
Abstract
In mammals, about 40 isoforms of voltage-gated potassium channels (KVs) have been found. To study such a variety of KVs, substances are needed that are able to selectively bind to them and change their properties. We have previously reported on the isolation and pharmacological characterization of MeKTx13-3, a peptide toxin from the venom of the scorpion Mesobuthus eupeus. The toxin has shown high affinity to a number of KVs, with little selectivity for the KV1.1 isoform. In this paper, we describe the production of an artificial derivative of MeKTx13-3, named MeKTx13-3_RMRH, using rational design. The selectivity of MeKTx13-3_RMRH in relation to KV1.1 is increased by an order of magnitude making it one of the most specific ligands of this KV isoform. Finally, using computer simulations, we demonstrate that the preference of the new ligand to KV1.1 can be realized through a specific positioning of the toxin in complex with the channel.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Voltage-gated potassium channels (KVs) are transmembrane (TM) proteins that provide passive selective current of potassium ions across the membrane in response to potential change [1]. Mature KV channel is a tetramer of α-subunits, each of which is formed by six TM segments (S1–S6) with one pore region (P) between S5 and S6 [2]. The channel pore domain is formed by S5 and S6 segments of all four α-subunits. In addition, KVs may contain auxiliary β-subunits that can modify the properties of the channel [3]. A distinctive feature of KVs is the presence of voltage-sensing domains formed by four TM segments (S1–S4) in each α-subunit [4, 5]. According to the latest guidelines of the International Union of Basic and Clinical Pharmacology (IUPHAR), about 40 genes of KV α-subunits are known in humans, which are referred to as isoforms [6]. Depending on the localization, as well as the functions performed, KVs can be formed by identical or different α-subunits, and so they can be homo- or heteromers [7, 8].
Investigation of the structural features, physiological and pharmacological characteristics of KVs is closely associated with the use of their ligands [9–11] that can be divided into several groups. The main ones are: (a) metal ions, for example, Cs+ and Ba2+ [12, 13]; (b) small organic molecules such as 4-aminopyridine and tetraethylammonium [14]; and (c) polypeptide toxins, for instance charybdotoxin (ChTx) and dendrotoxin [15]. Perhaps the richest source of KV ligands is the venoms of various animals, such as snakes, sea anemones, cone snails, spiders, and scorpions [16]. Scorpion toxins undoubtedly played a key role in the study of KVs: from the pioneering work on the inhibition of potassium current in the giant squid axon using noxiustoxin from the venom of the scorpion Centruroides noxius [17, 18] to the obtaining the crystal structure of KV in complex with ChTx from the venom of the scorpion Leiurus quinquestriatus [19, 20]. Currently, according to the Kalium database (https://kaliumdb.org/), approximately 350 polypeptide ligands of potassium channels are known and characterized, more than half (~200) of which are toxins isolated from scorpion venom [21, 22].
The majority of scorpion toxins acting on KVs consist of 30–50 amino acid residues, 6 or 8 of which are cysteine residues that form 3 or 4 intramolecular disulfide bonds respectively [16, 23]. These disulfides stabilize a characteristic polypeptide fold, which is called cysteine-stabilized α-helix / β-sheet (CSα/β) [24, 25]. Despite the common spatial structure, toxins can exhibit different pharmacological characteristics, selectively acting on particular KV isoforms. It is assumed that the selectivity is determined by the amino acid residues of the toxin that form specific contacts with the pore domain of the channel [26–28]. Therefore, these features can be used for the rational design of artificial derivatives with prescribed properties based on a common molecular framework [29–31].
We previously reported on the identification and characterization of the toxin MeKTx13-3 (Kalium ID: α-KTx 3.19, UniProt ID: C0HJQ6, 37 amino acid residues, three disulfide bonds) from the venom of the Central Asian scorpion Mesobuthus eupeus [32]. We performed a pharmacological characterization of this polypeptide and, using electrophysiological methods, found that it inhibits potassium current through some homotetrameric KVs, namely: KV1.1, 1.2, 1.3, and 1.6, with half-maximal inhibitory concentrations (IC50) of ~2, 100, 10, and 60 nM, respectively. The toxin was found to be selective to KV1.1, rather than KV1.2 or 1.3, which is a rather rare feature of polypeptide ligands of KVs [23]. Now we report that the introduction of a number of substitutions in the amino acid sequence of MeKTx13-3 allowed us to obtain a more selective peptide towards KV1.1—MeKTx13-3_RMRH. Using methods of molecular modeling of the structure and dynamics of complexes of this MeKTx13-3 derivative with KV1.1–1.3 homotetramers, we have established that the selectivity of its action can be realized due to the specific position of the toxin in the complex with the channel.
MATERIALS AND METHODS
Ethics Statement. This study strictly complied with the World Health Organization’s International Guiding Principles for Biomedical Research Involving Animals. The research was carried out in AAALAC accredited organization according to the standards of the Guide for Care and Use of Laboratory Animals (8th edition, Institute for Laboratory Research of Animals). The use of frogs was in accordance with the license number LA1210239 of Toxicology & Pharmacology, KU Leuven. The use of Xenopus laevis was approved by the Ethical Committee for animal experiments of KU Leuven (P186/2019). All animal care and experimental procedures agreed with the guidelines of the European Convention for the Protection of Vertebrate Animals used for Experimental and other Scientific Purposes (Strasbourg, 18.III.1986).
Recombinant Peptide Production. The procedure to obtain recombinant MeKTx13-3_RMRH was generally the same as described in our previous works [31]. The target peptide was produced in a bacterial expression system as a fusion with the carrier protein thioredoxin (Trx) [33] and cleaved by recombinant human enteropeptidase light chain [34]. DNA sequence encoding MeKTx13-3_RMRH was constructed from partially overlapping synthetic oligonucleotides by PCR in two steps. Firstly, all four oligonucleotides (F1, F2, R1 and R2; Table 1) were used for amplification in 5 cycles. As a result, a full gene sequence was obtained. Secondly, terminal oligonucleotides (F1 and R1) and diluted PCR mixture from the first step as a matrix were used for the second step of PCR.
The resulting PCR fragment was cloned into the expression vector pET-32b (Novagen) at KpnI and BamHI restriction sites. Escherichia coli SHuffle T7 Express cells (New England Biolabs) were transformed and cultured at 30°C in LB medium to an OD600 of ~0.6. Expression was induced by 0.2 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). Cells were cultured at room temperature (24°C) overnight (16 h) and harvested by centrifugation. The cell pellet was resuspended in 300 mM NaCl, 50 mM Tris-HCl buffer (pH 8.0) and ultrasonicated. The lysate was applied to a HisPur Cobalt Resin (ThermoFisher Scientific); and the Trx-containing fusion protein was purified according to the manufacturer’s protocol.
The fusion protein was dissolved in 50 mM Tris-HCl (pH 8.0) to 1 mg/ml concentration and hydrolyzed overnight (16 h) at 37°C by human enteropeptidase light chain (1 U of enzyme per 1 mg of substrate). MeKTx13-3_RMRH was purified by reversed-phase (RP) HPLC in a linear gradient of acetonitrile concentration (0–60% in 60 min) in the presence of 0.1% trifluoroacetic acid (TFA) on a Jupiter C5 column (4.6 × 250 mm; Phenomenex). The purity of the target peptide was checked by MALDI-TOF MS and analytical chromatography on a Vydac C18 column (4.6 × 250 mm; Separations Group) in the same acetonitrile gradient.
Mass spectrometry. Molecular mass measurements for natural and recombinant peptides were performed using MALDI on an Ultraflex TOF-TOF (Bruker Daltonik) spectrometer as described in our previous papers [35]. 2,5-Dihydroxybenzoic acid (Sigma-Aldrich) was used as a matrix. Measurements were carried out in both linear and reflector modes. Mass spectra were analyzed with the Data Analysis 4.3 and Data Analysis Viewer 4.3 software (Bruker).
Ion channel expression. Our general approach to ion channel expression in oocytes was described in detail previously [36]. Briefly, for the expression of KV genes (rat (r)KV1.1, KV1.2, human (h)KV1.3, rKV1.4, rKV1.5, and rKV1.6) in X. laevis oocytes, linearized plasmids containing the respective gene sequences were transcribed using the T7 mMESSAGE-mMACHINE transcription kit (Ambion). 50 nl of cRNA solution (1 ng/nl) were injected into oocytes using a micro-injector (Drummond Scientific). The oocytes were incubated in ND96 solution: 96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 2 mM MgCl2 and 5 mM HEPES, pH 7.4, supplemented with 50 mg/l gentamycin sulfate.
Electrophysiological recordings. Two-electrode voltage clamp recordings were performed at room temperature (18–22°C) using a Geneclamp 500 amplifier (Molecular Devices) controlled by a pClamp data acquisition system (Axon Instruments) as described previously [36]. Bath solution composition was ND96. KV currents were evoked by 250-ms depolarization to 0 mV from a holding potential of –90 mV, followed by 250-ms pulses to –50 mV. Concentration–response curves were plotted, in which the percentage of current inhibition was plotted as a function of toxin concentration. Data were fitted with the Hill equation:
y = 100/[1 + (IC50/[toxin])h],
where y is the amplitude of the toxin-induced effect, IC50 is half-maximal inhibitory concentration, [toxin] is toxin concentration, and h is the Hill coefficient. Comparison of two sample means was performed using a paired Student’s t-test (p-value of 0.05 was used as a threshold of significance). All data were obtained in at least three independent experiments (n ≥ 3) and are presented as mean ± standard error of the mean. The results obtained were processed using the program Origin (OriginLab Corporation).
Molecular modeling. The model of MeKTx13-3_RMRH structure was generated in the PyMOL Molecular Graphics System, version 1.8 (Schrödinger, LLC), using in silico mutagenesis option. Since the amino acid sequence of MeKTx13-3 is identical to that of BmKTX [37], the known 3D structure of the latter (PDB ID: 1BKT) [38] was used as a template. KV1.1 and KV1.3 models were generated previously [31, 39–41] in MODELLER [42] using the KV1.2 structure [43] as a template.
Complexes of MeKTx13-3_RMRH with potassium channels were modeled analogously to the procedure described in our previous works [31, 39–41], considering that the toxin interacts with KVs similarly to ChTx [44]. The model of the complex of MeKTx13-3_RMRH with KV1.2 was built on the basis of the KV1.2/2.1–ChTx complex crystal structure (PDB ID: 4JTA) [20]: the structure of the peptide was spatially aligned with the structure of channel-bound ChTx, which was subsequently replaced by the aligned toxin. Complexes with KV1.1 and KV1.3 were generated similarly, but the first step was spatial alignment of the channel models with the KV1.2/2.1 chimera [31, 39–41].
Molecular dynamics simulations. Preparation of the studied molecular systems for the molecular dynamics (MD) simulation was performed automatically using our in-house software package IMPULSE (Krylov et al., in preparation) and included the following steps. The resulting complexes of MeKTx13-3_RMRH with KVs were placed inside a lipid bilayer mimicking a neuronal membrane. We used a pre-equilibrated fragment of bilayer (7.0 × 7.0 × 13.5 nm3; 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine/1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine/cholesterol, POPC:POPE:Chl; 100:50:50 molecules, respectively, solvated with 14172 water molecules) that has been described in detail in our previous works [41, 45, 46]; some phospholipid and cholesterol molecules were removed to provide room for the protein. The TIP3P water model [47] and the required number of Na+/Cl– ions (to maintain electroneutrality) were used for resolvation.
All systems were equilibrated (heated up to 37°C) during 100 ps of MD simulation. Positions of the channel Cα atoms of residues not involved in the channel pore vestibule, as well as the Nε atom of Lys26 in MeKTx13-3_RMRH were restrained during the equilibration to prevent destabilization of the initial complex. Systems were then subjected to 500 ns of MD. All simulations were performed with the GROMACS software [48] (version 2018) using the AMBER99SB-ILDN parameters set [49]. Simulations were carried out with a time step of 2 fs, imposing 3D periodic boundary conditions, in the isothermal-isobaric (NPT) ensemble with a semi-isotropic pressure of 1 bar using the Berendsen pressure coupling algorithm [50], and at a constant temperature of 37°C using the V-rescale thermostat [51]. Van der Waals interactions were truncated using a 1.4-nm spherical cut-off function. Electrostatic interactions were treated with the PME algorithm. During the simulation, the position of the Nε atom of Lys26 in each complex was restrained inside the channel pore.
Analysis of intermolecular contacts and estimation of residual contributions to intermolecular interaction energy were performed based on MD trajectory using our in-house software package IMPULSE (Krylov et al., in preparation) analogously to the procedures described in our previous studies [31, 41]. Briefly, H-bonds were assigned using the parameters set from the hbond utility of GROMACS software [48] (the distance D—A ≤ 0.35 nm and the angle D—H—A ≥ 150° for the hydrogen bond D—H···A, where D and A are the hydrogen bond donor and acceptor, respectively); salt bridges, π–cation, stacking, and hydrophobic contacts were calculated using algorithms described in our previous works [52, 53]. The AMBER99SB-ILDN parameters set [49] and 1.4 nm cutoff distance for van der Waals/electrostatic interactions were used to estimate the energy of intermolecular non-bonded interactions. All drawings of 3D structures were prepared with the PyMOL. Graphical representation of the interaction energy profiles was performed using Python built-in libraries and NumPy package.
RESULTS AND DISCUSSION
New K V ligand design strategy. Previous investigations clarified a number of positions in the toxins of α-KTx3 family underlying its high affinity to KV1.3. Mutagenesis studies of BmKTx (possessing the same amino acid sequence as MeKTx13-3) demonstrated that substitution Asp33His enhances the activity on KV1.3 several times [54]. Another mutant of BmKTx named ADWX-1, comprising residues Arg11 and His33, was reported to block the channel currents with subnanomolar activity [29], while substitutions Arg11Ala or His33Ala result in significant activity decrease. Besides, AgTx-2 [30, 55], OSK1 [56] and some other toxins, which have residues Arg12, Met29, Arg31 and His34 (corresponding to Arg11, Met28, Arg30 and His33 in α-KTx3 toxins lacking N-terminal glycine), were also reported as highly efficient KV1.3 blockers (Table 2). We took an attempt to produce a new highly selective KV1.3 blocker by introducing these four substitutions (Gly11Arg, Ile28Met, Gly30Arg, and Asp33His) in MeKTx13-3. Surprisingly, the peptide obtained (MeKTx13-3_RMRH) demonstrated high affinity to KV1.1, while its activity against KV1.3 did not change.
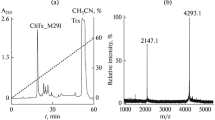
Recombinant toxin production. Recombinant MeKTx13-3_RMRH was produced in E. coli SHuffle B strain as an expression system according to our common protocol. The coding sequence was cloned into the pET-32b expression vector at KpnI and BamHI restriction sites. Trx was used as a folding companion to obtain the peptide with disulfide bonds like in the natural peptide. The target peptide was purified from hydrolyzed fusion protein by RP-HPLC (Fig. 1) and identified by using MALDI-TOF MS and comparison of the calculated and experimentally measured molecular mass. The final yield of the peptide was ~3.5 mg from 1 l of bacterial culture.
Electrophysiological profiling of MeKTx13-3_RMRH. Characterization of MeKTx13-3_RMRH pharmacological activity was performed by electrophysiology on a number of KV isoforms using the two-electrode voltage-clamp technique. Figure 2 demonstrates the recordings of currents through homotetrameric potassium channels of various isoforms (KV1.1–1.3 and 1.6) before and after application of the toxin at a concentration of 1 nM. It is obvious that MeKTx13-3_RMRH is highly selective to KV1.1. At higher concentrations, it also inhibited KV1.2, 1.3, and 1.6, while no effect on KV1.4 and 1.5 was observed up to 1 µM (data not shown). For all KV isoforms on which the effect was observed, concentration–response curves were plotted (Fig. 2, bottom panel) and the of the IC50 values were calculated: 0.11 ± 0.02 nM, 10.7 ± 0.8 nM, 8.1 ± 0.2 nM, and 16.3 ± 1.0 nM on KV1.1, 1.2, 1.3, and 1.6 respectively.
Molecular modeling. Recently we determined structural and dynamic details of MeKTx13-3 interaction with KVs to design a selective KV1.3 blocker [31]. In current work we have performed a computational study of its mutant MeKTx13-3_RMRH in complex with potassium channels to find out the cause of the unexpected specificity to KV1.1. We generated structure models of MeKTx13-3_RMRH complexes with KV1.1–1.3, computed MD trajectories, analyzed intermolecular contacts and residual contributions to interaction energy during the MD simulations, and compared the results with data on MeKTx13-3 (Figs. 3, 4b–4d, 5, Table 3).
According to the results of the computational study, the substitution Ile28Met does not lead to noticeable effects on intermolecular contacts or interaction energy (Fig. 3). The number and the quality of intermolecular contacts (hydrophobic) formed by the residues in complexes of MeKTx13-3 and MeKTx13-3_RMRH differ insignificantly (Table 3). This observation is well consistent with experimental data demonstrating that Ile28Met mutation does not result in functional differences between BmKTx and MeuKTx-3 (Table 2) [61].
As it was noted in our previous study [31], Asp33 in MeKTx13-3 makes significant positive contribution to the interaction energy (negatively affects the affinity) (Fig. 3) due to electrostatic repulsion with a conserved negatively charged residue in the channel vestibule (Asp377/375/399 in KV1.1/1.2/1.3, see Fig. 4a). It is not surprising that the substitution Asp33His in MeKTx13-3_RMRH “cancels out” this unfavorable energy contribution. In addition, His33 forms a hydrogen bond in the complex with KV1.1 (His33–Gly376, see Fig. 4c). Thus, benefits of the Asp33His substitution for stabilizing MeKTx13-3_RMRH–KV1.1 complex are beyond doubt. However, it is unlikely that this residue is essential for the increase of the specificity to KV1.1 observed when moving from MeKTx13-3 to MeKTx13-3_RMRH, since (a) His9 forms two hydrogen bonds with KV1.2, which might increase the affinity to this channel (Table 2); (b) the substitution Asp33Arg in MeKTx13-3_AAAR [31] (which adds favorable contribution to the interaction energy and provides multiple possibilities for polar contact formation) did not prevent the decrease of the affinity to KV1.1 caused by other substitutions.
Other substituted residues Arg11 and Arg30 in MeKTx13-3_RMRH make significant favorable contributions to the interaction energy (Fig. 3). Moreover, these structure modifications result in the formation of twelve (a quarter of the total number) intermolecular contacts (Table 3): three hydrogen bonds and three salt bridges (Arg11–Asp361, Arg11–Asp377, and Arg30–Asp361, see Figs. 4c, 4d), as well as three π–π and three π–cation interactions (Arg11–His355, Arg11–Phe356, and Arg30–Phe356) in the complex with KV1.1. Needless to say, analogous contacts are impossible in the complexes of MeKTx13-3 with KVs since Gly11 and Gly30 do not possess side chains.
Nevertheless, the contacts formed by Arg11 and Arg30 do not explain high MeKTx13-3_RMRH specificity to KV1.1. These substitutions do not cause such a prominent increase of affinity to KV1.2 and KV1.3, although they are involved in multiple interactions with these two channels as well. Evidently, the contacts of Arg11 and Arg30 provide optimal toxin positioning and orientation of channel regions to allow other interactions in the complex with KV1.1, which actually underlie the peptide specificity. Thus, contacts Arg30–Asp361 and Arg30–Phe356 fix a region of the receptor between the residues Phe356 and Asp361 next to the toxin in the MeKTx13-3_RMRH–KV1.1 complex. Such a fixation restricts the mobility of the adjacent fragment between the residues Glu353 and His355 and keeps an optimal position of Glu353 for the formation of a hydrogen bond and a salt bridge with the peptide residue Lys31 (Figs. 4, 5). Analogous contacts are not realized in the complexes with KV1.2/KV1.3 since side chains of the corresponding residues Asp351/Thr375 cannot reach the ε-amino group of Lys31.
Other interactions realized due to the toxin positioning sufficient for the contact formation is Pro36–Tyr379. Main chain of Pro36 in MeKTx13-3_RMRH forms a hydrogen bond with the side chain of KV1.1-specific residue Tyr379 (Fig. 4c), which does not appear in the MeKTx13-3–KV1.1 complex. Analogous contacts are not observed in the complexes with other channels: side chain of Val377 in KV1.2 is too short and lacks the appropriate functional group, while side chain of His401 in KV1.3 cannot reach the main chain of Pro36.
Obviously, two hydrogen bonds and two salt bridges Lys37–Asp361 and Lys37–Asp377, as well as medium-lived hydrogen bonds His9–Glu353 and Gln12–Glu353 (Fig. 4d), also become possible in the MeKTx13-3_RMRH–KV1.1 complex as a result of the specific positioning of the toxin relatively to the receptor. It could be noted that analogous contacts formed by Lys37 were observed in the complex of MeKTx13-3 with KV1.3 (Lys37–Asp383 and Lys37–Asp399), while analogous contacts formed by His9 and Gln12 were found in the complex of MeKTx13-3 with KV1.2 (His9–Asp351 and Gln12–Asp351) [31], but not in the complexes of MeKTx13-3_RMRH with any of these two channels. Such a loss of contacts can also be explained by slightly different positioning of the wild-type and mutant toxins in complexes with KV1.2 and 1.3.
Specific position of MeKTx13-3_RMRH stabilized by interactions of Arg11 and Arg30 in the studied complexes causes not only the disappearance of some contacts observed in the wild-type toxin, but also appearance of novel interactions. Thus, 45, 29, and 23 specific contacts are observed in the complexes of MeKTx13-3_RMRH with KV1.1, KV1.2, and KV1.3 respectively, while only 10, 8, and 3 of them are identical between MeKTx13-3 and MeKTx13-3_RMRH. Apparently, when comparing MeKTx13-3 to MeKTx13-3_RMRH the contacts number increase results in the increase of affinity to KV1.1 and KV1.2 (since the mutant forms more contacts with these two channels), but does not affect the affinity to KV1.3 (Tables 2, 3).
Analysis of intermolecular contacts shows that MeKTx13-3_RMRH forms 23 specific and 245 hydrophobic contacts with KV1.3, while MeKTx13-3 forms 30 specific and 263 hydrophobic contacts. However, the number of long-living hydrogen bonds and salt bridges in the complex with MeKTx13-3_RMRH (10 and 5, respectively) is greater than in complex with the wild-type toxin (8 and 4). Presumably, the similar potency of toxins to KV1.3 is due to the redistribution of the quantity and quality of the contacts (Table 3).
Summarizing the results of the computational analysis, we conclude that Arg11 and Arg30 play an essential role in the selective binding of MeKTx13-3_RMRH to KV1.1. In complexes with KVs these residues stabilize specific toxin position relatively to the receptor due to multiple intermolecular interactions. The toxin position provides the formation of “supporting” contacts (by Lys31, Pro36, and Lys 37, as well as His9 and Gln12) in the complex with KV1.1, which presumably underlie the high affinity to this particular channel isoform.
K V 1.1-selective ligands are uncommon among animal toxins. Using Kalium database [22] we have analyzed the data on known potassium channel ligands and found out that just a few natural polypeptide toxins possess KV1.1 selectivity. There are only three scorpion toxins (KTx) among them: hongotoxin-1 (α-KTx 2.5, P59847) from the venom of the Central American scorpion Centruroides limbatus, and MeKTx13-2 (α-KTx 3.18, C0HJQ4) and MeKTx13-3 (α-KTx 3.19, C0HJQ6), both of which are from M. eupeus [32, 62]. In the venom of other animals KV1.1-selective toxins are also rare components. Only two such molecules, i.e. BgK (κ-actitoxin-Bgr1a, P29186) and APEKTx1 (κPI-actitoxin-Ael3a, P86862), were found and purified from sea anemones Bunodosoma granuliferum and Anthopleura elegantissima, respectively [36, 63]. Currently, APEKTx1 is the undeniable leader among the KV1.1-selective polypeptides displaying an IC50 value of ~1 nM without cross-activity on other isoforms up to 1 µM concentration [36].
To estimate ligand specificity against different ion channel isoforms, for example KV1.1, we used the so-called “selectivity factor” representing the ratio of IC50 (or Kd) values for two channels [40]. This parameter can be applied to evaluate easily the specificity of each toxin to KV1.1 (Table 4). Expectedly, APEKTx1 displays the highest selectivity factor (more than 1000 for each isoform pair). MeKTx13-2 are MeKTx13-3 are natural scorpion toxins showing a modest preference to KV1.1 as compared with KV1.3, whereas this parameter for MeKTx13-3_RMRH is ten folds higher. Thus, currently, MeKTx13-3_RMRH is the most selective ligand of KV1.1 designed on the scaffold of a potassium channel scorpion toxin (Table 4).
A prevailing majority of polypeptide ligands are cross-active and simultaneously inhibit several similar isoforms of homotetrameric potassium channels [16, 64, 65]. Sometimes, toxin specificity is “shifted” towards one or two particular isoforms. For example, OSK1 (α-KTx 3.7, P55896) from Orthochirus scrobiculosus venom is more selective to KV1.3, whereas OSK3 (α-KTx 8.8, A0A1L2FZD4) from the venom of the same scorpion acts on both KV1.2 and 1.3 isoforms in comparable concentrations [39, 56]. Most of the work on identification and design of selective polypeptides acting on potassium channels is focused on KV1.3 ligands [10]. This KV isoform is considered an important pharmacological target in the development of a number of autoimmune diseases [66], and selective peptide inhibitors, such as moka1 [30], ShK-186 [67], and HsTX1 [R14A] [68], are suggested as potential therapeutic agents [69]. A number of polypeptides, such as conotoxin κM-RIIIJ (κ-conotoxin RIIIJ, P0CG45) [70] and actinotoxin BcsTx1 (κ-actitoxin-Bcs3a, C0HJC2) [71], as well as scorpion toxins MeKTx11-1 (α-KTx 1.16, C0HJQ7) [40] and MMTX (α-KTx 26.4, P0DL65) [72], present a high selectivity on KV1.2. Despite Kv1.1 isoform being structurally close to KV1.2, the list of its selective inhibitors is rather short (see above).
There are several general limitations to the research targeted to design and characterize novel isoform-specific ligands, including KTx derivatives. Firstly, subunit composition of KV tetramers can be very different and heteromeric channels are much more frequently expressed in vivo, including channels containing KV1.1 and KV1.2 subunits [73]. Moreover, the stoichiometry of such complexes has not yet been established, and it is still unclear, where exactly they are expressed in the organism. The second limitation is the deficiency of pharmacological data for a large number of known toxins. Unfortunately, full-scale measurements, including IC50 and Kd determination, were performed only for a minor pool of polypeptide toxins. Finally, further development of new KV ligands is significantly dependent upon the determination of the spatial structure of channel–toxin complexes [20], and therefore overcoming the corresponding challenges in structural biology is of prior importance. It should be noted that KV1.1 and 1.2 are considered among the most common KV isoforms in the central nervous system, where they are localized mainly in axons and nerve endings [7]. Application of selective ligands similar to MeKTx13-3_RMRH described here will shed light on specific functions of these channels.

Production of MeKTx13-3_RMRH. (a) RP-HPLC separation of recombinant MeKTx13-3_RMRH from fusion protein hydrolyzed by human enterokinase light chain. (b) Purification of the target peptide by RP-HPLC.

Pharmacological characterization of MeKTx13-3_RMRH. (a) Representative traces of currents through KVs in control (black) and after the application of 1 nM of peptide (red). (b) Concentration–response inhibition curves of the toxin on KV1.1–1.3 and 1.6 obtained by electrophysiological measurements.

Interaction energy profiles of peptide toxins in complexes with KV1.1–1.3. (a) Profile for MeKTx13-3; and (b) MeKTx13-3_RMRH. Bar charts show residual contributions to the interaction energy averaged over MD simulation. Error bars indicate standard deviations. Amino acid sequences are shown above the residue numbers; substitutions in MeKTx13-3_RMRH are shown in red font. Data for MeKTx13-3 were described in a previous study [31].

Modeled structure of MeKTx13-3_RMRH in complex with KV1.1. (a) Amino acid sequence alignment of the extracellular pore region of KV1.1–1.3 channels. The indices “h” and “r” in front of the channel names indicate the source organism (human and rat respectively). Residue numbering is above each sequence; different residues are shaded in gray. (b) Overall structure of the KV1.1–MeKTx13-3_RMRH complex after 500-ns MD simulation inside a hydrated lipid bilayer membrane. KV1.1 subunits are shown in cartoon representation (gray, brown, cyan, and blue); the pore domain helices of the channel subunit in the foreground and voltage-sensing domain of the adjacent subunit, as well as extended extracellular loops are omitted for clarity. Lipids are shown in a semi-transparent space-filling representation; atoms are colored: oxygen, red; phosphorus, orange; nitrogen, blue; hydrogen of amino and hydroxyl group, white; carbon of POPC, light-yellow; carbon of POPE, yellow; and carbon of cholesterol, beige. Some lipids are omitted for clarity. MeKTx13-3_RMRH is presented in pink; residue Lys26 (plugs the channel pore) is shown as sticks. (c, d) Close-up left-side and right-side views (respectively) on the channel pore vestibule shown in panel (b). The channel is in a semi-transparent representation. Lipids are omitted for clarity. Side chains of Lys26 and other residues involved in intermolecular contacts are shown as sticks; for residues Pro36, Lys37, and Gly376 some atoms of the main chain are also shown as sticks. Polar contacts (hydrogen bonds and salt bridges) are shown as dashed green lines. In the moment presented in panel (d), His9 does not form a contact with Glu353.

(a, b) Changes of the distance between channel and toxin fragments during MD simulation. (a) Distance between KV1.1 fragment Phe356–Asp361 and residue Gly30/Arg30 of MeKTx13-3/MeKTx13-3_RMRH. (b) Distance between KV1.1 fragment Glu353–His355 and residue Lys31 in MeKTx13-3/MeKTx13-3_RMRH. The distance was calculated between geometric centers of the polypeptide fragments of channel and the main chain of corresponding toxin residues. (c, d) Root mean deviation (RMSD) of the position of KV1.1 fragments during MD simulation of its complex with MeKTx13-3/MeKTx13-3_RMRH. (c) RMSD of Phe356–Asp361 fragment. (d) RMSD of Glu353–His355 fragment.
REFERENCES
Hille, B., Ion channels of excitable membranes, 3rd ed., Sinauer Associates: Sunderland MA, Chicago, 2001.
Long, S.B., Campbell, E.B., and MacKinnon, R., Crystal structure of a mammalian voltage-dependent Shaker family K+ channel, Science, 2005, vol. 309, pp. 897–903. https://doi.org/10.1126/science.1116269
Pongs, O., Leicher, T., Berger, M., Roeper, J., Bahring, R., Wray, D., Giese, K.P., Silva, A.J., and Storm, J.F., Functional and molecular aspects of voltage-gated K+ channel β subunits, Ann. N.Y. Acad. Sci., 1999, vol. e868, pp. 344–355. https://doi.org/10.1111/j.1749-6632.1999.tb11296.x
Catterall, W.A., Ion channel voltage sensors: Structure, function, and pathophysiology, Neuron, 2010, vol. 67, pp. 915–928. https://doi.org/10.1016/j.neuron.2010.08.021
Sansom, M.S.P., Potassium channels: Watching a voltage-sensor tilt and twist, Curr. Biol., 2000, vol. 10, pp. R206–R209. https://doi.org/10.1016/S0960-9822(00)00354-7
Alexander, S.P.H., Mathie, A., Peters, J.A., Veale, E.L., Striessnig, J., Kelly, E., Armstrong, J.F., Faccenda, E., Harding, S.D., Pawson, A.J., Sharman, J.L., Southan, C., Davies, J.A., Aldrich, R.W., Becirovic, E., Biel, M., Catterall, W.A., Conner, A.C., Davies, P., Delling, M., Virgilio, F. Di, Falzoni, S., George, C., Goldstein, S.A.N., Grissmer, S., Ha K., Hammelmann, V., Hanukoglu, I., Jarvis, M., Jensen, A.A., Kaczmarek, L.K., Kellenberger, S., Kennedy, C., King, B., Lynch, J.W., Perez-Reyes, E., Plant, L.D., Rash, L.D., Ren, D., Sivilotti, L.G., Smart, T.G., Snutch, T.P., Tian, J., Van, den Eynde, C., Vriens, J., Wei, A.D., Winn, B.T., Wulff, H., Xu, H., Yue, L., Zhang, X., and Zhu, M., The Concise Guide to Pharmacology 2019/20: Ion channels, Br. J. Pharmacol., 2019, vol. 176, pp. S142–S228. https://doi.org/10.1111/bph.14749
Vacher, H., Mohapatra, D.P., and Trimmer, J.S., Localization and targeting of voltage-dependent ion channels in mammalian central neurons, Physiol. Rev., 2008, vol. 88, pp. 1407–1447. https://doi.org/10.1152/physrev.00002.2008
Manganas, L.N. and Trimmer, J.S., Subunit composition determines Kv1 potassium channel surface expression, J. Biol. Chem., 2000, vol. 275, pp. 29685–29693. https://doi.org/10.1074/jbc.M005010200
Garcia, M.L., Galvez, A., Garcia-Calvo, M., King, V.F., Vazquez, J., and Kaczorowski, G.J., Use of toxins to study potassium channels, J. Bioenerg. Biomembr., 1991, vol. 23, pp. 615–646. https://doi.org/10.1007/BF00785814
Wulff, H., Castle, N.A., and Pardo, L.A., Voltage-gated potassium channels as therapeutic targets, Nat. Rev. Drug. Discov., 2009, vol. 8, pp. 982–1001. https://doi.org/10.1038/nrd2983
Norton, R.S. and Chandy, K.G., Venom-derived peptide inhibitors of voltage-gated potassium channels, Neuropharmacology, 2017, vol. 127, pp. 124–138. https://doi.org/10.1016/j.neuropharm.2017.07.002
Hagiwara, S., Miyazaki, S., and Rosenthal, N.P., Potassium current and the effect of cesium on this current during anomalous rectification of the egg cell membrane of a starfish, J. Gen. Physiol., 1976, vol. 67, pp. 621–638. https://doi.org/10.1085/jgp.67.6.621
Ludwig, J., Terlau, H., Wunder, F., Bruggemann, A., Pardo, L.A., Marquardt, A., Stuhmer, W., and Pongs, O., Functional expression of a rat homologue of the voltage gated ether a go-go potassium channel reveals differences in selectivity and activation kinetics between the Drosophila channel and its mammalian counterpart, EMBO J., 1994, vol. 13, pp. 4451–4458. https://doi.org/10.1002/j.1460-2075.1994.tb06767.x
Robertson, D.W. and Steinberg, M.I. Potassium channel modulators: Scientific applications and therapeutic promise, J. Med. Chem., 1990, vol. 33, pp. 1529–1541. https://doi.org/10.1021/jm00168a001
Moczydlowski, E., Lucchesi, K., and Ravindran, A., An emerging pharmacology of peptide toxins targeted against potassium channels, J. Membr. Biol., 1988, vol. 105, pp. 95–111. https://doi.org/10.1007/BF02009164
Kuzmenkov, A.I., Grishin, E.V., and Vassilevski, A.A., Diversity of potassium channel ligands: Focus on scorpion toxins. Biochemistry (Mosc.), 2015, vol. 80, pp. 1764–1799. https://doi.org/10.1134/S0006297915130118
Domingos Possani, L., Martin, B.M., and Svendsen, I.B., The primary structure of noxiustoxin: A K+ channel blocking peptide, purified from the venom of the scorpion Centruroides noxius Hoffmann, Carlsberg Res. Commun., 1982, vol. 47, pp. 285–289. https://doi.org/10.1007/BF02907789
Carbone, E., Wanke, E., Prestipino, G., Possani, L.D., and Maelicke, A., Selective blockage of voltage-dependent K+ channels by a novel scorpion toxin, Nature, 1982, vol. 296, pp. 90–91. https://doi.org/10.1038/296090a0
Miller, C., Moczydlowski, E., Latorre, R., and Phillips, M., Charybdotoxin, a protein inhibitor of single Ca2+-activated K+ channels from mammalian skeletal muscle, Nature, 1985, vol. 313, pp. 316–318. https://doi.org/10.1038/313316a0
Banerjee, A., Lee, A., Campbell, E., and Mackinnon, R., Structure of a pore-blocking toxin in complex with a eukaryotic voltage-dependent K(+) channel, Elife, 2013, vol. 2, pp. e00594. https://doi.org/10.7554/eLife.00594
Kuzmenkov, A.I., Krylov, N.A., Chugunov, A.O., Grishin, E.V., and Vassilevski, A.A., Kalium: a database of potassium channel toxins from scorpion venom, Database (Oxford), 2016, baw056. https://doi.org/10.1093/database/baw056
Tabakmakher, V.M., Krylov, N.A., Kuzmenkov, A.I., Efremov, R.G., and Vassilevski, A.A., Kalium 2.0, a comprehensive database of polypeptide ligands of potassium channels, Sci. Data, 2019, vol. 6, pp. 73. https://doi.org/10.1038/s41597-019-0074-x
Bergeron, Z.L. and Bingham, J.P., Scorpion toxins specific for potassium (K+) channels: A historical overview of peptide bioengineering, Toxins (Basel), 2012, vol. 4, pp. 1082–1119. https://doi.org/10.3390/toxins4111082
Mouhat, S., Andreotti, N., Jouirou, B., and Sabatier, J.-M., Animal toxins acting on voltage-gated potassium channels, Curr. Pharm. Des., 2008, vol. 14, pp. 2503–2518. https://doi.org/10.2174/138161208785777441
Mouhat, S., Jouirou, B., Mosbah, A., De Waard, M., and Sabatier, J.M., Diversity of folds in animal toxins acting on ion channels, Biochem. J., 2004, vol. 378, pp. 717–726. https://doi.org/10.1042/bj20031860
Jouirou, B., Mouhat, S., Andreotti, N., De Waard, M., and Sabatier, J.M., Toxin determinants required for interaction with voltage-gated K+ channels, Toxicon, 2004, vol. 43, pp. 909–914. https://doi.org/10.1016/j.toxicon.2004.03.024
Giangiacomo, K.M., Ceralde, Y., and Mullmann, T.J., Molecular basis of α-KTx specificity, Toxicon, 2004, vol. 43, pp. 877–886. https://doi.org/10.1016/j.toxicon.2003.11.029
Rodríguez De La Vega, R.C., Merino, E., Becerril, B., and Possani, L.D., Novel interactions between K+ channels and scorpion toxins, Trends Pharmacol. Sci., 2003, vol. 24, pp. 222–227. https://doi.org/10.1016/S0165-6147(03)00080-4
Han, S., Yi, H., Yin, S.J., Chen, Z.Y., Liu, H., Cao, Z.J., Wu, Y.L., and Li, W.X., Structural basis of a potent peptide inhibitor designed for Kv1.3 channel, a therapeutic target of autoimmune disease, J. Biol. Chem., 2008, vol. 283, pp. 19058–19065. https://doi.org/10.1074/jbc.M802054200
Takacs, Z., Toups, M., Kollewe, A, Johnson, E., Cuello, L.G., Driessens, G., Biancalana, M., Koide, A., Ponte, C.G., Perozo, E., Gajewski, T.F., Suarez-Kurtz, G., Koide, S., and Goldstein, S.A.N., A designer ligand specific for Kv1.3 channels from a scorpion neurotoxin-based library, Proc. Natl. Acad. Sci. USA, 2009, vol. 106, pp. 22211–22216. https://doi.org/10.1073/pnas.0910123106
Gigolaev, A.M., Kuzmenkov, A.I., Peigneur, S., Tabakmakher, V.M., Pinheiro-Junior, E.L., Chugunov, A.O., Efremov, R.G., Tytgat, J., and Vassilevski, A.A., Tuning scorpion toxin selectivity: Switching from KV1.1 to KV1.3, Front. Pharmacol., 2020, vol. 11, pp. 1010. https://doi.org/10.3389/fphar.2020.01010
Kuzmenkov, A.I., Peigneur, S., Tytgat, J., and Vassilevski, A.A., Pharmacological characterisation of MeKTx13-2 and MeKTx13-3, peptide ligands of potassium channels from the scorpion Mesobuthus eupeus venom, Russ. J. Physiol., 2019, vol. 105, pp. 1452–1462. https://doi.org/10.1134/S0869813919110074
McCoy, J. and Lavallie, E., Expression and purification of thioredoxin fusion proteins, Curr. Protoc. Mol. Biol., 2001, Chapter 16: Unit16.8. https://doi.org/10.1002/0471142727.mb1608s28
Gasparian, M.E., Bychkov, M.L., Dolgikh, D.A., and Kirpichnikov, M.P., Strategy for improvement of enteropeptidase efficiency in tag removal processes, Protein. Expr. Purif., 2011, vol. 79, pp. 191–196. https://doi.org/10.1016/j.pep.2011.04.005
Kuzmenkov, A.I., Sachkova, M.Y., Kovalchuk, S.I., Grishin, E.V., and Vassilevski, A.A., Lachesana tarabaevi, an expert in membrane-active toxins, Biochem. J., 2016, vol. 473, pp. 2495–2506. https://doi.org/10.1042/BCJ20160436
Peigneur, S., Billen, B., Derua, R., Waelkens, E., Debaveye, S., Béress, L., and Tytgat, J., A bifunctional sea anemone peptide with Kunitz type protease and potassium channel inhibiting properties, Biochem. Pharmacol., 2011, vol. 82, pp. 81–90. https://doi.org/10.1016/j.bcp.2011.03.023
Romi-Lebrun, R., Lebrun, B., Martin-Eauclaire, M.-F., Ishiguro, M., Escoubas, P., Wu, F.Q., Hisada, M., Pongs, O., and Nakajima, T., Purification, characterization, and synthesis of three novel toxins from the Chinese scorpion Buthus martensi, which act on K+ channels, Biochemistry, 1997, vol. 36, pp. 13473–13482. https://doi.org/10.1021/bi971044w
Renisio, J.G., Romi-Lebrun, R., Blanc, E., Bornet, O., Nakajima, T., and Darbon, H., Solution structure of BmKTX, a K+ blocker toxin from the Chinese scorpion Buthus martensi, Proteins, 2000, vol. 38, pp. 70–78. https://doi.org/10.1002/(SICI)1097-0134(20000101)38:1<70::AID-PROT8>3.0.CO;2-5
Kuzmenkov, A.I., Peigneur, S., Chugunov, A.O., Tabakmakher, V.M., Efremov, R.G., Tytgat, J., Grishin, E.V., and Vassilevski, A.A., C-Terminal residues in small potassium channel blockers OdK1 and OSK3 from scorpion venom fine-tune the selectivity, Biochim. Biophys. Acta. Proteins Proteomics, 2017, vol. 1865, pp. 465–472. https://doi.org/10.1016/j.bbapap.2017.02.001
Kuzmenkov, A.I., Nekrasova, O.V., Peigneur, S., Tabakmakher, V.M., Gigolaev, A.M., Fradkov, A.F., Kudryashova, K.S., Chugunov, A.O., Efremov, R.G., Tytgat, J., Feofanov, A.V., and Vassilevski, A.A., KV1.2 channel-specific blocker from Mesobuthus eupeus scorpion venom: Structural basis of selectivity, Neuropharmacology, 2018, vol. 143, pp. 228–238. https://doi.org/10.1016/j.neuropharm.2018.09.030
Berkut, A.A., Chugunov, A.O., Mineev, K.S., Peigneur, S., Tabakmakher, V.M., Krylov, N.A., Oparin, P.B., Lihonosova, A.F., Novikova, E.V., Arseniev, A.S., Grishin, E.V., Tytgat, J., Efremov, R.G., and Vassilevski, A.A., Protein surface topography as a tool to enhance the selective activity of a potassium channel blocker, J. Biol. Chem., 2019, vol. 294, pp. 18349–18359. https://doi.org/10.1074/jbc.RA119.010494
Webb, B. and Sali, A., Comparative protein structure modeling using MODELLER. In: Current Protocols in Bioinformatics, John Wiley & Sons, Inc., Hoboken, N.J., USA, 2016, 5.6.1-5.6.37. https://doi.org/10.1002/cpbi.3
Chen, X., Wang, Q., Ni, F, and Ma, J., Structure of the full-length Shaker potassium channel Kv1.2 by normal-mode-based X-ray crystallographic refinement, Proc. Natl. Acad. Sci. USA, 2010, vol. 107, pp. 11352–11357. https://doi.org/10.1073/pnas.1000142107
Goldstein, S.A., Pheasant, D.J., and Miller, C., The charybdotoxin receptor of a Shaker K+ channel: peptide and channel residues mediating molecular recognition, Neuron, 1994, vol. 12, pp. 1377–1388. https://doi.org/10.1016/0896-6273(94)90452-9
Lyukmanova, E.N., Shenkarev, Z.O., Shulepko, M.A., Paramonov, A.S., Chugunov, A.O., Janickova, H., Dolejsi, E., Dolezal, V., Utkin, Y.N., Tsetlin, V.I., Arseniev, A.S., Efremov, R.G., Dolgikh, D.A., and Kirpichnikov, M.P., Structural insight into specificity of interactions between nonconventional three-finger weak toxin from Naja kaouthia (WTX) and muscarinic acetylcholine receptors, J. Biol. Chem., 2015, vol. 290, pp. 23616–23630. https://doi.org/10.1074/jbc.M115.656595
Chugunov, A.O., Volynsky, P.E., Krylov, N.A., Nolde, D.E., and Efremov, R.G., Temperature-sensitive gating of TRPV1 channel as probed by atomistic simulations of its trans- and juxtamembrane domains, Sci. Rep., 2016, vol. 6, pp. 33112. ttps://doi.org/10.1038/srep33112
Jorgensen, W.L., Chandrasekhar, J., Madura, J.D., Impey, R.W., and Klein, M.L., Comparison of simple potential functions for simulating liquid water, J. Chem. Phys., 1983, vol. 79, pp. 926–935. https://doi.org/10.1063/1.445869
Abraham, M.J., Murtola, T., Schulz, R., Pall, S., Smith, J.C., Hess, B., and Lindahl, E., GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers, SoftwareX, 2015, vol. 1, pp. 19–25. https://doi.org/10.1016/j.softx.2015.06.001
Klepeis, J.L., Lindorff-Larsen, K., Shaw, D.E., Palmo, K., Dror, R.O., Maragakis, P., and Piana, S., Improved side-chain torsion potentials for the Amber ff99SB protein force field, Proteins, 2010, vol. 78, pp. 1950–1958. https://doi.org/10.1002/prot.22711
Berendsen, H.J.C., Postma, J.P.M., van Gunsteren, W.F., DiNola, A., and Haak, J.R., Molecular dynamics with coupling to an external bath, J. Chem. Phys., 1984, vol. 81, pp. 3684–3690. https://doi.org/10.1063/1.448118
Bussi, G., Donadio, D., and Parrinello, M., Canonical sampling through velocity rescaling, J. Chem. Phys., 2007, vol. 126, pp. 014101. https://doi.org/10.1063/1.2408420
Pyrkov, T.V., Chugunov, A.O., Krylov, N.A., Nolde, D.E., and Efremov, R.G., PLATINUM: a web tool for analysis of hydrophobic/hydrophilic organization of biomolecular complexes, Bioinformatics, 2009, vol. 25, pp. 1201–1202. https://doi.org/10.1093/bioinformatics/btp111
Pyrkov, T. and Efremov, R., A fragment-based scoring function to re-rank ATP docking results, Int. J. Mol. Sci., 2007, vol. 8, pp. 1083–1094. https://doi.org/10.3390/i8111083
Chen, Z., Hu, Y., Hu, J., Yang, W., Sabatier, J.M., De Waard, M., Cao, Z., Li, W., Han, S., and Wu, Y., Unusual binding mode of scorpion toxin BmKTX onto potassium channels relies on its distribution of acidic residues, Biochem. Biophys. Res. Commun., 2014, vol. 447, pp. 70–76. https://doi.org/10.1016/j.bbrc.2014.03.101
Garcia, M.L., Garcia-Calvo, M., Hidalgo, P., Lee, A., and MacKinnon, R., Purification and characterization of three inhibitors of voltage-dependent K+ channels from Leiurus quinquestriatus var. hebraeus venom, Biochemistry, 1994, vol. 33, pp. 6834–6839. https://doi.org/10.1021/bi00188a012
Mouhat, S., Visan, V., Ananthakrishnan, S., Wulff, H, Andreotti, N., Grissmer, S., Darbon, H., De Waard, M., and Sabatier, J.M., K+ channel types targeted by synthetic OSK1, a toxin from Orthochirus scrobiculosus scorpion venom, Biochem. J., 2005, vol. 385, pp. 95–104. ttps://doi.org/10.1042/BJ20041379
Abbas, N., Belghazi, M., Abdel-Mottaleb, Y., Tytgat, J., Bougis, P.E., and Martin-Eauclaire, M.F., A new Kaliotoxin selective towards Kv1.3 and Kv1.2 but not Kv1.1 channels expressed in oocytes, Biochem. Biophys. Res. Commun., 2008, vol. 376, pp. 525–530. https://doi.org/10.1016/j.bbrc.2008.09.033
Kozminsky-Atias, A., Somech, E., and Zilberberg, N., Isolation of the first toxin from the scorpion Buthus occitanus israelis showing preference for Shaker potassium channels, FEBS Lett., 2007, vol. 581, pp. 2478–2484. https://doi.org/10.1016/j.febslet.2007.04.065
Abdel-Mottaleb, Y., Vandendriessche, T., Clynen, E., Landuyt, B., Jalali, A., Vatanpour, H., Schoofs, L., and Tytgat, J., OdK2, a Kv1.3 channel-selective toxin from the venom of the Iranian scorpion Odonthobuthus doriae, Toxicon, 2008, vol. 51, pp. 1424–1430. https://doi.org/10.1016/j.toxicon.2008.03.027
Kuzmenkov, A.I., Vassilevski, A.A., Kudryashova, K.S., Nekrasova, O.V., Peigneur, S., Tytgat, J., Feofanov, A.V., Kirpichnikov, M.P., and Grishin, E.V., Variability of potassium channel blockers in Mesobuthus eupeus scorpion venom with focus on Kv1.1: An integrated transcriptomic and proteomic study, J. Biol. Chem., 2015, vol. 290(19), pp. 12195–12209. https://doi.org/10.1074/jbc.M115.637611
Gao, B., Peigneur, S., Tytgat, J., and Zhu, S., A potent potassium channel blocker from Mesobuthus eupeus scorpion venom, Biochimie, 2010, vol. 92, pp. 1847–1853. https://doi.org/10.1016/j.biochi.2010.08.003
Koschak, A., Bugianesi, R.M., Mitterdorfer, J., Kaczorowski, G.J., Garcia, M.L., and Knaus, H.G., Subunit composition of brain voltage-gated potassium channels determined by hongotoxin-1, a novel peptide derived from Centruroides limbatus venom, J. Biol. Chem., 1998, vol. 273, pp. 2639–2644. https://doi.org/10.1074/jbc.273.5.2639
Cotton, J., Crest, M., Bouet, F., Alessandri, N., Gola, M., Forest, E., Karlsson, E., Castaneda, O., Harvey, A.L., Vita, C., and Menez, A., A potassium-channel toxin from the sea anemone Bunodosoma granulifera, an inhibitor for Kv1 channels. Revision of the amino acid sequence, disulfidebridge assignment, chemical synthesis, and biological activity, Eur. J. Biochem., 1997, vol. 244:192–202. https://doi.org/10.1111/j.1432-1033.1997.00192.x
Peigneur, S., Orts, D.J.B., Prieto da Silva, A.R., Oguiura, N., Boni-Mitake, M., de Oliveira, E.B., Zaharenko, A.J., de Freitas, J.C., and Tytgat, J., Crotamine pharmacology revisited: Novel insights based on the inhibition of KV channels, Mol. Pharmacol., 2012, vol. 82, pp. 90–96. https://doi.org/10.1124/mol.112.078188
Grissmer, S., Nguyen, A.N., Aiyar, J., Hanson, D.C., Mather, R.J., Gutman, G.A., Karmilowicz, M.J., Auperin, D.D., and Chandy, K.G., Pharmacological characterization of five cloned voltage-gated K+ channels, types Kv1.1, 1.2, 1.3, 1.5, and 3.1, stably expressed in mammalian cell lines, Mol. Pharmacol., 1994, vol. 45(6), pp. 1227–1234.
Chandy, K.G. and Norton, R.S., Peptide blockers of Kv1.3 channels in T cells as therapeutics for autoimmune disease, Curr. Opin. Chem. Biol., 2017, vol. 38:97–107. https://doi.org/10.1016/j.cbpa.2017.02.015
Beeton, C., Pennington, M.W., Wulff, H., Singh, S., Nugent, D., Crossley, G., Khaytin, I., Calabresi, P.A., Chen, C.Y., Gutman, G.A., and Chandy, K.G., Targeting effector memory T cells with a selective peptide inhibitor of Kv1.3 channels for therapy of autoimmune diseases, Mol. Pharmacol., 2005, vol. 67, pp. 1369–1381. https://doi.org/10.1124/mol.104.008193
Rashid, M.H., Huq, R., Tanner, M.R., Chhabra, S., Khoo, K.K., Estrada, R., Dhawan, V., Chauhan, S., Pennington, M.W., Beeton, C., Kuyucak, S., and Norton, R.S., A potent and Kv1.3-selective analogue of the scorpion toxin HsTX1 as a potential therapeutic for autoimmune diseases, Sci. Rep., 2015, vol. 4, pp. 4509. https://doi.org/10.1038/srep04509
Tajti, G., Wai, D.C.C., Panyi, G., and Norton, R.S., The voltage-gated potassium channel KV1.3 as a therapeutic target for venom-derived peptides, Biochem. Pharmacol., 2020, vol. 181, p. 114146. https://doi.org/10.1016/j.bcp.2020.114146
Chen, P., Dendorfer, A., Finol-Urdaneta, R.K., Terlau, H., and Olivera, B.M., Biochemical characterization of κM-RIIIJ, a Kv1.2 channel blocker, J. Biol. Chem., 2010, vol. 285, pp. 14882–14889. https://doi.org/10.1074/jbc.M109.068486
Orts, D.J.B., Peigneur, S., Madio, B., Cassoli, J.S., Montandon, G.G., Pimenta, A.M.C., Bicudo, J.E.P.W., Freitas, J.C., Zaharenko, A.J., and Tytgat, J., Biochemical and electrophysiological characterization of two sea anemone type 1 potassium toxins from a geographically distant population of Bunodosoma caissarum, Mar. Drugs, 2013, vol. 11, pp. 655–679. https://doi.org/10.3390/md11030655
Wang, X., Umetsu, Y., Gao, B., Ohki, S., and Zhu, S., Mesomartoxin, a new Kv1.2-selective scorpion toxin interacting with the channel selectivity filter, Biochem. Pharmacol., 2015, vol. 93, pp. 232–239. https://doi.org/10.1016/j.bcp.2014.12.002
Koch, R.O., Wanner, S.G., Koschak, A., Hanner, M., Schwarzer, C., Kaczorowski, G.J., Slaughter, R.S., Garcia, M.L., and Knaus, H.G., Complex subunit assembly of neuronal voltage-gated K+ channels. Basis for high-affinity toxin interactions and pharmacology, J. Biol. Chem., 1997, vol. 272, pp. 27577–27581. https://doi.org/10.1074/jbc.272.44.27577
ACKNOWLEDGMENTS
Supercomputer calculations were sponsored in the framework of the HSE University Basic Research Program and Russian Academic Excellence Project “5-100”. The molecular dynamics simulations were carried out using the computational facilities of the Supercomputer Center “Polytechnical” at the St. Petersburg Polytechnic University and IACP FEB RAS Shared Resource Center “Far Eastern Computing Resource” equipment (https://cc.dvo.ru).
Funding
This work was supported by the Russian Scientific Foundation (grant no. 20-44-01015). E.L. Pinheiro-Junior was supported by São Paulo Research Foundation (FAPESP, Brazil), no. 2016/04761-4, and Coordination for the Improvement of Higher Education Personnel (CAPES, Brazil), no. 88881.186830/2018-01.
Author information
Authors and Affiliations
Contributions
A.I. Kuzmenkov and A.A. Vassilevski designed the research. A.I. Kuzmenkov and A.M. Gigolaev performed biochemical experiments and recombinant peptide production. S. Peigneur and E.L. Pinheiro-Junior carried out electrophysiological studies. V.M. Tabakmakher performed molecular modeling. A.A. Vassilevski supervised the biochemical experiments. J. Tytgat supervised the electrophysiological experiments. R.G. Efremov supervised the molecular modeling. V.M. Tabakmakher, A.I. Kuzmenkov, and A.A. Vassilevski wrote the manuscript.
Corresponding author
Ethics declarations
CONFLICT OF INTEREST
The authors declare no evident or potential competing interests related to the publication of this article.
Additional information
Russian Text © The Author(s), 2021, published in Rossiiskii Fiziologicheskii Zhurnal imeni I.M. Sechenova, 2021, Vol. 107, Nos. 4–5, pp. 584–604https://doi.org/10.31857/S0869813921040130.
Rights and permissions
About this article

Cite this article
Tabakmakher, V.M., Kuzmenkov, A.I., Gigolaev, A.M. et al. Artificial Peptide Ligand of Potassium Channel KV1.1 with High Selectivity. J Evol Biochem Phys 57, 386–403 (2021). https://doi.org/10.1134/S0022093021020186
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0022093021020186