under an optical microscope and glued to a thin glass fiber. The data collection was performed on a Rigaku Mercury CCD diffractometer with graphite-monochromatized MoKα radiation (λ = 0.71073 Å) at T = 293 K. The intensity data set was collected with the ω-scan technique and reduced using the Rigaku CRYSTALCLEAR software [20]. The structure was solved by direct methods and refined by full-matrix least-squares techniques. Non-hydrogen atoms were located by difference Fourier maps and subjected to the anisotropic refinement. All carbon-attached H atoms were allowed to ride on their respective parent C atoms with C–H distances of 0.93 Å or 0.96 Å, and were included in the structure factor calculations with assigned isotropic displacement parameters Uiso(H) = 1.2Ueq(C). All calculations were performed with the Siemens SHELXTL crystallographic software [21-23].
Crystallographic data for 1: C16H15CuF6N7P, Mr = 513.86, crystal size 0.28×0.21×0.17 mm, monoclinic space group, P21/c, a = 7.4008(11) Å, b = 20.408(3) Å, c = 14.879(2) Å, β = 111.380(11)°, V = 2092.6(6) Å3, T = 296(2) K, Z = 4, Dcalc = 1.63 g/cm3, μ = 1.2 mm–1, F(000) = 1032 e, 10836 reflections measured in the range of 2.940 < θ < 25.349°, 3815 unique (Rint = 0.0323), 3815 observed with I > 2σ(I). Structure solution and refinement based on 2556 reflections and 282 refined parameters, 0 restraints gave R1 = 0.0740, wR2 = 0.1496, S = 1.00 for all reflections, R1 = 0.0448, wR2 = 0.1297, and S = 1.00 for reflections with (I > 2σ(I), Δρ(max / min) = 0.35 / –0.49 e/Å3.
CCDC 2062437 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
RESULTS AND DISCUSSION
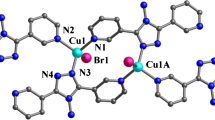
Crystal structure analysis. The single crystal X-ray diffraction analysis has revealed that compound 1 crystallized in the monoclinic space group P21/c with Z = 4. It displays a structure formed by cationic \([\text{Cu}(\text{Hbpt}){{(\text{C}{{\text{H}}_{\text{3}}}\text{CN})}_{\text{2}}}]_{n}^{n+}\) chains and \(\text{PF}_{6}^{-}\) anions with abundant supramolecular interactions. As shown in Fig. 1, the asymmetric unit is made up of one Cu(I) cation, one Hbpt ligand, two coordinated acetonitrile molecules, and one \(\text{PF}_{6}^{-}\) anion. The Cu(I) cation is located in a distorted tetrahedral coordination formed by two pyridyl N atoms from two Hbpt ligands, and two N atoms of two terminally bonded acetonitrile molecules. The Cu–N bond lengths vary from 1.973(3) Å to 2.387(4) Å, while the N–Cu–N bond angles range between 86.53(15)° to 142.36(12)° (Table 1). The two coordinated acetonitrile molecules display approximately linear structures with the C–C–N bond angles of 178.4(5)° and 178.6(5)°, respectively. Other bond lengths and angles are similar to those reported for the other Hbpt-based complexes [10-12, 16-19]. Each Hbpt ligand connects [Cu(CH3CN)2]+ moieties in bidentate coordination modes to form a cationic \([\text{Cu}(\text{Hbpt}){{(\text{C}{{\text{H}}_{\text{3}}}\text{CN})}_{\text{2}}}]_{n}^{n+}\) chain approximately along the [1 0 1] direction (Fig. 2). These cationic chains stack together in a space-filling fashion via abundant π⋯π stacking interactions (Table 3). The \(\text{PF}_{6}^{-}\) anions reside between these cationic chains connected via weak C–H⋯F hydrogen bonds (Table 2) and electrostatic attraction to form a stable structure (Fig. 3).

Coordination environment of the Cu(I) cation in the crystal of 1 (30% probability displacement ellipsoids). All hydrogen atoms are omitted for clarity. Symmetry code A: –1+x, y, –1+z.
Powder X-ray diffraction. A powder X-ray diffraction study was carried out for 1 to check the phase purity of the product. As shown in Fig. 4, all major peak positions of the experimental data of the bulk sample are in good agreement with those simulated using the single crystal X-ray diffraction data, revealing the presence of one main crystalline phase, and that the synthesized bulk material is identical with that of the single crystal.
Photoluminescence behaviour. Solid-state photoluminescence spectra show that compound 1 exhibits relatively strong red emission with a peak around 656 nm upon photoexcitation at 373 nm (Fig. 5), while the free Hbpt ligand displays an emission band around 450 nm upon maximum photoexcitation at 373 nm [12]. In comparison with the free Hbpt ligand, the band maximum of 1 displays a relatively large shift with a wider spectral range, as well as a slightly enhanced emission intensity. The large shift and different band ranges may suggest that the emission of 1 and the pure Hbpt ligand are based on different photoluminescence mechanisms.

View of a cationic \([\text{Cu}(\text{Hbpt}){{(\text{C}{{\text{H}}_{\text{3}}}\text{CN})}_{\text{2}}}]_{n}^{n+}\) chain approximately along the crystallographic [1 0 1] direction. Hydrogen atoms and \(\text{PF}_{6}^{-}\) counter-anions are omitted for clarity.
The density of states (DOS) calculation [24-26] of 1 (Fig. 6; Fig. S3 and its explanation in the Supplementary Materials) indicates that the top of valence bands (VBs) is relatively flat and the bottom of conduction bands (CBs) have a small dispersion. The top orbitals of VBs in the range between –1.40 eV and the Fermi level (0 eV) are mainly formed by Cu 3d states (41.5 electrons/eV), while the bottom orbitals of CBs between 0.80 eV and 2.20 eV are mainly the contributions from Hbpt-2p states (15.8 electrons/eV). Together with the structural features of 1, the origin of the maximum emission band of 1 can be ascribed to metal-to-ligand charge transfer (MLCT) where electrons are transferred from Cu(I) cations (Cu 3d states, VBs) to the unoccupied π* orbitals of the Hbpt ligands (Hbpt-2p states, CBs).

Packing diagram of cationic\([\text{Cu}(\text{Hbpt}){{(\text{C}{{\text{H}}_{\text{3}}}\text{CN})}_{\text{2}}}]_{n}^{n+}\) chains and \(\text{PF}_{6}^{-}\) anions in 1 along the a axis. Hydrogen atoms are omitted for clarity.

Measured (1) and simulated (2) powder XRD patterns of 1.

Solid-state photoluminescence spectra of 1 and the Hbpt ligand at room temperature. Inset: excitation spectrum of 1.

DOS (total and partial) of 1. The Fermi level is set at 0 eV.
CONCLUSIONS
In summary, we have synthesized a copper(I) coordination compound constructed by cationic \([\text{Cu}(\text{Hbpt}){{(\text{C}{{\text{H}}_{\text{3}}}\text{CN})}_{\text{2}}}]_{n}^{n+}\) chains and \(\text{PF}_{6}^{-}\) anions with abundant supramolecular interactions. The compound can emit strong red emission which probably originates from metal-to-ligand charge transfer.
ADDITIONAL INFORMATION
Additional plots of the structure, DFT calculation methodology, computational descriptions/explanations, and the FTIR spectrum are given as supplementary materials available online.
REFERENCES
M. P. Suh, Y. E. Cheon, and E. Y. Lee. Coord. Chem. Rev., 2008, 252, 1007-1026. https://doi.org/10.1016/j.ccr.2008.01.032
R. J. Kuppler, D. J. Timmons, Q. R. Fang, J. R. Li, T. A. Makal, M. D. Young, D. Yuan, D. Zhao, W. Zhuang, and H. C. Zhou. Coord. Chem. Rev., 2009, 253, 3042-3066. https://doi.org/10.1016/j.ccr.2009.05.019
Y. J. Cui, Y. F. Yue, G. D. Qian, and B. L. Chen. Chem. Rev., 2012, 112, 1126-1162. https://doi.org/10.1021/cr200101d
V. W. W. Yam and K. K. W. Lo. Chem. Soc. Rev., 1999, 28, 323-334. https://doi.org/10.1039/a804249g
X. Y. Tang, H. X. Li, J. X. Chen, Z. G. Ren, and J. P. Lang. Coord. Chem. Rev., 2008, 252, 2026-2049. https://doi.org/10.1016/j.ccr.2007.11.001
Z. N. Chen, N. Zhao, Y. Fan, and J. Ni. Coord. Chem. Rev., 2009, 253, 1-20. https://doi.org/10.1016/j.ccr.2007.11.015
W. Guo, S. Shu, T. Zhang, Y. Jian, and X. Liu. ACS Appl. Energy Mater., 2020, 3, 2983-2988. https://doi.org/10.1021/acsaem.0c00087
X. Liu, L. Li, Y. Z. Yang, and K. L. Huang. Dalton Trans., 2014, 43, 4086-4092. https://doi.org/10.1039/C3DT53219D
X. Liu and K. L. Huang. Inorg. Chem., 2009, 48, 8653-8655. https://doi.org/10.1021/ic900611u
X. Liu, Z. Zhao, C. H. Wang, S. Fu, and K. L. Huang. RSC Adv., 2017, 7, 40632-40639. https://doi.org/10.1039/C7RA07061F
L. Li, C. H. Wang, X. L. Zhang, and X. Liu. Eur. J. Inorg. Chem., 2015, 859-863. https://doi.org/10.1002/ejic.201403037
C. H. Wang, X. Liu, Y. Z. Yang, and K. L. Huang. Inorg. Chim. Acta, 2013, 407, 116-120. https://doi.org/10.1016/j.ica.2013.07.044
X. Hei and J. Li. Chem. Sci., 2021, 12, 3805-3817. https://doi.org/10.1039/D0SC06629J
A. V. Artemev, M. P. Davydova, X. Hei, M. I. Rakhmanova, D. G. Samsonenko, I. Y. Bagryanskaya, K. A. Brylev, V. P. Fedin, J. Chen, M. Cotlet, and J. Li. Chem. Mater., 2020, 32, 10708-10718. https://doi.org/10.1021/acs.chemmater.0c03984
A. V. Artemev, M. P. Davydova, A. S. Berezin, M. R. Ryzhikov, and D. G. Samsonenko. Inorg. Chem., 2020, 59, 10699-10706. https://doi.org/10.1021/acs.inorgchem.0c01171
T. Zhang, Z. Zhao, Y. Tang, J. S. Liu, G. L. Huang, and X. Liu. Z. Naturforsch. B, 2021, 76, 79-84. https://doi.org/10.1016/j.clinbiochem.2020.10.011
M. Shao, M. Li, Z. Wang, X. He, and H. Zhang. Cryst. Growth Des., 2017, 17, 6281-6290. https://doi.org/10.1021/acs.cgd.7b00967
S. Hu, D. Lin, Z. Xie, C. Zhou, W. He, and F. Yu. Transition Met. Chem., 2015, 40, 623-629. https://doi.org/10.1007/s11243-015-9955-9
F. P. Huang, J. L. Tian, G. J. Chen, D. D. Li, W. Gu, X. Liu, S. P. Yan, D. Z. Liao, and P. Cheng. CrystEngComm, 2010, 12, 1269-1279. https://doi.org/10.1039/B915506F
CrystalClear: Software Users Guide. Tokyo, Japan: Molecular Structure Corporation, Rigaku Corporation, 2002.
G. M. Sheldrick. SHELXTL, Reference Manual. Madison, Wisconsin: Siemens Analytical X-ray Instruments Inc., 1994.
G. M. Sheldrick. Acta Crystallogr., Sect. A, 2015, 71, 3-8. https://doi.org/10.1107/S2053273314026370
G. M. Sheldrick. Acta Crystallogr., Sect. C, 2015, 71, 3-8. https://doi.org/10.1107/S2053229614024218
X. Liu, K. L. Huang, G. M. Liang, M. S. Wang, and G. C. Guo. CrystEngComm, 2009, 11, 1615-1620. https://doi.org/10.1039/b902121c
X. Liu, W. J. Ru, and Z. Zhao. Z. Naturforsch., B, 2017, 72, 167-170. https://doi.org/10.1515/znb-2016-0192
S. Fu, B. Fu, Z. Zhao, and X. Liu. Z. Naturforsch., B, 2018, 73, 85-89. https://doi.org/10.1515/znb-2017-0171
Funding
We gratefully acknowledge the financial support of the National Natural Science Foundation of China (21071156) and the Natural Science Foundation of Chongqing (cstc2019jcyj-msxmX0170) and the National College Student′s Innovative Entrepreneurship Iraining Program (202110637007).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflict of interests.
Additional information
Russian Text © The Author(s), 2022, published in Zhurnal Strukturnoi Khimii, 2022, Vol. 63, No. 1, pp. 5-7.https://doi.org/10.26902/JSC_id84800
Supplementary material
Rights and permissions
About this article

Cite this article
Tao, Y.L., Wang, C.H., Xiang, H.Y. et al. A COPPER(I) COORDINATION COMPOUND WITH RED PHOTOLUMINESCENCE. J Struct Chem 63, 1–8 (2022). https://doi.org/10.1134/S0022476622010012
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0022476622010012