
Abstract
Neutrophils are one of the main cells of innate immunity that perform a key effector and regulatory function in the development of the human inflammatory response. Apoptotic forms of neutrophils are important for regulating the intensity of inflammation and restoring tissue homeostasis. This review summarizes current data on the molecular mechanisms of modulation of neutrophil apoptosis by the main regulatory factors of the inflammatory response—cytokines, integrins, and structural components of bacteria. Disturbances in neutrophil apoptosis under stress are also considered, molecular markers of changes in neutrophil lifespan associated with various diseases and pathological conditions are presented, and data on pharmacological agents for modulating apoptosis as potential therapeutics are also discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
During the evolution of the human innate immunity system, a mechanism for recognizing the signals of potential damage to structural tissue components was formed, represented by evolutionarily conserved pattern recognition receptors (PRR) of immunocompetent cells. The ability of these receptors to recognize pathogen-associated molecular patterns (PAMP), as well as danger-associated molecular patterns (DAMP), released from the destroyed extracellular matrix and damaged or dying cells, is closely related to the formation of an acute inflammatory response at the site of infection or damage [1–3]. Unlike other granular leukocytes, neutrophils express a huge variety of PRRs [2], and it is these effector cells that are one of the first to respond to danger signals, migrate from the blood vascular system to the site of damage, identify the stimulus, and choose between the optimal ways to neutralize it, such as a generation of reactive oxygen species (ROS), degranulation, phagocytosis, or the formation of neutrophil extracellular traps (NETs, i.e. NETosis) [1]. At each stage of the acute inflammatory response, neutrophils play one of the key roles as effector and regulatory cells in the process, while the time and form of their death are critical for a full-fledged resolution of the inflammatory process and restoration of homeostasis [1, 4].
Apoptosis is the main form of neutrophil death, and the mechanism of its induction is not unique, retaining the basic features inherent to all cells [5]. For example, binding of extracellular ligands FASL, TNF or TRAIL to the corresponding membrane “death receptors” FAS, TNF-R1, TRAIL-R1/2 activates cytoplasmic “death domains” FADD or FADD/TRADD and triggers a multistep cascade of intracellular molecular interactions, the key role in which is assigned to caspases-8 and -3, protein kinases of the MAPK family, and regulatory proteins of the Bcl-2 family, some of which exhibit proapoptotic properties (Bad, Bim, Bax, Bid, Bak), while others have a protective effect (Mcl-1, A1, Bcl-XL) [6–8] (Fig. 1).
Intracellular triggers induce the apoptosis pathways mediated by the endoplasmic reticulum or mitochondria [7–10]. Mitochondria-mediated apoptosis depends on the dominance of pro- apoptotic Bcl-2 proteins in the cytoplasm, the functional state of mitochondrial membranes, and the activity of caspase-9 [6, 8, 9]. In apoptosis mediated by the endoplasmic reticulum (ER), the pivotal role is played by the so-called “unfolded protein response” which triggers a caspase-independent specific pathway of programmed neutrophil death [10]. Like other phagocytic cells, neutrophils are able to provide the phagocytosis-induced apoptotic program [7] (Fig. 1). Apoptotic forms of neutrophils lose their functional activity, acquire specific morphological characteristics, and are eliminated from the circulation by phagocytic cells of the bone marrow, lungs, or liver [1, 11].
Stress is a highly conserved biological response to alarm or threat signals, related with an evolutionarily ancient system of innate immunity and crucial for organism’s survival [12]. Differently directed changes in the neutrophil lifespan, associated with the implementation of the protection program, occur during both inflammation and stress. Disturbances in neutrophil apoptosis as a factor that regulates their functional activity are of pathogenetic significance during the formation of chronic diseases of various etiologies.

Molecular regulation of neutrophil apaptosis medated by death receptors (FAS, TNF-R1, TRAILR), phagocytosis, and endoplasmic reticulum stress. Apoptosis suppression pathways are highlighted in red.
REGULATION OF NEUTROPHIL APOPTOSIS IN ACUTE INFLAMMATORY PROCESSES
Under physiological conditions, the lifespan of neutrophils is limited to 12–18 h in the circulatory system and 1–4 days in tissues [1, 8, 11, 13, 14]. With the development of an inflammatory response characterized by neutrophil activation, the lifespan can increase several times, allowing the immune system to make the most of the functional potential of neutrophils [1]. After completion of the acute phase of inflammation, spent neutrophils undergo apoptosis, and the engulfment of apoptotic bodies by macrophages directly at the site of inflammation serves as a signal for switching from a pro-inflammatory to an anti-inflammatory program with subsequent restoration of tissue homeostasis [4, 15]. In this case, the lifespan of neutrophils is of critical importance and is modulated by a complex of various signals, including the factors of the cellular metabolic status and extracellular milieu [9, 16, 17].
It has been shown that the molecular mechanisms for apoptosis suppression are already activated during the influx of neutrophils into the site of damage. It was noted that transepithelial migration of these cells is accompanied by a decrease in the expression of procaspases-3, -6, -7, and -8, as well as procaspase-3 and -8 mRNA [18], and direct in vitro contact of neutrophils with the endothelial monolayer inhibits the development both of spontaneous and Fas- or TNFR-mediated apoptosis [19]. During transendothelial migration toward the site of inflammation, neutrophils receive an apoptosis suppression signal through integrin interaction, while pro-inflammatory cytokines, growth factors, PAMP, DAMP and other molecular regulators serve as additional stimuli to prolong the lifespan of neutrophils in the acute phase of inflammation [1, 2, 8]. In patients with severe pneumonia, a comparison of BAL fluid neutrophils with their circulatory counterparts revealed a quantitative predominance of antiapoptotic proteins Bcl-XL and A1 of the Bcl-2 family in BAL fluid neutrophils [20]. At the resolution phase of inflammation, cytokines with anti-inflammatory properties make a significant contribution to the modulation of neutrophil apoptosis [1, 17].
Integrins as factors of apoptosis regulation
Integrins are transmembrane heterodimeric receptors for adhesion molecules and extracellular matrix proteins. Different α and β subunits are expressed on the cell surface, forming integrins with different specificity for ligands. αLβ2, αMβ2, αVβ3, and α9β1 integrins are expressed on the plasma membrane of neutrophils [21] with a predominance of αMβ2 or Mac-1 (CD11b/CD18, ITAM antigen) belonging to the β2 integrin subfamily [22, 23]. Mac-1 activation by intercellular adhesion molecules-1 (ICAM-1) located on the surface of capillary endothelial cells, makes a significant contribution to increasing the viability of neutrophils during their transendothelial migration to the site of inflammation [23]. Binding of Mac-1 to such proteins of neutrophil azurophilic granules as fibrinogen, plasminogen and myeloperoxidase is also accompanied by a delay in spontaneous apoptosis [22, 24, 25]. It has been shown that a co-stimulatory signal of β2-integrin is required to activate the transcriptional activity of NF-κB upon exposure of neutrophils to inflammatory cytokines IL-8 and GM-CSF [26]. The mechanism of signal transduction from ligated integrins is associated with the activation of tyrosine kinases from the Src and Syk families and focal adhesion kinases, specifically Pyk2 [27]. In neutrophils, the kinases Syk and Pyk2 modulate the activity of the kinases Akt and p38 MAPK [28]. A ligand interaction with Mac-1 causes its clustering and activation of Akt and ERK1/2, as well as factors that block mitochondrial sequestration [24, 29]. In addition, it has been shown that soluble fibrinogen promotes the translocation of NF-κB into the nucleus by activating a cytoplasmic inhibitor of the degradation of this factor [29]. It has also been found that fibrinogen- or plasminogen-induced activation of the ERK1/2- and Akt-mediated cell survival pathways requires a recruitment of both αMβ2 integrin subunits [24]. While binding Mac-1 to myeloperoxidase, in addition to the activation of ERK1/2 and Akt, there has been noted an accumulation of the antiapoptotic factor Mcl-1 of the Bcl-2 family in the cytoplasm, a prevention of mitochondrial dysfunction, and a maintenance of effector caspase-3 in an inactive state [25]. Nevertheless, it has been shown that thioglycollate-induced neutrophils isolated from the mice deficient in the CD11b gene (CD11b–/–) exhibit a delayed apoptosis [30], which suggests the existence of other integrin-mediated pathways for regulating this process. For example, it is currently known that binding of integrin α9β1, belonging to the β1 subfamily, by vascular cell adhesion molecules-1 (VCAM-1 or CD106) leads to a delay not only in spontaneous but also Fas-induced neutrophil apoptosis, and is accompanied by stimulation of the PI3K-mediated cell survival pathway and activation of the transcription factor NF-κB [21].
The involvement of neutrophils in β2-integrin interaction can not only suppress but also activate neutrophil apoptosis. It was established that, in this case, the lifespan of cells depends on the functional activity of the integrin receptor and co-stimulatory effects of some cytokines. Binding of the β2 integrin subunit to ligands at a simultaneous action of such proapoptotic factors as FasL, TNF, or ultraviolet radiation can accelerate neutrophil apoptosis [31]. Fibronectin-induced enhancement of TNFR-mediated neutrophil apoptosis depends on phosphorylation of the Ly-GDI protein, which is a GTPase regulator [32]. The possibility of Mac-1 binding to the iC3b component of the complement or the Fc receptor determines the interaction of this integrin with opsonized bacteria, which is accompanied by their phagocytosis [33] and, as a consequence, the induction of apoptosis [7]. It has been experimentally established that antibodies to Mac-1 block apoptosis mediated by phagocytosis of opsonized particles [30]. Integrins are involved in the intercellular interaction of neutrophils and macrophages. This interaction can lead both to the activation of apoptosis in neutrophils and their subsequent phagocytosis by macrophages [34, 35]. Thus, through the integrin receptors, neutrophil apoptosis can be modulated by various ligands—expressed on the surface of adjacent cells, located in the extracellular matrix, or dissolved.
Cytokines as factors of apoptosis regulation
Apart from the CR family of the TNF receptor superfamily (FAS, TNF-R1, and TRAIL-R1/2) associated with the “death domains” FADD and FADD/TRADD, the neutrophil plasma membrane has numerous receptors, the activation of which does not affect the “death domains” but exerts a multidirectional influence on various types of apoptosis during transduction of the intracellular signal on its way to effector caspase-3, a key “death molecule” [1, 36].
Many cytokines and growth factors playing an important role in the development of the inflammatory response can alter the lifespan of neutrophils in different ways. It was demonstrated that IL-1, IL-2, IL-3, IL-6, IL-8, IL-15, IL-18, IL-32γ, TNF, IFNg, IFNα, IFNβ, GM-CSF, G-CSF, MIF and IGF-1 are able to delay apoptosis, having an anti-inflammatory effect in the acute phase of inflammation [5, 11, 20, 36–45]. Of these factors, TNF and IL-6 have bifunctional properties, i.e. can activate neutrophil apoptosis under certain conditions [6, 8, 16, 41].
TNF is a ligand of the TNFR family, the molecular complex of which triggers both a proapoptotic pathway, mediated by caspase-8 and associated exclusively with TNF-R1, and an antiapoptotic pathway mediated by both types of receptors TNF-R1 and TNF-R2 [16, 36]. TNF-mediated modulation of neutrophil survival is accomplished by various factors [46], which eventually shift the balance of pro- and antiapoptotic signals in one or another direction, but to a greater extent depends on the TNF concentration in the incubation medium and oxygen tension in the extracellular milieu [8, 16].
The effect of most cytokines is mediated by type I and type II CRs. Growth factors G-CSF and GM-CSF are among the most studied type I CR ligands [1, 11, 36, 47]. The lifespan of neutrophils isolated from the blood of volunteers after a course of recombinant human G-CSF administration (10 µg/kg subcutaneously, once a day, for 7 days) was significantly higher than in the control group [48]. Both G-CSF and GM-CSF suppress Fas-mediated apoptosis of human neutrophils [49, 50], although G-CSF has no effect on mouse neutrophils [51].
Presumably, the main mechanism of the antiapoptotic effect of cytokines on neutrophils consists in altering the expression ratio of the Bcl-2 family proteins toward protective proteins. For example, GM-CSF, G-CSF, IL-3, IL-6, IL-15, and IFN-γ reduce Bax expression [5, 52]. GM-CSF and TNF cause PI3K-dependent phosphorylation and cytosolic Bad translocation [53]. G-CSF and GM-CSF maintain the expression of the cell survival proteins A1 and Mcl-1 [20, 47], but do not affect expression levels of other Bcl-2 family proteins (Bcl-XL, Bax, Bcl-w) both in mouse and human neutrophils [47, 51]. It is noteworthy that the data on the induction of Bim, Bax and Bcl-XL expression by the growth factors G-CSF and GM-CSF are contradictory [20, 47, 51], and it appears that the ambiguity of these results is due to such nuts and bolts of in vitro studies as the dose and duration of exposure to the above factors, as well as the degree of purification of the neutrophil population.
The antiapoptotic effects of pro-inflammatory cytokines on neutrophils can be mediated not only by a balance of the Bcl-2 family proteins but also by the functional activity of key protein-kinase cascades. Type I CRs (IL-4R, IL-6R, IL-12R, IL-15R, G-CSFR, GM-CSFR) and type II CRs (IFNAR, IFNBR, IFNGR, IL-10R) are constitutively associated with JAK, the substrate for which is are STAT proteins. Ligand binding to this type of receptors induces the activation of protein kinase cascades in the JAK-STAT signaling pathway, which regulates many cellular processes, including apoptosis [36]. Among the proteins of the JAK family, the key role in transduction of an antiapoptotic signal belongs to JAK2 [54] which triggers the JAK2 → STATs signaling pathway. The activation of this pathway has been shown for many type I and II CR ligands, including G-CSF, GM-CSF [54, 55], IL-15 [56], IFNα, and IFNγ [36, 57]. The features of the activation pathways for a particular type of CRs appear to be determined by a combination of receptor-specific variants of JAK and STAT due to a wide range of their molecular diversity. An important complementary role in the regulation of postreceptor signal transduction mechanisms is played by PI3K and its substrate Akt, nuclear transcription factor NF-κB, Bcl-2 family proteins, IAP-family inhibitors of apoptosis, and a number of other auxiliary enzymes [6, 36]. For example, when treating neutrophils with G-CSF, activation of the G-CSFR → JAK2 → STAT3/5 signaling pathway is accompanied by an increase in cIAP2 expression [55]. GM-CSF induces a delay in apoptosis through the signaling pathways mediated by ERK and PI3K/Akt [45], but not p38 MAPK [58]. At the same time, another type I CR ligand, IL-15, activates not only JAK2 and ERK but also p38 MAPK, which also leads to a delay in apoptosis [56].
Among the type II CR ligands, IFNα/β and IFNγ are of greatest interest. The IFNα- and IFNγ-induced prolongation of neutrophil survival is associated with the increased expression of cIAP2, but not cIAP1, Mcl-1, and A1 [57]. The antiapoptotic effect of IFNβ on neutrophils is due to the activation of PI3 and Cd protein kinases, as well as the translocation of NF-κB into the nucleus [59, 60]. It has also been shown that IFNγ inhibits Fas-mediated apoptosis in neutrophils in a healthy person [44].
Some interleukins that activate other types of cytokine receptors also have an antiapoptotic potential. They include primarilly IL-1α/β and IL-18, which are ligands of the IL-1R/TLR superfamily [36], actively involved in the regulation of the inflammatory process. In contrast to type I and type II CRs, upon activation of the IL-1R/TLR receptor complex, the signal is initiated by the adapter protein MyD88 and kinases of the IRAK family with subsequent transduction to the transcription factor NF-κB and MAPK [36]. It was found that IL-1β has a suppressive effect both on spontaneous [38] and Fas-induced apoptosis [50]. Incubation of a purified population of neutrophils from the blood of a healthy person with IL-18 is also accompanied by apoptosis suppression, and the mechanism that increases the lifespan of neutrophils is associated with an increase in the activity of the survival factor A1, but not Mcl-1, and is mediated by PI3K and ERK, but not p38MAPK pathways [61].
Some proteins with inflammatory cytokine properties are ligands for non-cytokine receptors. One of them is represented by the growth hormone IGF-1. It was established that IGF-1 causes a pronounced delay both in spontaneous and FAS-induced neutrophil apoptosis in a healthy person under in vitro conditions [44], while the mechanism of IGF-1 action is not concerned with Fas expression and caspase-8 activity but is associated with PI3K and the inhibitory, but not blocking, effect on mitochondrial membrane depolarization. In this case, the IGF-1 effectiveness is comparable to the effect of GM-CSF and IFN-γ, which inhibit Fas-mediated apoptosis by almost 50% [44].
The well-studied cytokine IL-8, which functionally belongs to the group of chemokines and is a ligand for the CXCR chemoreceptor of the GPCR family [36], not only enhances the influx of neutrophils into the site of inflammation, but also prolongs their survival [6]. It was revealed that exposure of cultured neutrophil to IL-8 delays both spontaneous and Fas- or TNF-induced apoptosis [6, 45, 50], and the cell survival signaling pathways are mediated by ERK/Akt [45]. Another, no less important, pleiotropic cytokine with chemokine-like activity and antiapoptotic effect, is MIF. It has been shown that the mechanism of the MIF effect on apoptosis in human neutrophils is associated with the prevention of cytochrome C and Smac release from mitochondria, which are involved in the activation of effector caspase-3, as well as with the maintenance of the proapoptotic factors Bid and Bax in an inactive state [40].
Among the cytokines released during the development of the resolution phase of inflammatory response, IL-4 and IL-10 are of particular interest as the most studied in terms of their effect on apoptosis [1, 17]. Neutrophils enter this phase in an already activated state, and their lifespan is increased by a complex of modulators. It has been shown that IL-10 abolishes in vitro suppression of spontaneous apoptosis mediated by the pro-inflammatory factors TNF, GM-CSF, G-CSF, IFNγ, or LPS but does not affect the survival of neutrophils being in the basal state [62, 63]. When studying the molecular mechanism of delayed neutrophil apoptosis reversal, S. Ward et al. concluded that transduction of the IL-10-mediated signal is associated with suppression of ERK activity but differs from the signal transduction pathways to ERK from the receptor complexes activated by GM-CSF or TNF [64]. The proapoptotic effect was also demonstrated when studying the effect of IL-4 on activated LPS and human neutrophils cultured under hypoxic conditions [17]. IL-10 and IL-4 are ligands for IL-10R and IL-4R, and most authors emphasize the importance of Jak1/STAT3 interactions in the transduction of a signal mediated by these interleukins [1, 17, 65].
Apoptosis modulation by bacterial components
Among several types of PRRs, one of the most studied are transmembrane TLRs, which are the members of the IL-1R/TLR receptor family. They are of great importance for recognizing various structural components of microorganisms with subsequent activation of a pronounced inflammatory response [36, 66]. By binding to TLRs, components of microorganisms can influence neutrophil apoptosis even in the absence of direct cell infection. All 10 types of the known TLRs are represented in human neutrophils, excluding TLR3 [7, 36], three of which are located in intracellular compartments (TLR-7, -8, -9), while the rest are expressed on the plasma membrane [66]. The most studied PAMPs of bacterial origin are considered to be membrane LPSs, LPs, PGs, and lipoteichoic acids, which have an antiapoptotic potential of a varying degree of manifestation [6, 7, 22, 61, 64, 67, 68]. It has been experimentally demonstrated that LPS, which is a component of the outer membrane of most gram-negative bacteria and, at the same time, a TLR4 ligand, delays spontaneous neutrophils apoptosis in vitro and in vivo [6, 22, 61, 64, 68], and that the extent of this effect is comparable to that of such inflammatory mediators as IL-18, G-CSF or GM-CSF [6, 61]. It has been shown that after LPS administration, rat lung neutrophils exhibit increased activities of PI3K, Akt, p38 MAPK, and ERK, as well as increased transcriptional activity of NF-κB and expression of the survival factor Mcl-1 [69].
LPs and PGs of bacterial cell membranes are the main TLR2 agonists that form a signal in a complex with TLR1 or TLR6 [36, 67]. TLR2 activation is typically accompanied by delayed apoptosis of varying severity [22, 68]. For example, LPs of gram-negative F. tularensis or PGs of gram-positive S. aureus, by binding to TLR2, can significantly increase the lifespan of neutrophils by modulating the apoptotic pathways [67, 68]. Currently, there are data indicating an interaction of bacterial PGs not only with TLR2 but also with intracellular PRRs of the NOD family. Specifically, muramyldipeptide, a NOD2 receptor agonist of gram-negative bacteria, has been shown to activate the classic pro-inflammatory neutrophil survival pathways mediated by the activation of NF-κB and MAPK [70].
Bacterial DNA, released during lysis or proliferation of bacteria, has all the immunological properties of PAMPs, while short Cp-DNA sequences are selectively recognized by neutrophil TLR9. TLR9-induced activation of bacterial DNA stimulates the functional activity of neutrophils, as manifested in the induction of the expression of chemokines and the integrin Mac-1, regulation of adhesion molecules, enhancement of phagocytic activity, and suppression of spontaneous apoptosis [66], which eventually contributes to the maintenance of the acute phase of the inflammatory response. The molecular mechanisms of antiapoptotic signaling of TLR agonists to neutrophils are mediated by the adapter protein MyD88 [66, 71] and associated with the activation of the PI3K/Akt and MEK/ERK pathways, triggering of NF-κB-mediated transcription of pro-inflammatory cytokine genes, as well as increasing the expression of survival factors A1 and Mcl-1 and decreasing the effector caspase-3 activity [8, 61, 64, 66, 68]. The mechanism of suppression of FasL-induced apoptosis by TLR ligands is less studied. We know that in mouse neutrophils, the cytoprotective effect caused by an interaction with TLR1/2, TLR2/6, or TLR4 is associated with inhibition of caspase-8 cleavage and does not affect the balance of Bcl-2 family proteins [71].
It is also worth noting that, in addition to the antiapoptotic effect of the bacterial structural components, toxins of some microorganisms, e.g., E. coli verotoxin, Shiga toxin, and phenol-soluble modulins, are also capable of prolonging the lifespan of neutrophils [11], thereby effectively increasing their count during the development of the acute phase of inflammation.
APOPTOSIS DISTURBANCES IN PATHOLOGY
The pathogenesis of most diseases is underlain by inflammation, which is an immunological response aimed at localizing and neutralizing the damaging agent [72]. In most cases, acute inflammatory responses are resolved by removing the tissue provocative and gradual decreasing the leukocyte count at the focus of inflammation as a result of the cessation of their recruitment from the circulatory system, as well as due to their apoptosis followed by phagocytosis and the restoration of homeostasis at the site of damage. Dysregulation of apoptosis during the development of inflammatory responses is associated with a failure in algorithm of the inflammatory response; it also contributes to the development of the chronic pathological process and disease progression [4, 7, 73].
For example, it is well known that in inflammations of an infectious etiology, an increase in the lifespan of activated neutrophils is accompanied by an excessive local release of a complex of cytotoxic bactericidal substances and regulatory cytokines; however, in the case of a severe inflammatory process, the production of cytokines often gets out of control and makes a significant contribution to the development of a systemic inflammatory response [72]. At the same time, untimely acceleration of apoptosis in acute infectious diseases is accompanied by incomplete phagocytosis and often provokes the development of secondary necrosis due to excessive local damaging effects of the products of cellular degradation on tissues [7, 11]. Such disorders facilitate the elusion of microorganisms from lysis and favor their dissemination in host tissues [7, 67], thus promoting the maintenance of recurrent inflammatory processes and the development of chronic pathology. For example, a persistent respiratory syncytial virus infection causes an exacerbation of bronchiolits associated with the pathogenesis of bronchial asthma [74].
Some researchers believe assume that an increase in Fas expression plays an important role in the mechanism of accelerating apoptosis of human neutrophils in inflammatory processes. It has been shown that in patients with frequent acute respiratory diseases and clinical signs of intoxication, there is an increase in Fas expression in neutrophils and an elevated count of apoptotic forms of neutrophils [75].
Multidirectional disorders of the apoptosis process in neutrophils occur not only in the development of infectious processes but also in many noninfectious diseases and pathological conditions [5, 15, 40, 76–105] (Table 1). It is noteworthy that some molecular regulators of neutrophil apoptosis, such as CD69, SAA, and SHP, are practically not detectable in healthy people, but their count increases dramatically in some diseases. At the same time, increased CD69 and SAA expression is associated with suppression of apoptosis [91, 96, 97], while SHP, on the contrary, with its activation [76, 46] (Table 1). Local tissue lesions associated with acute circulatory disorders or trauma is the cause of the development of sterile inflammation accompanied by DAMP release and neutrophilic infiltration. A DAMP-mediated increase in the duration of active neutrophil functioning in sterile inflammation can cause damage to tissues and organs, acting as an additional pathogenetic factor of the disease [1]. For example, based on experimental data, some authors believe that in acute heart failure, neutrophils can exert a direct damaging effect on cardiomyocytes in the site of ischemia, expanding the infarction zone by activating integrin interactions [3]. In patients with chronic heart failure, myocarditis may also be partially associated with a decrease in the rate of spontaneous neutrophil apoptosis [106]. This mechanism of apoptosis modulation is also one of the pathophysiological factors in the development of the so-called multiple organ dysfunction syndrome during endogenous intoxication of various origins [62]. A quantitative assessment of the count of apoptotic neutrophil forms in the patient’s peripheral blood can serve as a criterion for the severity of endogenous intoxication [75]. In diseases accompanied by uremia, an acceleration of neutrophil apoptosis is usually observed [81, 90], however, in conditions when the intoxication of the body reaches a significant intensity, for example, in sepsis or burn injury, an increase in the neutrophil lifespan is observed [78, 84, 85, 87, 92].
Antimicrobial activity of neutrophils combined with inherited features of host immune system’s functioning makes an appreciable contribution to the development of widespread diseases of the autoimmune genesis. A genetically determined decrease in ROS production by the NADPH-oxidase neutrophil complex is due to the attenuation of their functional activity and disturbed apoptosis, which significantly increases the risk of systemic lupus erythematosus, rheumatoid arthritis, chronic inflammatory bowel diseases [107–110], and leads to severe infectious complications in patients with chronic granulematosis [111]. In patients with systemic lupus erythematosus, an increase in the blood count of apoptotic neutrophil forms was found, and this feature correlated with the severity of the disease [112]. In active ANCA-associated vasculitis, primed neutrophils are also characterized by accelerated apoptosis [113]. At the same time, ANCA-associated vasculitis in remission, as well as rheumatoid arthritis and polycythemia vera, are characterized by delayed neutrophil apoptosis, as revealed in an in vitro experiment [95]. In Kostmann’s syndrome and glycogen storage disease type 1b, mutations associated with impaired neutrophil metabolism were found (Table 1), which promoted the premature activation of intracellular inducers of apoptosis and cell death at the stage of maturation, which is the main etiological factor in the pathogenesis of chronic neutropenia that accompanies these diseases [76, 101, 102]. Currently, several groups of congenital neutropenias are known, developing due to genetically determined premature apoptosis of myeloid neutrophil precursors of [14].
Factors that increase the lifespan of neutrophils appear in the blood serum of patients with inflammatory processes of various etiologies. Such regulators have been found in patients with burn injuries, sepsis, systemic inflammatory response syndrome, ischaemia/reperfusion injury, Crohn’s disease, ulcerative colitis, rheumatoid arthritis, as well as after extensive abdominal surgery [13, 80, 97, 114–117]. Factors of multidirectional regulation of apoptosis are revealed in the synovial fluid of patients with rheumatoid arthritis, and their quantitative analysis can be of clinical significance. For example, at an early stage of rheumatoid arthritis (disease duration less than 3 months), neutrophils in the synovial fluid and peripheral blood demonstrate delayed apoptosis, which is associated with a high level of antiapoptotic cytokines IL-2, IL-4, IL-15, GM-CSF and G-CSF. At the same time, with active rheumatoid arthritis, soluble FasL appears in the synovial fluid, and the local count of apoptotic neutrophil forms increases despite an increased G-CSF level [13]. Apparently, this is due to a decrease in the response of neutrophils in patients with rheumatoid arthritis to inflammatory cytokines GM-CSF, G-CSF, and TNF, accompanied by an increase in the basal phosphorylation level of ERK and p38MAPK [118]. It was experimentally established that the development of arthritis induced by immune complexes is mediated by the activation of Fcγ receptors and associated tyrosine kinase Syk, which modulates the rate of neutrophil apoptosis. A low production of inflammatory cytokines and accelerated apoptosis are the hallmarks of Syk-deficient synovial area neutrophils [119]. A similar mechanism for the suppression of neutrophil apoptosis was found in binding FcγIIα immunoreceptors to anti-IL-8/IL-8 immune complexes isolated from the pulmonary edema fluid of patients with ARDS [120]. The authors demonstrated that this interaction involved the proteins of the Srk tyrosine kinase family, Syk tyrosine kinase, ERK, and PI3 kinase, and due to the activation of cell survival pathways, the balance of the proteins of the Bcl-2 family shifts toward antiapoptotic Bcl-XL, decreasing thereby the activity of effector caspases -3 and -9. Given the obtained data and reports on the detection of other autoantibodies in the pulmonary edema fluid of patients with ARDS, Fudala et al. suggest that autoimmune mechanisms significantly contribute to increasing the lifespan of functionally active neutrophils and are of great importance in the pathogenesis of this syndrome [120].
STRESS AS A FACTOR OF APOPTOSIS REGULATION
The organism’s response to psychoemotional or physiological stress is accompanied by an increase in the intake of catecholamines and GCs into the circulatory system [121]. The activation of the sympathetic system under stress affects immunoregulatory mechanisms through the synthesis and release of a number of pro-inflammatory mediators, including IL-6, IL-1β, and TNF [121], known as modulators of neutrophil apoptosis [5, 8, 36, 38, 52]. At the same time, stimulation of the parasympathetic system, as well as the intake of GCs, make a significant contribution to the suppression of inflammation [121]. The feature and duration of stress exposure act as factors of reciprocal regulation of the activity of the neuroendocrine and immune systems, which manifests itself in the ambiguous effect of stress on the viability of neutrophils. Thus, in persons influenced by prolonged examination stress or with adjustment disorders, activation of spontaneous neutrophil apoptosis was found to be activated [8, 122]. However, in healthy people who underwent short-term stress in the form of starvation, increased sports exercises, or sleep disturbances, a delay in apoptosis was revealed [8].
In vitro studies have shown that drugs belonging to the GC group increase the viability of neutrophils obtained from healthy donors [123]. Β2-adrenergic receptor agonists (salbutamol, formoterol, salmeterol) potentiate the antiapoptotic effect of GCs (budesonide, fluticasone) under similar experimental conditions but do not exert their own effect on spontaneous neutrophil apoptosis [124]. Among the patent medicines of the GC group, dexamethasone is the most studied in terms of its effect on apoptosis. During the first 24 hours of incubation, dexamethasone reduces spontaneous neutrophil apoptosis by 50%, while a combined treatment of neutrophils with dexamethasone and leukotriene B4 inhibits their apoptosis by 90% [125]. At the same time, the antiapoptotic effect of GM-CSF on neutrophils is insignificantly enhanced by glucocorticoids, including dexamethasone [123, 124]. Interestingly, dexamethasone increases the expression of the leukotriene B4 receptor on neutrophils [125], which has its own antiapoptotic potential [124]. This drug also suppresses neutrophil apoptosis induced by oxidative stress (cell incubation in the glucose/glucose oxidase enzymatic system) [126], which is apparently due to its ability to suppress NADPH-dependent ROS production [127]. GC-mediated inhibition of apoptosis is accompanied by an increase in the expression of the inhibitors of apoptosis proteins (IAPs) and survival factor Mcl-1 in human neutrophils, while in the experiments on bovine neutrophils, an increase in the expression of the survival factor A1, as well as a decrease in the expression of the proapoptotic factor Bak and the number of Fas receptors on the cell surface, was observed [127]. It should be noted that chronic stress is accompanied by a resistance to the GC effects due to inhibition of the functional activity of GC receptors [128].
Despite the in vitro antiapoptotic effect of GCs on neutrophils, which is able to maintain to a certain extent the development of the acute phase of the inflammatory response, synthetic GCs are being actively used in clinical practice as anti-inflammatory drugs demonstrating a wide range of regulatory effects on the intensity of the immune response. By enhancing the phagocytic activity of macrophages, which is targeted at eliminating apoptotic forms of neutrophils, GCs make a significant contribution to the unfolding of the resolution phase of inflammation [129]. By regulating the transcriptional activity with a large number of genes, GCs induce in neutrophils the expression of the two proteins: one with a proapoptotic potential and pronounced anti-inflammatory effect (annexin 1, also known as lipocortin-1 [130]), and another encoded by the GILZ gene [127]. These peptides seem to be able to smooth out the direct antiapoptosis effect of GCs by stimulating neutrophil apoptosis.
Thus, stress, through the hormones and appropriate neurotransmitters, brings about multidirectional disturbances in the system that provides complex regulation of the duration of neutrophils’ functionally active lifespan, and thereby influences the organism’s susceptibility to infections and the ability to restore homeostasis at the site of damage.
PHARMACOLOGICAL MODULATION OF NEUTROPHIL APOPTOSIS
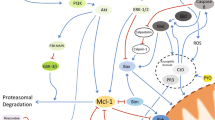
Disorders of neutrophil function, including apoptotic disturbances, are reported for diseases of varied etiology (Table 1), and this attracts attention to these cells as an object of therapeutic exposures [22, 23, 130]. The key inducers and modulators of neutrophil apoptosis can be considered as potential targets of pharmacological treatment (Fig. 1). To date, on various mouse models of inflammation, it has been shown that some agents have an anti-inflammatory effect accompanied by an increase in neutrophil apoptosis. Specifically, a similar therapeutic effect was found for R-roscovitine (a CDK inhibitor) in models of carrageenan-induced pleurisy, bleomycin-induced lung injury and pneumococcal meningitis [132, 133], for rolipram (a PDE-4 inhibitor) in a model of LPS-induced pleurisy [134], and for rTRAIL (a recombinant TRAIL-R ligand) in models of zymosan-induced peritonitis and LPS-induced acute lung injury [135]. The endogenous mediators resolvin E1, annexin A1, and epimer of lipoxin A4, demonstrating therapeutic efficacy in experimental models of inflammation, are of interest as potential anti-inflammatory drugs that enhance both apoptosis and efferocytosis of neutrophils [15, 23]. The main molecular messengers responsible for the proapoptotic effect of the aforementioned agents are proteins of the Bcl-2 family, regulatory kinases, and effector caspase-3 (Fig. 2).
Currently, a number of agents with a pleiotropic effect, including a proapoptotic effect on neutrophils, are at various phases of clinical trials. For example, roscovitin is passing clinical trials not only as a drug against certain types of cancer but also as a potential anti-inflammatory drug for the treatment of rheumatoid arthritis, glomerulonephritis, and cystic fibrosis complicated by chronic infection [136]. In other pathologies, on the contrary, the therapeutic effect of pharmacological agents is associated with the prolongation of neutrophil viability. For example, chemotherapy treatment of tumors often promotes the development of neutropenia. In these cases, the use of a recombinant GM-CSF (sargamostim) leads to a delay in neutrophil apoptosis caused by the effect of anti-cancer drugs. A similar effect has been reported for synthetic corticosteroids, such as fluticasone and prednisolone [137] (Fig. 2).
It is also noteworthy that apoptotic forms of neutrophils have their own anti-inflammatory potential, perhaps due to the release from them of α-defensims, small cationic peptides that suppress the production of proinflammatory cytokines by macrophages [138]. When apoptotic forms of neutrophils were administered to experimental animals with LPS-induced sepsis, the level of circulating proinflammatory cytokines was found to decrease, preventing thereby the endotoxic shock [139], which allows these cells to be considered as a potential treatment against the “cytokine storm”.

Molecular mechanisms for neutrophil apoptosis modulation by potential pharmacological agents. Anti-inflammatory effects of rolipram, R-roscovitine, and endogenous mediators of the resolution phase of inflammation (annexin A1, resolving E1, lipoxyn A4) are mediated by a shift in the balance of the Bcl-2 proteins toward a predominance of proapoptotic factors, as well as by a blockade of cell survival signal transduction via PI3 kinase and transcription factor NF-κB. A recombinant ligand of the death receptor TRAILR (rTRAIL) induces apoptosis via the caspase pathway (see Fig. 1). The effect of sargamostime is induced by binding to the cytokine GM-CSF receptor (GM-CSFR) and is analogous to the anti-apoptotic effect of GM-CSF. Expression of the cell survival factor Mcl-1 increases under the effect of glucocorticoids.
OTHER FORMS OF NEUTROPHIL DEATH IN INFLAMMATION
In terms of the regulation of the inflammatory response intensity, the form of neutrophil death in the focus of inflammation is a no less important immunomodulatory factor than the degree of their functional activity. Lytic forms of cell death (netosis, pyroptosis, necrosis, and necroptosis) are often accompanied by an increase in the intensity of the acute phase of inflammation due to the release of DAMP, ROS, and proteases from dying cells [1, 140, 141]. Apoptosis, on the other hand, is a nonlytic evolutionary conserved form of cell death that exhibits no inflammatory potential [7, 140]. In contrast to other forms of cell death, netosis is characterized by the formation of extracellular antimicrobial networks from decondensed nuclear DNA strands, citrullinated histones, and granular bactericidal proteases leaving the cell. It should be noted that netosis is not always accompanied by lysis of the plasma membrane, and the enucleated forms of neutrophil retains a number of effector functions, including phagocytosis [1, 7, 140]. Pyroptosis is targeted at eliminating intracellular pathogens using intracellular antimicrobial traps, the production of which is mediated by the formation of specific membrane pores [140, 142]. Necroptosis of neutrophils (regulated necrosis) is induced by various signals and appears to represent an additional factor of antiviral defense [140, 142].
Autophagy is an extremely conserved protective response of a cell to stress, including a deficiency of nutrients [7, 141, 143]. With an excessive stress intensity, autophagy can lead to cell death [141, 144]. The morphological marker of autophagy is the presence of large double-membrane bound cytoplasmic vacuoles, or autophagosomes, which, after fusion with lysosomes, transformed into autolysosomes, where lytic degradation of their own macromolecules and damaged organelles takes place [7, 141, 143]. Autophagy often accompanies apoptosis, netosis, and necrosis of neutrophils [143, 145], and in some infections, it incorporates into the immune defense system against a pathogen, ensuring its elimination in autolysosomes (a xenophagy phenomenon) [144, 145].
The mechanisms underlying lytic forms of neutrophil death in the pathogenesis of varied diseases have not been sufficiently studied, but nevertheless, it was established that they exhibit some considerable differences from apoptosis, in which proteolysis of substrates is mainly provided by the activation of effector caspases [6–8, 140] (Fig. 1). Except for necrosis, which is a process of uncontrolled cell swelling followed by membrane destruction and disintegration [141], all other forms of cell death are programmed in the form of specific sequences of molecular events [6, 140, 142], providing for intracellular cross-interactions between different pathways of terminal signal transduction [136, 142, 143, 146]. The data obtained to date form a general idea that, in contrast to other neutrophil death programs, it is the apoptosis program that is integrated into the mechanism of complex suppression of the acute inflammatory response as its main regulator.
CONCLUSION
During evolution, several mechanisms have been formed to induce apoptosis, which are maintained by multidirectional regulatory pathways involved in convergent interactions in transducing external and internal signals to effector caspases. For example, GM-CSF and TNF are able, on the one hand, to significantly suppress spontaneous apoptosis of human peripheral blood neutrophils, and, on the other hand, to accelerate mitochondria-mediated apoptosis via a caspase-9-dependent mechanism [20]. It was also found out that phagocytosis-induced apoptosis abolishes the increase in neutrophil lifespan mediated by proinflammatory cytokines [7], while neutrophil resistance to intrinsic apoptotic signaling found in critically ill patients is overcome by the activation of Fas receptors [98]. Interestingly, neutrophils in the blood of patients with septic shock are characterized by delayed apoptosis, despite a noticeable overexpression of the proapoptotic protein Bim [47]. Multifactorial regulation of neutrophil lifespan makes it possible, in some cases, to level off the impact of an infectious agent on apoptosis. IL-6 and GM-CSF have been shown to abolish the acceleration of neutrophil death induced by influenza A (H1N1) virus and Epstein-Barr virus, respectively [42, 147]. It has also been shown that LPSs, on the one hand, increase the lifespan of neutrophils, and on the other hand, stimulate the synthesis of the IL-10 receptor, thus inducing the acceleration of apoptosis in response to IL-10 [148]. Despite the fact that all phases of the inflammatory response are strictly regulated, chronic pathological process may be associated with a change in the basal activity of neutrophils and be accompanied by a disturbance of the main algorithm for the regulation of apoptosis [73, 118]. For example, it was found that neutrophils in the synovial fluid of patients with rheumatoid arthritis have an initially high level of NF-κB activation, and their TNF-induced stimulation is not accompanied by the induction of apoptosis, in contrast to neutrophils in blood [149]. Thus, the fate of a neutrophil is determined by a resultant shift, in one or another direction, in the intracellular balance of pro- and antiapopotic factors under the effect of a complex of regulatory signals modulated by the basal cell activity level.
A better insight into the mechanisms of multimodal apoptosis regulation as a factor of integral functional activity of neutrophils is of great importance for developing a therapeutic strategy and identifying key regulatory molecules as novel pharmacological drug targets.
Abbreviations
- BAL:
-
bronchoalveolar lavage
- G6PC3:
-
glucose-6-phosphatase complex type 3
- GC:
-
glucocorticoids
- LP:
-
lipoproteins
- LPS:
-
lipopolysaccharide
- ARDS:
-
acute respiratory distress syndrome
- PG:
-
peptidoglycans
- AK2:
-
adenylate kinase 2
- Akt:
-
protein kinase B
- ANCA:
-
antineutrophil cytoplasmic antibody
- Apaf-1:
-
apoptosis protease activating factor 1
- Bcl-2:
-
B-cell lymphoma protein 2
- CDK:
-
cyclin-dependent kinase
- cIAP:
-
cellular inhibitor of apoptosis proteins
- Cp-DNA:
-
unmethylated Сp dinucleotides
- CR:
-
cytokine receptor
- DAMP:
-
danger-associated molecular patterns
- ER-stress:
-
endoplasmic reticulum stress
- ERK:
-
extracellular signal regulated kinase
- FADD:
-
Fas-associated death domain protein
- Fas:
-
Fas receptor (FasR), or apoptosis antigen 1 (APO-1 or APT), or cluster of differentiation 95 (CD95)
- FasL:
-
Fas ligand
- FcRγ:
-
immunoglobulin Fc receptor gamma-chain
- G-CSF:
-
granulocyte colony-stimulating factor
- GILZ:
-
glucocorticoid-induced leucine zipper
- GM-CSF:
-
granulocyte macrophage-colony stimulating factor
- GPCR:
-
G-protein-coupled receptor
- HAX1:
-
hematopoietic cell-specific Lyn substrate 1-associated protein X-1
- Hsp:
-
heat shock proteins
- IAP:
-
inhibitor of apoptosis proteins
- ICAM-1:
-
intercellular adhesion molecule 1
- IFN:
-
interferon
- IFNAR, -BR, -GR:
-
receptor for interferon alpha, -beta, -gamma
- IGF-1:
-
insulin-like growth factor-1
- IkBα:
-
inhibitor of nuclear factor-kB alpha
- IL:
-
interleukin
- IRAK:
-
IL-1 receptor-associated kinase
- JAK:
-
Janus kinase
- Mac-1:
-
macrophage-1 antigen
- MAPK:
-
mitogen-activated protein kinase
- MIF:
-
macrophage migration inhibitory factor
- MyD88:
-
myeloid differentiation primary-response protein 88
- NF-κB:
-
nuclear factor “kappa-light-chain-enhancer” of activated B-cells
- NOD:
-
nucleotide-binding oligomerization domain
- PAMP:
-
pathogen-associated molecular patterns
- PDE-4:
-
phosphodiesterase-4
- PI3K:
-
phosphatidylinositol 3-kinase
- PRR:
-
pattern recognition receptors
- Pyk2:
-
pyruvate kinase 2
- RIP:
-
receptor-interacting protein
- ROS:
-
reactive oxygen species
- SAA:
-
serum amyloid A
- SHP:
-
Src homology domain 2 (SH2)-containing tyrosine phosphatase
- Smac:
-
second mitochondria-derived activator of caspase
- Src:
-
Rous sarcoma virus proto-oncogene
- STAT:
-
signal transducer and activator of transcription
- Syk:
-
spleen tyrosine kinase
- TLR:
-
Toll-like receptor
- TNF:
-
tumor necrosis factor
- TNFR:
-
TNF receptor
- TRADD:
-
TNF receptor-associated death domain
- TRAF:
-
TNF-receptor-associated factor
- TRAIL:
-
TNF-related apoptosis-inducing ligand
- VCAM-1:
-
vascular cell adhesion molecule-1
- XIAP:
-
X-linked inhibitor of apoptosis protein
REFERENCES
Liew PX, Kubes P (2019) The Neutrophil’s Role During Health and Disease. Physiol Rev 99(2):1223–1248.
Geering B, Stoeckle C, Conus S, Simon HU (2013) Living and dying for inflammation: neutrophils, eosinophils, basophils. Trends Immunol 34(8):398–409.
Frangogiannis NG (2014) The immune system and the remodeling infarcted heart: cell biological insights and therapeutic opportunities. J Cardiovasc Pharmacol 63(3):185–195.
Schett G, Neurath MF (2018) Resolution of chronic inflammatory disease: universal and tissue-specific concepts. Nat Commun 9(1):3261. https://doi.org/10.1038/s41467-018-05800-6
Dibbert B, Weber M, Nikolaizik WH, Vogt P, Schöni MH, Blaser K, Simon HU (1999) Cytokine-mediated Bax deficiency and consequent delayed neutrophil apoptosis: a general mechanism to accumulate effector cells in inflammation. Proc Natl Acad Sci U S A 96(23):13330–13335.
Akgul C, Moulding DA, Edwards SW (2001) Molecular control of neutrophil apoptosis. FEBS Lett 487 (3): 318–322.
Kennedy AD, DeLeo FR (2009) Neutrophil apoptosis and the resolution of infection. Immunol Res 43(1-3): 25–61.
Luo HR, Loison F (2008) Constitutive neutrophil apoptosis: mechanisms and regulation. Am J Hematol 83(4):288–295.
Kumar S, Dikshit M (2019) Metabolic Insight of Neutrophils in Health and Disease. Front Immunol 10:2099. https://doi.org/10.3389/fimmu.2019.02099
Binet F, Chiasson S, Girard D (2010) Evidence that endoplasmic reticulum (ER) stress and caspase-4 activation occur in human neutrophils. Biochem Biophys Res Commun 391(1):18–23.
Kobayashi SD, Malachowa N, DeLeo FR (2017) Influence of Microbes on Neutrophil Life and Death. Front Cell Infect Microbiol 7:159. https://doi.org/10.3389/fcimb.2017.00159
Slavich GM, Irwin MR (2014) From stress to inflammation and major depressive disorder: a social signal transduction theory of depression. Psychol Bull 140(3):774–815.
Cascão R, Rosário HS, Souto-Carneiro MM, Fonseca JE (2010) Neutrophils in rheumatoid arthritis: More than simple final effectors. Autoimmun Rev 9(8):531–535.
Bartels M, Murphy K, Rieter E, Bruin M (2016) Understanding chronic neutropenia: life is short. Br J Haematol 172(2):157–169.
Barnig C, Bezema T, Calder P C, Charloux A, Frossard N, Garssen J, Haworth O, Dilevskaya K, Levi-Schaffer F, Lonsdorfer E, Wauben M, Kraneveld AD, Te Velde AA (2019) Activation of Resolution Pathways to Prevent and Fight Chronic Inflammation: Lessons From Asthma and Inflammatory Bowel Disease. Front Immunol 10:1699. https://doi.org/10.3389/fimmu.2019.01699
Dyugovskaya L, Polyakov A, Ginsberg D, Lavie P, Lavie L (2011) Molecular pathways of spontaneous and TNF-{alpha}-mediated neutrophil apoptosis under intermittent hypoxia. Am J Respir Cell Mol Biol 45(1):154–162.
Harris AJ, Mirchandani AS, Lynch RW, Murphy F, Delaney L, Small D, Coelho P, Watts ER, Sadiku P, Griffith D, Dickinson RS, Clark E, Willson JA, Morrison T, Mazzone M, Carmeliet P, Ghesquiere B, O’Kane C, McAuley D, Jenkins SJ, Whyte MKB, Walmsley SR (2019) IL4Rα Signaling Abrogates Hypoxic Neutrophil Survival and Limits Acute Lung Injury Responses In Vivo. Am J Respir Crit Care Med 200(2):235–246.
Le’Negrate G, Rostagno P, Auberger P, Rossi B, Hofman P (2003) Downregulation of caspases and Fas ligand expression, and increased lifespan of neutrophils after transmigration across intestinal epithelium. Cell Death Differ 10(2):153–162.
Tennenberg SD, Finkenauer R, Wang T (2002) Endothelium down-regulates Fas, TNF, and TRAIL-induced neutrophil apoptosis. Surg Infect (Larchmt) 3(4):351–357.
Cowburn AS, Summers C, Dunmore BJ, Farahi N, Hayhoe RP, Print CG, Cook SJ, Chilvers ER (2011) Granulocyte/macrophage colony-stimulating factor causes a paradoxical increase in the BH3-only proapoptotic protein Bim in human neutrophils. Am J Respir Cell Mol Biol 44(6):879–887.
Ross EA, Douglas MR, Wong SH, Ross EJ, Curnow SJ, Nash GB, Rainger E, Scheel-Toellner D, Lord JM, Salmon M, Buckley CD (2006) Interaction between integrin alpha9beta1 and vascular cell adhesion molecule-1 (VCAM-1) inhibits neutrophil apoptosis. Blood 107(3): 1178–1183.
Filep JG, El Kebir D (2009) Neutrophil apoptosis: a target for enhancing the resolution of inflammation. J Cell Biochem 108(5):1039–1046.
El Kebir D, Filep JG (2013) Targeting neutrophil apoptosis for enhancing the resolution of inflammation. Cells 2(2):330–348.
Pluskota E, Soloviev DA, Szpak D, Weber C, Plow EF (2008) Neutrophil apoptosis: selective regulation by different ligands of integrin alphaMbeta2. J Immunol 181(5):3609–3619.
El Kebir D, József L, Pan W, Filep JG (2008) Myeloperoxidase delays neutrophil apoptosis through CD11b/CD18 integrins and prolongs inflammation. Circ Res 103(4):352–359.
Kettritz R, Choi M, Rolle S, Wellner M, Luft FC (2004) Integrins and cytokines activate nuclear transcription factor-kappaB in human neutrophils. J Biol Chem 279(4):2657–2665.
Gu J, Nada S, Okada M, Sekiguchi K (2003) Csk regulates integrin-mediated signals: involvement of differential activation of ERK and Akt. Biochem Biophys Res Commun 303(3):973–977.
Avdi NJ, Nick JA, Whitlock BB, Billstrom MA, Henson PM, Johnson GL, Worthen GS (2001) Tumor necrosis factor-alpha activation of the c-Jun N-terminal kinase pathway in human neutrophils. Integrin involvement in a pathway leading from cytoplasmic tyrosine kinases apoptosis. J Biol Chem 276(3):2189–2199.
Rubel C, Gomez S, Fernandez GC, Isturiz MA, Caamano J, Palermo MS (2003) Fibrinogen-CD11b/CD18 interaction activates the NF-κB pathway and delays apoptosis in human neutrophils. Eur J Immunol 33:1429–1438.
Coxon A, Rieu P, Barkalow F J, Askari S, Sharpe AH, von Andrian UH, Arnaout MA, Mayadas TN (1996) A novel role for the beta 2 integrin CD11b/CD18 in neutrophil apoptosis: a homeostatic mechanism in inflammation. Immunity 5(6):653–666.
Whitlock BB, Gardai S, Fadok V, Bratton D, Henson PM (2000) Differential roles for alpha(M)beta(2) integrin clustering or activation in the control of apoptosis via regulation of akt and ERK survival mechanisms. J Cell Biol 151(6):1305–1320.
Kettritz R, Xu YX, Faass B, Klein JB, Müller EC, Otto A, Busjahn A, Luft F C, Haller H (2000) TNF-alpha-mediated neutrophil apoptosis involves Ly-GDI, a Rho GTPase regulator. J Leukoc Biol 68(2):277–283.
Yuki K, Hou L (2020) Role of β2 Integrins in Neutrophils and Sepsis. Infect Immun 88(6):e00031. https://doi.org/10.1128/IAI.00031-20
Meszaros AJ, Reichner JS, Albina JE (2000) Macrophage-induced neutrophil apoptosis. J Immunol 165(1):435–441.
Kourtzelis I, Hajishengallis G, Chavakis T (2020) Phagocytosis of Apoptotic Cells in Resolution of Inflammation. Front Immunol 11:553. https://doi.org/10.3389/fimmu.2020.00553
Futosi K, Fodor S, Mócsai A (2013) Neutrophil cell surface receptors and their intracellular signal transduction pathways. Int Immunopharmacol 17(3):638–650.
Schepetkin IA, Cherdyntseva NV, Vasil’ev NV (1994) Regulation of functional activity of neutrophils by cytokines (review). Immunologiya/Immunology (Russia) 15(1):4–6.
McNamee JP, Bellier PV, Kutzner BC, Wilkins RC (2005) Effect of pro-inflammatory cytokines on spontaneous apoptosis in leukocyte sub-sets within a whole blood culture. Cytokine 31(2):161–167.
Pericle F, Liu JH, Diaz JI, Blanchard DK, Wei S, Forni G, Djeu JY (1994) Interleukin-2 prevention of apoptosis in human neutrophils. Eur J Immunol 24(2):440–444.
Baumann R, Casaulta C, Simon D, Conus S, Yousefi S, Simon HU (2003) Macrophage migration inhibitory factor delays apoptosis in neutrophils by inhibiting the mitochondria-dependent death pathway. FASEB J 17(15):2221–2230.
Biffl WL, Moore EE, Moore FA, Barnett CC Jr (1995) Interleukin-6 suppression of neutrophil apoptosis is neutrophil concentration dependent. J Leukoc Biol 58(5):582–584.
Dienz O, Rud JG, Eaton SM, Lanthier PA, Burg E, Drew A, Bunn J, Suratt BT, Haynes L, Rincon M (2012) Essential role of IL-6 in protection against H1N1 influenza virus by promoting neutrophil survival in the lung. Mucosal Immunol 5(3):258–266.
Allaeys I, Gymninova I, Canet-Jourdan C, Poubelle PE (2014) IL-32γ delays spontaneous apoptosis of human neutrophils through MCL-1, regulated primarily by the p38 MAPK pathway. PLoS One 9(10):e109256. https://doi.org/10.1371/journal.pone.0109256
Himpe E, Degaillier C, Coppens A, Kooijman R (2008) Insulin-like growth factor-1 delays Fas-mediated apoptosis in human neutrophils through the phosphatidylinositol-3 kinase pathway. J Endocrinol 199(1):69–80.
Klein JB, Rane MJ, Scherzer JA, Coxon PY, Kettritz R, Mathiesen JM, Buridi A, McLeish KR (2000) Granulocyte-macrophage colony-stimulating factor delays neutrophil constitutive apoptosis through phosphoinositide 3-kinase and extracellular signal-regulated kinase pathways. J Immunol 164(8):4286–4291.
Schepetkin IA, Budanova OP, Malyshev IYu, Atochin DN (2018) Molecular mechanisms of neutrophil apoptosis (review). Patogenez/Pathogenesis (Russia) 16(4): 5–18.
Andina N, Conus S, Schneider EM, Fey MF, Simon HU (2009) Induction of Bim limits cytokine-mediated prolonged survival of neutrophils. Cell Death Differ 16(9):1248–1255.
Leavey PJ, Sellins KS, Thurman G, Elzi D, Hiester A, Silliman CC, Zerbe G, Cohen JJ, Ambruso DR (1998) In vivo treatment with granulocyte colony-stimulating factor results in divergent effects on neutrophil functions measured in vitro. Blood 92(11):4366–4374.
Liles WC, Kiener PA, Ledbetter JA, Aruffo A, Klebanoff SJ (1996) Differential expression of Fas (CD95) and Fas ligand on normal human phagocytes: implications for the regulation of apoptosis in neutrophils. J Exp Med 184(2):429–440.
Iwase M, Takaoka S, Uchida M, Kondo G, Watanabe H, Ohashi M, Nagumo M (2006) Accelerative effect of a selective cyclooxygenase-2 inhibitor on Fas-mediated apoptosis in human neutrophils. Int Immunopharmacol 6(3):334–341.
Villunger A, O'Reilly L A, Holler N, Adams J, Strasser A (2000) Fas ligand, Bcl-2, granulocyte colony-stimulating factor, and p38 mitogen-activated protein kinase: Regulators of distinct cell death and survival pathways in granulocytes. J Exp Med 192(5):647–658.
Ottonello L, Frumento G, Arduino N, Bertolotto M, Dapino P, Mancini M, Dallegri F (2002) Differential regulation of spontaneous and immune complex-induced neutrophil apoptosis by proinflammatory cytokines. Role of oxidants, Bax and caspase-3. J Leukoc Biol 72(1):125–132.
Cowburn AS, Cadwallader KA, Reed BJ, Farahi N, Chilvers ER (2002) Role of PI3-kinase-dependent Bad phosphorylation and altered transcription in cytokine-mediated neutrophil survival. Blood 100(7):2607–2616.
Sakamoto C, Suzuki K, Hato F, Akahori M, Hasegawa T, Hino M, Kitagawa S (2003) Antiapoptotic effect of granulocyte colony-stimulating factor, granulocyte-macrophage colony-stimulating factor, and cyclic AMP on human neutrophils: protein synthesis-dependent and protein synthesis-independent mechanisms and the role of the Janus kinase-STAT pathway. Int J Hematol 77(1):60–70.
Hasegawa T, Suzuki K, Sakamoto C, Ohta K, Nishiki S, Hino M, Tatsumi N, Kitagawa S (2003) Expression of the inhibitor of apoptosis (IAP) family members in human neutrophils: up-regulation of cIAP2 by granulocyte colony-stimulating factor and overexpression of cIAP2 in chronic neutrophilic leukemia. Blood 101(3):1164–1171.
Pelletier M, Ratthé C, Girard D (2002) Mechanisms involved in interleukin-15-induced suppression of human neutrophil apoptosis: role of the antiapoptotic Mcl-1 protein and several kinases including Janus kinase-2, p38 mitogen-activated protein kinase and extracellular signal-regulated kinases-1/2. FEBS Lett 532(1-2):164–170.
Sakamoto E, Hato F, Kato T, Sakamoto C, Akahori M, Hino M, Kitagawa S (2005) Type I and type II interferons delay human neutrophil apoptosis via activation of STAT3 and up-regulation of cellular inhibitor of apoptosis 2. J Leukoc Biol 78(1):301–309.
Aoshiba K, Yasui S, Hayashi M, Tamaoki J, Nagai A (1999) Role of p38-mitogen-activated protein kinase in spontaneous apoptosis of human neutrophils. J Immunol 162(3):1692–1700.
Scheel-Toellner D, Wang K, Henriquez NV, Webb PR, Craddock R, Pilling D, Akbar AN, Salmon M, Lord JM (2002) Cytokine-mediated inhibition of apoptosis in non-transformed T cells and neutrophils can be dissociated from protein kinase B activation. Eur J Immunol 32(2):486–493.
Wang K, Scheel-Toellner D, Wong SH, Craddock R, Caamano J, Akbar AN, Salmon M, Lord JM (2003) Inhibition of neutrophil apoptosis by type 1 IFN depends on cross-talk between phosphoinositol 3-kinase, protein kinase C-delta, and NF-kappa B signaling pathways. J Immunol 171(2):1035–1041.
Hirata J, Kotani J, Aoyama M, Kashiwamura S, Ueda H, Kuroda Y, Usami M, Okamura H, Marukawa S (2008) A role for IL-18 in human neutrophil apoptosis. Shock 30(6):628–633.
Keel M, Ungethüm U, Steckholzer U, Niederer E, Hartung T, Trentz O, Ertel W (1997) Interleukin-10 counterregulates proinflammatory cytokine-induced inhibition of neutrophil apoptosis during severe sepsis. Blood 90(9):3356–3363.
Cox G (1996) IL-10 enhances resolution of pulmonary inflammation in vivo by promoting apoptosis of neutrophils. Am J Physiol 271(4 Pt 1):L566–L571.
Ward C, Murray J, Clugston A, Dransfield I, Haslett C, Rossi AG (2005) Interleukin-10 inhibits lipopolysaccharide-induced survival and extracellular signal-regulated kinase activation in human neutrophils. Eur J Immunol 35(9):2728–2737.
Bazzoni F, Tamassia N, Rossato M, Cassatella MA (2010) Understanding the molecular mechanisms of the multifaceted IL-10-mediated anti-inflammatory response: lessons from neutrophils. Eur J Immunol 40(9):2360–2368.
El Kebir D, József L, Filep JG (2008) Neutrophil recognition of bacterial DNA and Toll-like receptor 9-dependent and -independent regulation of neutrophil function. Arch Immunol Ther Exp (Warsz) 56(1):41–53.
Kinkead LC, Whitmore LC, McCracken JM, Fletcher JR, Ketelsen BB, Kaufman JW, Jones BD, Weiss DS, Barker JH, Allen LH (2018) Bacterial lipoproteins and other factors released by Francisella tularensis modulate human neutrophil lifespan: Effects of a TLR1 SNP on apoptosis inhibition. Cell Microbiol 20(2): e12795. https://doi.org/10.1111/cmi.12795
François S, El Benna J, Dang PM, Pedruzzi E, Gougerot-Pocidalo MA, Elbim C (2005) Inhibition of neutrophil apoptosis by TLR agonists in whole blood: involvement of the phosphoinositide 3-kinase/Akt and NF-kappaB signaling pathways, leading to increased levels of Mcl-1, A1, and phosphorylated Bad. J Immunol 174(6):3633–3642.
Sunil VR, Connor AJ, Lavnikova N, Gardner CR, Laskin JD, Laskin DL (2002) Acute endotoxemia prolongs the survival of rat lung neutrophils in response to 12-O-tetradecanoyl-phorbol 13-acetate. J Cell Physiol 190(3):382–389.
Jeong YJ, Kang MJ, Lee SJ, Kim CH, Kim JC, Kim TH, Kim DJ, Kim D, Núñez G, Park JH (2014) Nod2 and Rip2 contribute to innate immune responses in mouse neutrophils. Immunology 143(2):269–276.
O’Donnell JA, Kennedy CL, Pellegrini M, Nowell CJ, Zhang JG, O’Reilly LA, Cengia L, Dias S, Masters SL, Hartland EL, Roberts AW, Gerlic M, Croker BA (2015) Fas regulates neutrophil lifespan during viral and bacterial infection. J Leukoc Biol 97(2):321–326.
Chereshnev VA, Gusev EYu (2012) Immunological and Pathophysiological mechanisms of Systemic Inflammation. Meditsinskaya Immunologiya/Med Immunol (Russia) 14(1-2): 9–20.
Wright HL, Moots RJ, Edwards SW (2014) The multifactorial role of neutrophils in rheumatoid arthritis. Nat Rev Rheumatol 10(10):593–601.
Wang SZ, Forsyth KD (2000) The interaction of neutrophils with respiratory epithelial cells in viral infection. Respirology 5(1):1–10.
Poniakina ID (2003) Activation of apoptosis of the peripheral blood neutrophils as an indicator of the body autointoxication. Klin Lab Diagn/Clin Lab Diagn (Russia) 7: 19–21.
Tidow N, Kasper B, Welte K (1999) SH2-containing protein tyrosine phosphatases SHP-1 and SHP-2 are dramatically increased at the protein level in neutrophils from patients with severe congenital neutropenia (Kostmann’s syndrome). Exp Hematol 27(6):1038–1045.
Altznauer F, Conus S, Cavalli A, Folkers G, Simon HU (2004) Calpain-1 regulates Bax and subsequent Smac-dependent caspase-3 activation in neutrophil apoptosis. J Biol Chem 279(7):5947–5957.
Abraham E (2003) Nuclear factor-kappaB and its role in sepsis-associated organ failure. J Infect Dis 187 (Suppl 2):S364–369.
Aprikyan AA, Liles WC, Park JR, Jonas M, Chi EY, Dale DC (2000) Myelokathexis, a congenital disorder of severe neutropenia characterized by accelerated apoptosis and defective expression of bcl-x in neutrophil precursors. Blood 95(1):320–327.
Brannigan AE, O’Connell PR, Hurley H, O’Neill A, Brady HR, Fitzpatrick JM, Watson RW (2000) Neutrophil apoptosis is delayed in patients with inflammatory bowel disease. Shock 13(5):361–366.
Cohen G, Rudnicki M, Hörl WH (2001) Uremic toxins modulate the spontaneous apoptotic cell death and essential functions of neutrophils. Kidney Int Suppl 78:S48–52.
Delogu G, Moretti S, Famularo G, Antonucci A, Signore L, Marcellini S, Lo Bosco L, De Simone C (2001) Circulating neutrophils exhibit enhanced apoptosis associated with mitochondrial dysfunctions after surgery under general anaesthesia. Acta Anaesthesiol Scand 45(1):87–94.
Gamonal J, Sanz M, O’Connor A, Acevedo A, Suarez I, Sanz A, Martínez B, Silva A (2003) Delayed neutrophil apoptosis in chronic periodontitis patients. J Clin Periodontol 30(7):616–623.
Härter L, Mica L, Stocker R, Trentz O, Keel M (2003) Mcl-1 correlates with reduced apoptosis in neutrophils from patients with sepsis. J Am Coll Surg 197(6):964–673.
Hashiguchi N, Ogura H, Tanaka H, Koh T, Nakamori Y, Noborio M, Shiozaki T, Nishino M, Kuwagata Y, Shimazu T, Sugimoto H (2001) Enhanced expression of heat shock proteins in activated polymorphonuclear leukocytes in patients with sepsis. J Trauma 51(6):1104–1109.
Heeney MM, Ormsbee SM, Moody MA, Howard TA, DeCastro CM, Ware RE (2003) Increased expression of anti-apoptosis genes in peripheral blood cells from patients with paroxysmal nocturnal hemoglobinuria. Mol Genet Metab 78(4):291–294.
Hu Z, Sayeed MM (2004) Suppression of mitochondria-dependent neutrophil apoptosis with thermal injury. Am J Physiol Cell Physiol 286(1):C170–178.
Ina K, Kusugami K, Hosokawa T, Imada A, Shimizu T, Yamaguchi T, Ohsuga M, Kyokane K, Sakai T, Nishio Y, Yokoyama Y, Ando T (1999) Increased mucosal production of granulocyte colony-stimulating factor is related to a delay in neutrophil apoptosis in Inflammatory Bowel disease. J Gastroenterol Hepatol 14(1):46–53.
Kobayashi S, Yamashita K, Takeoka T, Ohtsuki T, Suzuki Y, Takahashi R, Yamamoto K, Kaufmann SH, Uchiyama T, Sasada M, Takahashi A (2002) Calpain-mediated X-linked inhibitor of apoptosis degradation in neutrophil apoptosis and its impairment in chronic neutrophilic leukemia. J Biol Chem 277(37):33968–33977.
Majewska E, Baj Z, Sulowska Z, Rysz J, Luciak M (2003) Effects of uraemia and haemodialysis on neutrophil apoptosis and expression of apoptosis-related proteins. Nephrol Dial Transplant 18(12):2582–2588.
Nopp A, Stridh H, Grönneberg R, Lundahl J (2002) Lower apoptosis rate and higher CD69 expression in neutrophils from atopic individuals. Inflamm Res 51(11):532–540.
Ogura H, Hashiguchi N, Tanaka H, Koh T, Noborio M, Nakamori Y, Nishino M, Kuwagata Y, Shimazu T, Sugimoto H (2002) Long-term enhanced expression of heat shock proteins and decelerated apoptosis in polymorphonuclear leukocytes from major burn patients. J Burn Care Rehabil 23(2):103–109.
O’Neill S, O’Neill AJ, Conroy E, Brady HR, Fitzpatrick JM, Watson RW (2000) Altered caspase expression results in delayed neutrophil apoptosis in acute pancreatitis. J Leukoc Biol 68(1):15–20.
Tsujimoto H, Takeshita S, Nakatani K, Kawamura Y, Tokutomi T, Sekine I (2001) Delayed apoptosis of circulating neutrophils in Kawasaki disease. Clin Exp Immunol 126(2):355–364.
Abdgawad M, Pettersson Å, Gunnarsson L, Bengtsson AA, Geborek P, Nilsson L, Segelmark M, Hellmark T (2012) Decreased neutrophil apoptosis in quiescent ANCA-associated systemic vasculitis. PloS one 7(3):e32439. https://doi.org/10.1371/journal.pone.0032439
Atzeni F, Del Papa N, Sarzi-Puttini P, Bertolazzi F, Minonzio F, Capsoni F (2004) CD69 expression on neutrophils from patients with rheumatoid arthritis. Clin Exp Rheumatol 22(3):331–334.
Christenson K, Björkman L, Tängemo C, Bylund J (2008) Serum amyloid A inhibits apoptosis of human neutrophils via a P2X7-sensitive pathway independent of formyl peptide receptor-like 1. J Leukoc Biol 83(1):139–148.
Paunel-Görgülü A, Zörnig M, Lögters T, Altrichter J, Rabenhorst U, Cinatl J, Windolf J, Scholz M (2009) Mcl-1-mediated impairment of the intrinsic apoptosis pathway in circulating neutrophils from critically ill patients can be overcome by Fas stimulation. J Immunol 183(10):6198–6206.
Garlichs CD, Eskafi S, Cicha I, Schmeisser A, Walzog B, Raaz D, Stumpf C, Yilmaz A, Bremer J, Ludwig J, Daniel WG (2004) Delay of neutrophil apoptosis in acute coronary syndromes. J Leukoc Biol 75(5):828–835.
Taneja R, Parodo J, Jia SH, Kapus A, Rotstein OD, Marshall JC (2004) Delayed neutrophil apoptosis in sepsis is associated with maintenance of mitochondrial transmembrane potential and reduced caspase-9 activity. Crit Care Med 32(7):1460–1469.
Kim SY, Jun HS, Mead PA, Mansfield BC, Chou JY (2008) Neutrophil stress and apoptosis underlie myeloid dysfunction in glycogen storage disease type Ib. Blood 111(12):5704–5711.
Klein C (2011) Genetic defects in severe congenital neutropenia: emerging insights into life and death of human neutrophil granulocytes. Annu Rev Immunol 29:399–413.
Tsai CY, Li KJ, Hsieh SC, Liao HT, Yu CL (2019) What's wrong with neutrophils in lupus? Clin Exp Rheumatol 37(4):684–693.
Midgley A, McLaren Z, Moots RJ, Edwards SW, Beresford MW (2009) The role of neutrophil apoptosis in juvenile-onset systemic lupus erythematosus. Arthritis Rheum 60(8):2390–2401.
Midgley A, Mayer K, Edwards SW, Beresford MW (2011) Differential expression of factors involved in the intrinsic and extrinsic apoptotic pathways in juvenile systemic lupus erythematosus. Lupus 20(1):71–79.
Rumalla VK, Calvano SE, Spotnitz AJ, Krause TJ, Hilkert RJ, Lin E, Lowry SF (2002) Alterations in immunocyte tumor necrosis factor receptor and apoptosis in patients with congestive heart failure. Ann Surg 236(2):254–260.
Olsson LM, Lindqvist AK, Källberg H, Padyukov L, Burkhardt H, Alfredsson L, Klareskog L, Holmdahl R (2007) A case-control study of rheumatoid arthritis identifies an associated single nucleotide polymorphism in the NCF4 gene, supporting a role for the NADPH-oxidase complex in autoimmunity. Arthritis Res Ther 9(5):R98. https://doi.org/10.1186/ar2299
Olsson LM, Johansson ÅC, Gullstrand B, Jönsen A, Saevarsdottir S, Rönnblom L, Leonard D, Wetterö J, Sjöwall C, Svenungsson E, Gunnarsson I, Bengtsson AA, Holmdahl R (2017) A single nucleotide polymorphism in the NCF1 gene leading to reduced oxidative burst is associated with systemic lupus erythematosus. Ann Rheum Dis 76(9):1607–1613.
Zhao J, Ma J, Deng Y, Kelly JA, Kim K, Bang SY, Lee HS, Li QZ, Wakeland EK, Qiu R, Liu M, Guo J, Li Z, Tan W, Rasmussen A, Lessard CJ, Sivils KL, Hahn BH, Grossman JM, Kamen DL, Gilkeson GS, Bae SC, Gaffney PM, Shen N, Tsao BP (2017) A missense variant in NCF1 is associated with susceptibility to multiple autoimmune diseases. Nat Genet 49(3):433–437.
Dhillon SS, Fattouh R, Elkadri A, Xu W, Murchie R, Walters T, Guo C, Mack D, Huynh HQ, Baksh S, Silverberg MS, Griffiths AM, Snapper SB, Brumell JH, Muise AM (2014) Variants in nicotinamide adenine dinucleotide phosphate oxidase complex components determine susceptibility to very early onset inflammatory bowel disease. Gastroenterology 147(3):680–689.
Nauseef WM, Clark RA (2019) Intersecting Stories of the Phagocyte NADPH Oxidase and Chronic Granulomatous Disease. Methods Mol Biol 1982:3–16.
Courtney PA, Crockard AD, Williamson K, Irvine A E, Kennedy RJ, Bell AL (1999) Increased apoptotic peripheral blood neutrophils in systemic lupus erythematosus: relations with disease activity, antibodies to double stranded DNA, and neutropenia. Ann Rheum Dis 58(5):309–314.
Harper L, Cockwell P, Adu D, Savage CO (2001) Neutrophil priming and apoptosis in anti-neutrophil cytoplasmic autoantibody-associated vasculitis. Kidney Int 59(5):1729–1738.
Chen MF, Chen JC, Chiu DF, Ng CJ, Shyr MH, Chen HM (2001) Prostacyclin analogue (OP-2507) induces delayed ex vivo neutrophil apoptosis and attenuates reperfusion-induced hepatic microcirculatory derangement in rats. Shock 16(6):473–478.
Chitnis D, Dickerson C, Munster AM, Winchurch RA (1996) Inhibition of apoptosis in polymorphonuclear neutrophils from burn patients. J Leukoc Biol 59(6):835–839.
Ertel W, Keel M, Infanger M, Ungethüm U, Steckholzer U, Trentz O (1998) Circulating mediators in serum of injured patients with septic complications inhibit neutrophil apoptosis through up-regulation of protein-tyrosine phosphorylation. J Trauma 44(5):767–775.
Jimenez MF, Watson RW, Parodo J, Evans D, Foster D, Steinberg M, Rotstein OD, Marshall JC (1997) Dysregulated expression of neutrophil apoptosis in the systemic inflammatory response syndrome. Arch Surg 132(12):1263–1269.
Inaba M, Takahashi T, Kumeda Y, Kato T, Hato F, Yutani Y, Goto H, Nishizawa Y, Kitagawa S (2008) Increased basal phosphorylation of mitogen-activated protein kinases and reduced responsiveness to inflammatory cytokines in neutrophils from patients with rheumatoid arthritis. Clin Exp Rheumatol 26(1):52–60.
Elliott ER, Van Ziffle JA, Scapini P, Sullivan BM, Locksley RM, Lowell CA (2011) Deletion of Syk in neutrophils prevents immune complex arthritis. J Immunol 187(8):4319–4330.
Fudala R, Krupa A, Matthay MA, Allen TC, Kurdowska AK (2007) Anti-IL-8 autoantibody:IL-8 immune complexes suppress spontaneous apoptosis of neutrophils. Am J Physiol Lung Cell Mol Physiol 293(2):L364–L374.
Raison CL, Lowry CA, Rook GA (2010) Inflammation, sanitation, and consternation: loss of contact with coevolved, tolerogenic microorganisms and the pathophysiology and treatment of major depression. Arch Gen Psychiatry 67(12):1211–1224.
Chernykh EI, Yazykov KG, Semke VY (2002) Apoptosis in peripheral blood leukocytes induced by hyperthermia and prednisolone in patients with dysadaptation. Bull Exp Biol Med 134(6):531–533.
Zhang X, Moilanen E, Kankaanranta H (2001) Beclomethasone, budesonide and fluticasone propionate inhibit human neutrophil apoptosis. Eur J Pharmacol 431(3):365–371.
Perttunen H, Moilanen E, Zhang X, Barnes PJ, Kankaanranta H (2008) Beta2-agonists potentiate corticosteroid-induced neutrophil survival. COPD 5(3):163–169.
Stankova J, Turcotte S, Harris J, Rola-Pleszczynski M (2002) Modulation of leukotriene B4 receptor-1 expression by dexamethasone: potential mechanism for enhanced neutrophil survival. J Immunol 168(7):3570–3576.
Ruiz LM, Bedoya G, Salazar J, García de OD, Patiño PJ (2002) Dexamethasone inhibits apoptosis of human neutrophils induced by reactive oxygen species. Inflammation 26(5):215–222.
Ronchetti S, Ricci E, Migliorati G, Gentili M, Riccardi C (2018) How Glucocorticoids Affect the Neutrophil Life. Int J Mol Sci 19(12):4090. https://doi.org/10.3390/ijms19124090
Pace TW, Hu F, Miller AH (2007) Cytokine-effects on glucocorticoid receptor function: relevance to glucocorticoid resistance and the pathophysiology and treatment of major depression. Brain Behav Immun 21(1):9–19.
Heasman SJ, Giles KM, Ward C, Rossi AG, Haslett C, Dransfield I (2003) Glucocorticoid-mediated regulation of granulocyte apoptosis and macrophage phagocytosis of apoptotic cells: implications for the resolution of inflammation. J Endocrinol 178(1):29–36.
Sugimoto MA, Vago JP, Teixeira MM, Sousa LP (2016) Annexin A1 and the Resolution of Inflammation: Modulation of Neutrophil Recruitment, Apoptosis, and Clearance. J Immunol Res 2016:8239258. https://doi.org/10.1155/2016/8239258
Chellappan DK, Yee LW, Xuan KY, Kunalan K, Rou LC, Jean LS, Ying LY, Wie LX, Chellian J, Mehta M, Satija S, Singh SK, Gulati M, Dureja H, Da Silva MW, Tambuwala MM, Gupta G, Paudel KR, Wadhwa R, Hansbro PM, Dua K (2020) Targeting neutrophils using novel drug delivery systems in chronic respiratory diseases. Drug Dev Res 81(4):419–436.
Rossi AG, Sawatzky DA, Walker A, Ward C, Sheldrake TA, Riley NA, Caldicott A, Martinez-Losa M, Walker TR, Duffin R, Gray M, Crescenzi E, Martin MC, Brady HJ, Savill JS, Dransfield I, Haslett C (2006) Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nat Med 12(9):1056–1064.
Koedel U, Frankenberg T, Kirschnek S, Obermaier B, Häcker H, Paul R, Häcker G (2009) Apoptosis is essential for neutrophil functional shutdown and determines tissue damage in experimental pneumococcal meningitis. PLoS Pathog 5(5):e1000461. https://doi.org/10.1371/journal.ppat.1000461
Sousa LP, Lopes F, Silva DM, Tavares LP, Vieira AT, Rezende BM, Carmo AF, Russo RC, Garcia CC, Bonjardim CA, Alessandri AL, Rossi AG, Pinho V, Teixeira MM (2010) PDE4 inhibition drives resolution of neutrophilic inflammation by inducing apoptosis in a PKA-PI3K/Akt-dependent and NF-kappaB-independent manner. J Leukoc Biol 87(5):895–904.
McGrath EE, Marriott HM, Lawrie A, Francis SE, Sabroe I, Renshaw SA, Dockrell DH, Whyte MK (2011) TNF-related apoptosis-inducing ligand (TRAIL) regulates inflammatory neutrophil apoptosis and enhances resolution of inflammation. J Leukoc Biol 90(5):855–865.
Meijer L, Nelson DJ, Riazanski V, Gabdoulkhakova AG, Hery-Arnaud G, Le Berre R, Loaëc N, Oumata N, Galons H, Nowak E, Gueganton L, Dorothée G, Prochazkova M, Hall B, Kulkarni AB, Gray RD, Rossi AG, Witko-Sarsat V, Norez C, Becq F, Ravel D, Mottier D, Rault G (2016) Modulating Innate and Adaptive Immunity by (R)-Roscovitine: Potential Therapeutic Opportunity in Cystic Fibrosis. J Innate Immun 8(4):330–349.
Brostjan C, Oehler R (2020) The role of neutrophil death in chronic inflammation and cancer. Cell Death Discov 6:26. https://doi.org/10.1038/s41420-020-0255-6
Miles K, Clarke D J, Lu W, Sibinska Z, Beau-mont PE, Davidson DJ, Barr TA, Campopiano DJ, Gray M (2009) Dying and necrotic neutrophils are anti-inflammatory secondary to the release of alpha-defensins. J Immunol 183(3):2122–2132.
Ren Y, Xie Y, Jiang G, Fan J, Yeung J, Li W, Tam PK, Savill J (2008) Apoptotic cells protect mice against lipopolysaccharide-induced shock. J Immunol 180(7):4978–4985.
Lawrence SM, Corriden R, Nizet V (2020) How Neutrophils Meet Their End. Trends Immunol 41(6):531–544.
Kroemer G, Levine B (2008) Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol 9(12):1004–1010.
Jorgensen I, Rayamajhi M, Miao EA (2017) Programmed cell death as a defence against infection. Nat Rev Immunol 17(3):151–164.
Germic N, Frangez Z, Yousefi S, Simon HU (2019) Regulation of the innate immune system by autophagy: neutrophils, eosinophils, mast cells, NK cells. Cell Death Differ 26(4):703–714.
Chargui A, Cesaro A, Mimouna S, Fareh M, Brest P, Naquet P, Darfeuille-Michaud A, Hébuterne X, Mograbi B, Vouret-Craviari V, Hofman P (2012) Subversion of autophagy in adherent invasive Escherichia coli-infected neutrophils induces inflammation and cell death. PLoS One 7(12):e51727. https://doi.org/10.1371/journal.pone.0051727
Ullah I, Ritchie ND, Evans TJ (2017) The interrelationship between phagocytosis, autophagy and formation of neutrophil extracellular traps following infection of human neutrophils by Streptococcus pneumoniae. Innate Immun 23(5):413–423.
Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, Brunner T, Simon HU (2006) Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol 8(10):1124–1132.
Larochelle B, Flamand L, Gourde P, Beauchamp D, Gosselin J (1998) Epstein-Barr virus infects and induces apoptosis in human neutrophils. Blood 92(1):291–299.
Crepaldi L, Gasperini S, Lapinet JA, Calzetti F, Pinardi C, Liu Y, Zurawski S, de Waal Malefyt R, Moore KW, Cassatella MA (2001) Up-regulation of IL-10R1 expression is required to render human neutrophils fully responsive to IL-10. J Immunol 167(4):2312–2322.
Hotta K, Niwa M, Hara A, Ohno T, Wang X, Matsuno H, Kozawa O, Ito H, Kato K, Otsuka T, Matsui N, Uematsu T (2001) The loss of susceptibility to apoptosis in exudated tissue neutrophils is associated with their nuclear factor-kappa B activation. Eur J Pharmacol 433(1):17–27.
Funding
This work was supported by the Program for Increasing the Competitiveness of the Tomsk Polytechnic University, by the Russian Science Foundation Grant No. 17-15-01111 (as part of the writing of the section “Pharmacological modulation of neutrophil apoptosis”).
Author information
Authors and Affiliations
Contributions
E. M. N. and I. A. S. reviewed the literature and prepared a manuscript. I. A. S. proposed the concept and edited the manuscript. D.N.A. carefully edited the manuscript, presented a critical edition, and contributed to the final version of the manuscript. All authors have read and agreed with the published version of the manuscript.
Corresponding author
Ethics declarations
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
Additional information
Translated by A. Polyanovsky
Russian Text © The Author(s), 2021, published in Zhurnal Evolyutsionnoi Biokhimii i Fiziologii, 2021, Vol. 57, No. 3, pp. 183–201https://doi.org/10.31857/S0044452921030086.
Rights and permissions
About this article

Cite this article
Noseykina, E.M., Schepetkin, I.A. & Atochin, D.N. Molecular Mechanisms for Regulation of Neutrophil Apoptosis under Normal and Pathological Conditions. J Evol Biochem Phys 57, 429–450 (2021). https://doi.org/10.1134/S0022093021030017
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0022093021030017