
Abstract
A combined experimental and theoretical study on a new complex [NiL(NCS)], derived from the Schiff base ligand HL, (E)-2-methoxy-6-(((pyridin-2-ylmethyl)imino)methyl)phenol is described herein. X-ray structure reveals that metal center of the complex has a square planar environment with a N3O donor set from the ligand and terminal thiocyanate anion. In the solid state, π–π stacking interactions lead to a one-dimensional arrangement with alternating short and long Ni…Ni distances. The different isomeric forms of the ligand can provide alternative molecular structures for the nickel(II) complex. Density functional theory calculations at B3LYP/6-311+G(d,p) level were performed to elucidate the structure and stability of the possible isomeric forms of the complex.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
The chemistry of nickel complexes plays a significant role in bioinorganic chemistry providing biomimic models for active sites of metalloenzymes such as carbon monoxide dehydrogenase, cytochrome-c3 hydrogenase, urease etc [1-3]. Such complexes have been extensively studied in connection to catalytic redox reactions, catechol oxidase activity, and alkaline phosphatase reactivity [4-6]. Synthesis of mononuclear Ni(II) complexes of Schiff base ligands are relevant since they can function as efficient antimicrobial, antifungal and anticancer agents [7-9]. In this regard, monocondensed NOO or NNO donor ligands react readily with nickel salts to form square planar nickel(II) complexes occupying three of their equatorial coordination sites. The fourth coordination site of the square plane can be coordinated to several neutral or anionic ligands. Pseudohalides, especially azide, thiocyanate and cyanate, act as suitable coligands utilizing their versatile coordination modes [10]. These mononuclear complexes are considered as “metalloligand” which can coordinate to another metal ion through oxygen atoms leading to phenoxo-bridged binuclear and trinuclear complexes.
The o-hydroxy aromatic imines a represent a leading group of ligands in coordination chemistry, producing very stable complexes with fascinating physicochemical, biological, and pharmacological properties. The keto-amine/enolimine tautomerism appears in o-hydroxy Schiff bases due to the intramolecular proton transfer from the oxygen to the nitrogen atom [11, 12]. Identification of the influence of isomerism on chemical and biological properties of molecules is a major task in pharmacophore-based virtual screening and drug discovery programs. Recently, computational approaches, including density functional theory (DFT) methods have been used for predicting electronic structures, geometrical parameters, and spectral properties since they can reproduce efficiently the experimental structures and provide vital information. These theoretical methods have gained considerable importance for understanding the tautomeric forms of DNA and RNA bases, heterocycles and Schiff bases, both in solution and solid state [13-16].
Combined experimental study and DFT insight on isomeric forms of metal chelates has become an emerging field of research [17-21]. However, such studies are still scarce for metal chelates as compared to the organic molecules, and this has prompted us to undertake a research in this area. In continuation of our previous work [22], we investigate here the structural aspects of a nickel(II) complex with particular emphasis on the stability of possible isomeric structures. Herein, we report the synthesis of a new nickel(II) complex [NiL(NCS)] (1) from (E)-2-methoxy-6-(((pyridin-2-ylmethyl)imino)methyl)phenol (HL) and thiocyanate co-ligand (Scheme 1). The square-planar geometry around the central Ni(II) atom in complex 1 is confirmed by spectroscopic characterization and X-ray crystal structure. Molecular geometries of all the possible structures of ligand and complex are fully optimized using B3LYP/6-311+G(d,p) theoretical level, both in gas phase and methanol solution, to evaluate their stability and the energy gap between the frontier molecular orbitals.

Synthesis of the ligand and the complex.
EXPERIMENTAL
Materials. All the chemicals and solvents employed for the syntheses were of analytical grade. 2-Picolylamine (or 2-aminomethylpyridine) and o-vanillin were purchased from Aldrich Chemical Company. Nickel(II) perchlorate hexahydrate was prepared by treating an aqueous suspension of nickel carbonate with 80% perchloric acid (E. Merck, India). It was then filtered through a fine glass-frit and preserved in a CaCl2 desiccator.
Caution! Perchorate salts are potentially explosive and should be handled with much care and in small amount, though no problem was encountered.
Physical measurements. The FTIR spectra of the ligand and the complex were recorded on a PerkinElmer RX I FTIR spectrometer with KBr pellets in the range 4000-400 cm–1. The electronic spectrum of the complex in HPLC grade acetonitrile was recorded at 300 K on a PerkinElmer LAMBDA 40 UV-Vis spectrophotometer in a 1 cm quartz cuvette in the range 200-800 nm. C, H, and N microanalyses were carried out with a PerkinElmer 2400 II elemental analyzer.
Synthesis of [NiL(NCS)] (1). HL was prepared following a reported method [23]. A methanolic solution of HL (0.291 g, 1 mmol) was added dropwise to a clear solution of Ni(ClO4)2·6H2O (0.366 g, 1 mmol) in 25 mL methanol and immediately an intense brown color developed. The solution was heated to boiling followed by the addition of sodium thiocyanate (0.081 g, 1 mmol) in a minimum volume of methanol. The solution, on being concentrated in the open atmosphere, yielded brown rectangular plate-shaped single crystals after 5 days. Anal. calcd (%) for C15H13N3NiO2S (FW: 358.05 g/mol) C 50.32, H 3.66, N 11.74. Found (%): C 50.28, H 3.71, N 11.58.
Crystal structure determination. Intensity data of 1 were collected at 133(2) K using MoKα radiation (λ = 0.71069 Å with a Bruker APEX-II CCD diffractometer. Cell refinement and data reduction were performed with the Bruker SAINT program [24]. The data set was collected using the ω scan mode over the 2θ range up to 53°. The structure was solved by direct methods using the SHELXS-97 program [25] and refined by full-matrix least-squares methods with SHELXL-2018/3 programs [26]. The non-hydrogen atoms were refined with anisotropic factors. All hydrogen atoms were positioned geometrically and treated as riding on the bound atom. Selected crystallographic data and structural refinements for the complex are summarized in Table 1. CCDC 2013418 contains the crystallographic data for complex 1. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html, or from the Cambridge Crystallographic Data Center, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: (+44) 1223 336 033; or E-mail: deposit@ccdc.cam.ac.uk.
Computational methods. The density functional theory (DFT) with Becke′s three parameter (B3) [27] exchange functional, along with the Lee–Yang–Parr (LYP) [28, 29] nonlocal correlation functional (B3LYP), was applied in this investigation. The basis set used was the standard valence triple zeta basis set, augmented by diffuse functions, d-type polarization functions for heavy elements, and p-type polarization functions for H, noted as 6-311+G(d,p) [30]. The molecular geometries of the studied species have been fully optimized by analytical gradient techniques. In the analysis of harmonic vibrational frequencies, the force constants were determined analytically for all the considered species. The stationary structures were confirmed by harmonic frequencies being all positive. The polarizable continuum model (PCM) self-consistent reaction field of Tomasi and co-workers [31] was employed to evaluate the solvent effects (with a dielectric constant of 32.613 to mimic the solvent methanol) at the same calculation level. The Gaussian-16 A03 package of programs [32] was used for all the computations.
RESULTS AND DISCUSSION
The crystal structure of the Schiff base ligand has been previously established, which shows the molecule to exist in the enol-imine form rather than in the keto-amine form in the solid state [23]. Interestingly, we find that another structural isomer of the ligand, denoted as HLb is also possible (Scheme 2). All these isomeric forms of the ligand can provide alternative molecular structures for the nickel(II) complex described here. Therefore, it was of importance to perform a comparative theoretical study on the stability of different isomeric forms of this metal complex.

Different isomeric forms of the ligand.
IR and UV-Vis spectra. In the IR spectra of the ligand the characteristic azomethine absorption band is obtained at 1640 cm–1 but for the complex this band is observed at 1611 cm–1. The shift of this band upon complexation towards lower wavenumbers is due to the coordination of the imine nitrogen atom to the metal ion [33]. The lowering of the phenolic C–O stretching frequency to 1225 cm–1 in the spectrum of the complex provides evidence for coordination to the metal ion through the deprotonated phenolic oxygen atom. New bands at 452 cm–1 and 377 cm–1 are observed only in the spectra of the complex, which can be attributed to ν(Ni–N) and ν(Ni–O), respectively [34]. The sharp single peak at 2084 cm–1 corresponds to the N-bonded thiocyanate as the terminal ligand [35].
The electronic spectrum of complex 1 in the HPLC grade acetonitrile solvent shows a low-intensity broad band around 550 nm, which is a typical d–d band for Ni(II) in the square-planar environment with a diamagnetic nature [36]. The intense band at 380 nm region can be attributed to a ligand→metal charge transfer transition (LMCT) [37]. The bands in the region 245-290 nm are due to the π–π* electronic transition within the ligand.
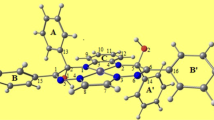
Crystal structure of [Ni(L)NCS] (1). The perspective view of complex 1 with the atom labeling scheme is shown in Fig. 1 and selected bond lengths and angles are summarized in Table 2. The crystal structure has two independent molecules (A and B) in the asymmetric unit, with similar geometrical features. During complexation, yhe enol form of the Schiff base ligand (L–) coordinates central Ni(II) (Ni1 or Ni2) using the NNO donor set. A terminal thiocyanato ligand completes the coordination around the nickel(II) center giving rise to a square planar geometry. Slight distortion in the square plane is manifested in the deviations of the trans angles from the ideal value of 180° (Table 2). The dihedral angle between Nimine/Ni/NPy and O/Ni/Nthio planes is 3.62° for A and 4.94° for B as compared with 0° for a perfect square-planar geometry, which indicates a minor “twist” in the square towards the tetrahedral arrangement. The deviations of the central nickel and other concerned atoms from the mean N3O plane are statistically insignificant (within 0.038 Å for A and within 0.052 Å for B). The terminal thiocyanate ligand is almost linear and shows a bent coordination mode with metal. The five-membered and six-membered rings formed by HL at the nickel(II) center lie almost in the same plane (intraplanar angle: 3.07° for A and 3.4° for B), with chelate bite angles of ≈85° and ≈95° respectively [38]. The Ni–N(Py) bond length in the present complex is longer than the Ni–N(azomethine) bonds, comparing well with those observed for other four-coordinate nickel(II) complexes of related ligands [39]. This variation in bond lengths despite of presence of sp2 N atoms as donors can be attributed to considerable π-back donation ability of the azo function. The Ni–N(thiocyanato) bond length agrees with the reported literature values for similar complexes [40].

ORTEP view of complex 1 (A and B) with the atom labelling scheme. Thermal ellipsoids are drawn at the 40% probability level. Dashed lines represent hydrogen bonds.
The two crystallographically independent molecules form a dimeric unit through C–H⋯O intermolecular hydrogen bonding via the uncoordinated –OCH3 side arms of the ligands (Fig. 1). The hydrogen bonding parameters are listed in Table 3. In the solid state, π–π stacking interactions involving the pyridine and phenolic rings of the neighbouring molecules give rise to a one-dimensional supramolecular substructure (Fig. 2). In the case of molecule A, two types of stacks with alternating short and long interplanar distances are observed with Ni⋯Ni distances 3.554(4) Å and 4.870(4) Å, respectively and the corresponding centroid-to-centroid ring distances are 3.510(8) Å and 3.781(8) Å, respectively. For molecule B, the inter-unit Ni⋯Ni distances are similar (3.638(5) Å and 3.654(5) Å) and the centroid-to-centroid ring distances (3.831(11) Å and 3.527(11) Å) are as expected for stacks [41].

Unit cell packing diagram of 1 showing one-dimensional arrangements via π–π stacking.
Theoretical calculations. The three possible isomeric forms for the Schiff base ligand (including enol-imine forms: HL and HLb, and keto-amine form: HLa) were fully optimized at the B3LYP/6-311+G(d,p) level in both gas phase and methanol solution (PCM model) (as shown in Fig. 3). Our calculation results suggest that the enol form HL is most stable in both gas phase and methanol solvent. In the gas phase, HL is 3.5 kcal/mol and 5.6 kcal/mol more stable than HLa and HLb, respectively. In the methanol solvent, HL is 5.1 kcal/mol more stable than HLb and slightly (0.3 kcal/mol) more stable than HLa. All the energies discussed here are zero-point corrected.
For the ligand-nickel compounds, two structures were explored by a computational study (Fig. 4). The HL–Ni complex is the compound corresponding to the HL ligand and it assumes a planar structure. The C1C2O3Ni4 dihedral angle is 180° in both gas phase and methanol solvent. The HLb–Ni complex that corresponds to the HLb ligand adopts a non-planar structure. Its C1C2O3Ni4 dihedral angle of atoms is predicted to be 133° and 136° in gas phase and methanol solvent, respectively. Moreover, the HL–Ni complex with the enol form ligand is more stable than its isomeric HLb–Ni complex by 15.9 kcal/mol and 13.3 kcal/mol, in gas phase and methanol solution, respectively. Hence, HL–Ni is the favoured form of the Schiff base ligand binding the nickel(II) complex, which also agrees well with the experimentally determined crystallographic structure.

Optimized structures of the ligands at the B3LYP/6-311+G(d,p) level (PCM model, solvent = methanol).

Optimized structures of the ligand-nickel(II) compounds at the B3LYP/6-311+G(d,p) level (PCM model, solvent = methanol).

Frontier molecular orbitals in the ligands and the complexes with B3LYP/6-311+G(d,p) optimized geometries.
The frontier orbital gap helps to characterize the chemical reactivity and kinetic stability of the molecules. The HOMO, LUMO, and HOMO–LUMO gap energies for the optimized forms of the ligand and the complex are calculated at the B3LYP/6-311+(d,p) level. The HOMO–LUMO gap was calculated to be 4.23 eV and 4.33 eV for HL ligand in gas phase and solvent, respectively. However, the HOMO–LUMO gaps for HL–Ni are significantly altered after complexation. The calculated values amount to 2.89 eV in gas phase and 3.59 eV in solvent.
It can be seen from the plot of HOMO and LUMO in Fig. 5 that the electrons reside around the methoxy phenol ring for both HOMO and LUMO of the Schiff base ligands HL. The HOMO of the nickel-ligand complexes (HL–Ni) is similar to that of the ligands with electrons occupying the methoxy phenol ring. However, it is noted that the electrons shifted to the pyridine rings at the LUMO of the complex. This provides an evidence of the electron transfer upon the complexation of nickel(II).
CONCLUSIONS
The Schiff base ligand may exist in two enol-imine forms (HL and HLb) and one keto-amine form (HLa). The enol form HL is most stable in both gas phase and methanol solvent, The HL ligand containing the NNO donor set was successfully coordinated to nickel(II). The corresponding crystallographic data show that the chelate [Ni(L)NCS] has a square planar coordination structure and this is supported through the theoretical calculations.
ACKNOWLEDGMENTS
S. Thakurta gratefully acknowledges Jadavpur University, Kolkata, India for infrastructural support.
REFERENCES
H. Dobbek, V. Svetlitchnyi, L. Gremer, R. Huber, and O. Meyer. Science, 2001, 293, 1281-1285.
A. Volbeda, M. H. Charon, C. Piras, E. C. Hatchikian, M. Frey, and J. C. Fontecilla-Camps. Nature, 1995, 373, 580-587.
E. Jabri, M. B. Carr, R. P. Hausinger, and P. A. Karplus. Science, 1995, 268, 998-1004.
Ş. Özalp-Yaman, V. T. Kasumov, and A. M. Önal. Polyhedron, 2005, 24, 1821-1828.
R. Sanyal, S. K. Dash, P. Kundu, D. Mandal, S. Roy, and D. Das. Inorg. Chim. Acta, 2016, 453, 394-401.
V. K. Bhardwaj and A. Singh. Inorg. Chem., 2014, 53, 10731-10742.
J. Adhikary, P. Kundu, S. Dasgupta, S. Mukherjee, S. Chattopadhyay, G. Aullón, and D. Das. Polyhedron, 2015, 101, 93-102.
H. Bahron, S. S. Khaidir, A. M. Tajuddin, K. Ramasamy, and B. M. Yamin. Polyhedron, 2019, 161, 84-92.
M. Salehi, F. Rahimifar, M. Kubicki, and A. Asadi. Inorg. Chim. Acta, 2016, 443, 28-35.
S. K. Dey, N. Mondal, M. S. El Fallah, A. Escuer, X. Solans, T. Matsushita, V. Gramlich, and S. Mitra. Inorg. Chem., 2004, 43, 2427-2434.
E. Temel, Ç. Albayrak, O. Büyükgüngör, and M. Odabaşoğlu. Acta Crystallogr., Sect. E, 2006, 62, o4484-o4486.
J. W. Ledbetter. J. Phys. Chem., 1966, 70, 2245-2249.
M. K. Shukla and J. Leszczynski. Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2013, 3, 637-649.
K. Jana and B. Ganguly. ACS Omega, 2018, 3, 8429-8439.
A. Kanaani, D. Ajloo, G. Grivani, A. Ghavami, and M. Vakili. J. Mol. Struct., 2016, 1112, 87-96.
I. Khosravi, F. Hosseini, M. Khorshidifard, M. Sahihi, and H. A. Rudbari. J. Mol. Struct., 2016, 1119, 373-384.
A. Karahan, S. Karabulut, H. Dal, R. Kurtaran, and J. Leszczynski. J. Mol. Struct., 2015, 1093, 1-7.
M. Kaur, H. Kaur, A. Kapila, and Reenu. J. Mol. Struct., 2019, 1185, 57-68.
C.-H. Wei, C.-E. Wu, Y.-L. Huang, R.G. Kultyshev, and F.-E. Hong. Chem. Eur. J., 2007, 13, 1583-1593.
S. Bhikraj, O. Q. Munro, and M. P. Akerman. Polyhedron, 2017, 124, 22-29.
M. Enamullah, and C. . Inorg. Chim. Acta, 2020, 501, 119245.
S. Thakurta, R. J. Butcher, A. Frontera, and S. Mitra. J. Coord. Chem., 2017, 70, 3715-3726.
S. Chithiraikumar and M. A. Neelakantan. J. Mol. Struct., 2016, 1108, 654-666.
Bruker SAINT and SHELXTL. Bruker AXS Inc.: Madison, Wisconsin, USA, 1999.
G. M. Sheldrick. SHELXS-97: Program for the Solution of Crystal Structures. University of Göttingen: Germany, 1997.
G. M. Sheldrick. Acta Crystallogr., Sect. C, 2015, 71, 3-8.
A. D. Becke. J. Chem. Phys., 1993, 98, 5648-5652.
C. Lee, W. Yang, and R. G. Parr. Phys. Rev. B, 1988, 37, 785-789.
B. Miehlich, A. Savin, H. Stoll, and H. Preuss. Chem. Phys. Lett., 1989, 157, 200-206.
W. J. Hehre, L. Radom, P. R. Schleyer, and J. A. Pople. Ab Initio Molecular Orbital Theory. Wiley: New York, 1986.
M. Cossi, V. Barone, R. Cammi, and J. Tomasi. Chem. Phys. Lett., 1996, 255, 327-335.
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox. Gaussian 16, Revision A.03. Gaussian, Inc.: Wallingford, CT, 2016.
M. T. H. Tarafder, A. Kasbollah, K. A. Crouse, A. M. Ali, B. M. Yamin, and H. K. Fun. Polyhedron, 2001, 20, 2363-2370.
S. Thakurta, J. Chakraborty, G. Rosair, R. J. Butcher, and S. Mitra. Inorg. Chim. Acta, 2009, 362, 2828-2836.
R. Kapoor, A. Kataria, A. Pathak, P. Venugopalan, G. Hundal, and P. Kapoor. Polyhedron, 2005, 24, 1221-1231.
P. Seth, L. K. Das, M. G. B. Drew, and A. Ghosh. Eur. J. Inorg. Chem., 2012, 2012, 2232-2242.
M. Rajasekar, S. Sreedaran, R. Prabu, V. Narayanan, R. Jegadeesh, N. Raaman, and A. K. Rahiman. J. Coord. Chem., 2010, 63, 136-146.
R. Kannappan, S. Tanase, I. Mutikainen, U. Turpeinen, and J. Reedijk. Inorg. Chim. Acta, 2005, 358, 383-388.
S. Seth and S. Chakraborty. Acta Crystallogr., Sect. C, 1984, 40, 1530-1533.
S. Shit, S. Sen, S. Mitra, and D. L. Hughes. Transition Met. Chem., 2009, 34, 269-274.
C. Janiak. J. Chem. Soc., Dalton Trans., 2000, 3885-3896.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflict of interests.
Additional information
Text © The Author(s), 2021, published in Zhurnal Strukturnoi Khimii, 2021, Vol. 62, No. 6, pp. 1007-1015.https://doi.org/10.26902/JSC_id74431
Rights and permissions
About this article

Cite this article
Thakurta, S., Maiti, M., Butcher, R.J. et al. COMBINED EXPERIMENTAL AND COMPUTATIONAL APPROACH TO THE STRUCTURE OF A NEW NICKEL(II) COMPLEX WITH TRIDENTATE SCHIFF BASE LIGAND. J Struct Chem 62, 938–946 (2021). https://doi.org/10.1134/S0022476621060135
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0022476621060135