
Abstract
Stable carbon isotopes of long-chain n-alkanes in lake sediments have been used for reconstructing past environmental changes. Gas chromatography isotope ratio mass spectrometry coupled to a combustion unit is the commonly applied technique for compound-specific carbon isotope analysis. This paper shows that sulfur in the lacustrine sediment can cause severe interference during the mass spectrometric measurement, leading to significant biases in δ13C of n-alkane. The δ13C deviation could be resulted from additional 12C33S+, 13C32S+ at mass-to-charge ratio (m/z) 45, and 12C34S+, H2C32S+at m/z 46, respectively. Such elemental sulphur interference can be eliminated by the activated copper in the pretreatment procedure.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
INTRODUCTION
The leaf waxes of vascular plants are widely used to reconstruct environmental change because they are usually preserved in sediments from which they are easily extracted and analysed (Cranwell, 1981; Rieley et al., 1991). For example, stable carbon isotopic composition (δ13C) analysis of n-alkanes has been widely used to characterize past changes in the carbon cycle and vegetation ( Castañeda et al., 2009; Diefendorf and Freimuth, 2017; Schouten et al., 2007; Smith et al., 2007). Such advances are based on the new techniques for measuring δ13C. Automated online measurement using gas chromatography/combustion/isotope ratio mass spectrometry (GC/C/IRMS) is a valuable technique for studying stable organic compounds, such as n-alkane (Reinnicke et al., 2012). It can measure isotopic composition at low enrichment and natural abundance level, and obtain reliable δ13C values with an accuracy of 0.3‰ (Meier-Augenstein, 2004).
Despite the high sensitivity and accuracy, some uncertainties remain. For example, the concurrent presence of N2O (m/z 44, 45, 46) in the ion source of the IRMS instrument could heavily disturb the CO2 (m/z 44, 45, 46) ion current, resulting in anomalous δ13C values (Craig and Keeling, 1963). Schmitt et al. (2013) also reported the interference from 86Kr2+ during carbon isotope analysis and proved that it comes about by the lateral tailing of the peak of doubly charged 86Kr in the neighboring m/z 44, 45, and 46 Faraday cups.
Among them, the interference of elemental sulfur has been studied since 1950s (e.g. Blumer, 1957). Sulfur is readily dissolved in most common organic solvents used during extraction of organic matter from sediments and soils, interfering significantly in GC-MS and GC-ECD (Jin et al., 2007). Metallic copper treatment is widely used as an effective method. Blumer (1957) explored the removal efficiency of different types of copper wires for elemental sulfur. Jin et al. (2007) confirmed that copper powder has an apparent catalysis on chloroform during conventional Soxhlet Extraction. There are also some other studies that referred to the elemental sulfur removal in pretreatment process (e.g. Logan et al., 2001; Summons et al., 2002). With the improvement of solvent extraction efficiency, we require less and less organic matter content in the samples. This means that we have access to a broader archive of paleoclimates, such as loess or ancient sedimentary rocks. However, previous studies have been conducted mainly in samples with extremely high organic matter content and with high maturity associated with petroleum geochemistry, less has been reported on sulfur interference in sedimentary rocks or modern lake sediments with relatively low organic matter content. Neither the visual performance during the pretreatment process nor the detailed characteristics showed during the measuring of compound specific isotopes have been reported. Desulfurization was also not included as a standard experimental procedure in many studies (e.g. Castañeda and Schouten, 2011; Collins et al., 2013; Sachse et al., 2006; Schwab et al., 2015; Sinninghe Damsté et al., 2011; Tierney et al., 2011).
During the processing of a batch of Oligocene Lake sediments samples on the Tibetan Plateau, we stumbled upon the effect of elemental sulfur on the carbon isotope testing of specific compound of plant leaf wax n-alkane. In this study, we investigated the influence of sulfur on the δ13C values and explored the possible mechanisms of this unusual disturbance.
MATERIALS AND METHODS
Our samples were collected from the Dingqing Formation in the Lunpola Basin in central Tibet and are characterized by fine-grained lake sediments (Fig. 1). 147 samples were rinsed before grinding to powder. 30 g of each sample was extracted in a Dionex accelerated solvent extractor (ASE 200) with a dichloromethane (DCM): methanol (MeOH) 9 : 1 reagent mix. The extracts were separated into nonpolar and polar fractions through silica gel flash column chromatography using n-Hexane and MeOH as eluents, respectively. The nonpolar fraction was purified using urea adduction. The purified nonpolar part was divided into two equal volume control groups. One group was directly measured for carbon isotopes, and another group was further purified using activated Cu granules (HCL soaked, and DCM rinsed).

Map showing the Tibetan Plateau, the Lunpola Basin, and the Cenozoic strata in the study area. Q: Quaternary.
To confirm the mineral, morphological and elemental characteristics of elemental sulfur, measurements of the N2-dried nonpolar fractions (Group A) are performed using a ZEISS EVO-18 scanning electron microscope (SEM) equipped with a Bruker XFlash 6130 energy dispersive spectroscope (EDS). X-ray diffraction (XRD) patterns were measured on an X’pert Pro powder diffractometer (PANalytical Corp., the Netherlands) using Cu Kα radiation (λ = 0.15406 nm) at a scan rate of 0.017° min–1 (Zhu et al., 2019).
The concentrations of n-alkanes were determined using an Agilent 6890 gas chromatography (GC) instrument with a flame ionization detector. The samples were injected in a split mode, with an inlet temperature of 310°C and a flow rate of the carrier gas of 1.2 mL/min. The GC oven temperature program was: 40°C (1 min) to 150°C at 10°C/min, and then to 310°C (20 min) at 6°C/min.
Compound-specific stable carbon isotope analysis was carried out using a TRACE GC instrument coupled via a combustion reactor to the Thermo Finnigan Delta V Plus isotope ratio mass spectrometer. The temperature program was the same as the GC analysis. Isotope values were reported in per mil (‰) vs. Vienna Peedee Belemnite (VPDB). The standard deviations of duplicate analyses were <0.5‰ for the standard and selected sample. All the measurements were made at the Institute of Earth Environment, Chinese Academy of Science, Xi’an.
RESULTS AND DISCUSSION
The Identification of Elemental Sulfur (S8)
During the experiment, several anomalous samples with much more negative δ13C values were found in our section (Fig. 2). These samples produced 45/44 and 46/44 signals that differed by a factor of 10 compared to other samples in the record (Fig. 3). To identify the cause of the discrepancy, we repeated the whole experiment and found that some yellowish solids precipitated after N2-drying of the extracted total lipid (Fig. 4a). Additionally, the XRD results show sharp peaks at 2θ angles of 23.2°, 25.9°, 26.8°. 27.9°, 28.8°, and 31.4°, indicating a highly crystallized orthorhombic sulfur (S8) structure (Wadi et al., 2020) (Fig. 4b). The SEM images show that they are all characterized by euhedral rectangular crystals with variable sizes (Figs. 4c, 4d). The EDS spectra images demonstrate that they consist mainly of S (Fig. 4e). Based on these chemical and morphological results, we ensure that these solids are elemental sulfur (S8).

Lithology showed the type and color of sediments and δ13C record of Chebuli Section. The red diamond boxes highlight the abnormal samples.

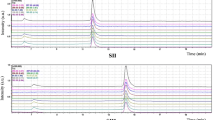
Online signals of the representative sample. (a) The original signal of the sulfur-contained sample. The signal intensity baseline of 46, 45 and 44 decreased gradually. (b) The sample signal after desulphurization. The difference of 46/44 and 45/44 was significantly reduced, and the baseline difference of signal intensity of 46, 45 and 44 was too small to be recognized.

Main characteristics of elemental sulfur in LPL27 (11.1 m). (a) Deposits after N2-drying of total extracts in room temperature; (b) XRD result. The red diamond boxes highlight the main 2θ angles of elemental sulfur; (c) SEM image (150x); (d) SEM image (1.15kx), the yellow plus symbol denote the location of the beam used in the EDS analysis; (e) EDS analysis result.
Elemental sulfur (S8) is insoluble in water but can be extracted with most organic solvents used in sample preparation, such as benzene and toluene, and is easily be chromatographed in the liquid phases (Andersson and Holwitt, 1994). This explains why elemental sulfur can also be recovered in our extraction procedures.
The Influence of Elemental Sulfur (S8) on Compound-Specific Carbon Isotope (δ13C) Measurements
The removal of elemental sulfur from hydrocarbon fractions by chemical leaching through a column of activated copper powder was first proposed by (Blumer, 1957), which can convert sulfur and copper into the black copper sulfide. Wu et al. (2011) further found that incubating 60 mL extracts with 12 g copper granules at 60°C for 2 h is the optimal procedure for removing interfering components. Because of the large amounts of each sample, we added 15 g active copper granules (DCM rinsed) to the extracts (dissolved in DCM), then placed them in a water bath at 30°C until there was no color change. Afterwards, we eluted the hydrocarbon fraction from the mixture.
During the analysis of the n-alkane carbon isotope ratios, different carbon length n-alkane molecules were separated using GC then combusted in an online reactor. Combustion took place in an O2 atmosphere to produce CO2, which is subsequently measured in the IRMS. Here, the most abundant isotopologues of CO2 falling into Faraday cups are the mass to charge ratios (m/z) 44, 45, and 46 (Schmitt et al., 2013).
The results of these untreated and S-removed samples are shown in Fig. 5. Obviously, ion current ratios of samples containing S deviate from those of pure n-alkane. Firstly, the 46/44 ratios of the treated samples were significantly lower, with a maximal value of 10, which was 3 to 5 times lower than that of the untreated samples (Fig. 5b). The 45/44 ratios show comparable results (Fig. 5a). Moreover, the δ13C values of the untreated samples can be up to ~3‰ negative and highly dispersed compared to that of the treated samples (Fig. 5c). Even for peaks which generated 4–6 V signals, the differences were as high as 1.3~2.8‰ (Table 1).

Comparison of the 46/44, 45/44 ion current ratios and the δ13C values between treated and untreated sample (LPL27).
The anomalies in the “45/44 ratio” should result in positive deviations in the δ13C values of the untreated samples. However, the observed δ13C values of the S containing samples are much lower. This is due to the 17O correction algorithm (Craig, 1957; Santrock et al., 1985), through which the 46/44 anomaly alters δ13C.
The measured ion currents at m/z 44, 45, 46 are dominated by 12C16\({\text{O}}_{2}^{ + }\), 13C16\({\text{O}}_{2}^{ + }\) and 12C16O18O+, respectively, as the abundances of 12C, 13C,16O, 17O and 18O are about 98.9, 1.1, 99.76, 0.04 and 0.2% (Coplen et al., 2002). The presence of 12C17O16O isotopologues accounts approximately 6~7% of mass 45 CO2 isotopologues (Kaiser, 2008). To determine the desired 13C/12C ratio, a 17O correction was applied to calculate the molecular ion-current ratio. According to Santrock et al. (1985), carbon δ values can be calculated from the carbon isotope ratios (13R, 17R, 18R) by the following simplified conversion equations.
The symbols 13R, 17R, and 18R refer to the abundance ratios 13C/12C, 17O/16O and 18O/16O, respectively. a is an empirical parameter used to express the observed relationship between 17R and 18R, and is expected to have a value of about 0.516 ± 0.015 (Kaiser, 2008; Young et al., 2002). K is a constant of proportional (Kaiser, 2008).
The calculation of 13R can be refer to in the following order:
1). 18R is calculated from 45R and 46R (eq. 4).
2). 17R is calculated from 18R (eq. 3).
3). 13R is calculated from 17R and 45R (eq. 1).
Following this sequence of calculations, if m/z 46 is anomaly high, the inaccuracy will be transferred to the ultimate carbon isotope value. Because 46R is primarily dependent on 18R, the solution of eq. (4) will yield a roughly correct value for 18R. However, the value of 17R will be overestimated by eq. (3). This will lead to an incorrectly low value for 13R (eq. (1)) and the derived δ13C value. Moreover, the use of eq. (3) in eq. (4) causes the initially calculated value of 18R (termed “roughly correct” above) to be underestimated. As a result, the δ18O values of the S containing samples are negatively biased (Table 1).
The SEM-EDS, XRD identifications, and the comparison of treated and untreated S-containing samples suggest elemental sulfur is the source of interference. However, it remains unclear how S interferes with the compound-specific carbon isotope (δ13C) measurements. Generally, there are three types of interference most encountered in IRMS analysis. Firstly, isobaric interference occurs when gas molecules giving rise to ions of the same mass/charge ratio as the analytic gas are present in the ion source at the same time (Meier-Augenstein et al., 2009). Example of isobaric interference is that between 14N16\({\text{O}}_{2}^{ + }\) and 12C16O18O+ affecting the abundant ions at m/z 46. Secondly, the formation of polyatomic interferences is induced by the reactions of the sample matrix, precursors in the plasma gas argon, the ambient air, and water (Pick et al., 2010). Pressurizing the reaction cell with a reaction gas like ammonia, methane, or oxygen leads to various ion-molecule reactions in the gas phase. Thus, three reaction schemes are possible: charge transfer, atom transfer, and collision-induced dissociation (Hattendorf and Günther, 2003; Pick et al., 2010). Finally, doubly charged ion interferences are due to doubly charged element isotopes with twice the mass of the analytic isotope, e.g. the alkaline and rare earth elements (Kohler and A. Leary, 1997).
One possible mechanism inducing these deviations is that the existence of S in the ion source changes the relative ionization efficiency of the carbon isotope isotopologues in the sense of “Ionization Quench” coined by Meier-Augenstein et al. (2009). This matrix interactions of the gasses in the ion source is commonly termed “chemical slope” (Severinghaus et al., 2003), referring to differences in the composition of the sample and standard gas mixture. However, in our results, all relevant signals (m/z 44, 45, 46) have increased by varying degrees, turning against lower ionization efficiency.
Alternatively, a more direct effect of S may be involved if the induced S ions fall into the Faraday cups. However, a direct isobaric effect seems unlikely as none of the S isotopes or combustion products are expected to fall into the given Faraday cups at their nominal m/z or m/2z (doubly charged ions) ratios. There may be other chemical reaction products that enter the Faraday cups.
When a certain amount of elemental sulfur is added into the combustion/isotope ratio mass spectrometry system, the most possible interference is isobaric interference. Mass interferences of 12C33S+ and 13C32S+ at m/z 45, and 12C34S+ at m/z 46 were detected from xylene blanks (Hausler, 1987). Moreover, Doddipatla et al. (2020) have proven that the reaction of the methylidyne radical (12C1H) with hydrogen sulfide (H232S) could make a reaction product with the chemical formula 1H212C32S plus atomic hydrogen (1H) in a single-collision event of two neutral reactants. The m/z 45 signal originated from dissociative electron-impact ionization of the parent molecule (H2C32S+; m/z 46) in the ionizer. All these ionic fragments can explain the elevated signals at m/z 46, 45 in our measurements.
At present, our mechanism about the behavior of sulfur ions in ion chromatography systems is only a conceptual model, which needs to be tested by further research. However, sulfur interference with monomeric carbon isotopes does exist, and as molecular level isotopes become more widely used as climate proxies, a sulfur removal procedure in the pretreatment process is necessary as a standard process.
The Sources of Sulfur in the Analysed Samples
Our samples were collected from lacustrine sediments with low redness values and high organic matter contents. Redness is associated with the number of iron oxides. In a lacustrine environment, iron oxides concentrations influenced by redox conditions which was primarily controlled by lake water depth (Kemp and Coe, 2009). This implies that the lake level was relatively high. The elemental sulfur (S8) in our samples may come from certain species of bacteria. For example, photosynthetic bacteria such as the green and purple sulfur bacteria (Chlorobium and Chrom atium, respectively) live within the anaerobic zone, where there is sufficient light penetration for them to make use of the products of anaerobic degradation processes (e.g., H2S and CO2). Although these phototrophic Sulphur bacteria can oxidize S2– to elemental Sulphur (during anoxygenic photosynthesis), they cannot oxidize S further to S\({\text{O}}_{4}^{{2 - }}\) (Killops and Killops, 2013). In fact, elemental sulfur (S8) is ubiquitous. It is extensively distributed in sediments and soils and can reach up to 1% or more (dry weight) in samples from non-pristine coal or harbor sediments (Andersson and Holwitt, 1994). It has also been found in matrices as diverse as lichen and air (Morosini et al., 1993), and has been shown to be endogenous in the epicuticular wax of many higher plants as a part of the antifungal defense system (Kylin et al., 1994). In such cases, it can dominate many environmentally important organic compounds by a wide margin and be a major disruption. However, most compound-specific carbon isotope pretreatment processes of n-alkanes did not involve the removal of elemental sulfur (Collister et al., 1994; Diefendorf and Freimuth, 2017; Huang et al., 1997; Rieley et al., 1991; Schefuß et al., 2003; Tipple and Pagani, 2010). Although these samples may not contain Sulphur, a clean-up procedure of remove sulfur is necessary to ensure the data accuracy.
CONCLUSIONS
This paper identified that elemental sulfur (S8) can severely interfere the δ13C measurement of n-alkanes. The interference for δ13C can be as large as 3‰. These disturbances may come from the lateral ion fragments produced in the combustion-isotope ratio mass spectrometry (GC/C/IRMS) system, e.g., 12C33S+, 13C32S+ at m/z 45, and 12C34S+, H2C32S+at m/z 46. To avoid this interference, we can utilize Cu to remove contaminating elemental sulfur (S8) in the pretreatment stage. Although we have repeated experiments many times to prove that this interference exists, in order to prevent errors caused by different experimental environments, further studies and cross-checks among different laboratories still needed.
REFERENCES
J. T. Andersson, and U. Holwitt, “An advantageous reagent for the removal of elemental sulfur from environmental samples,”. Fresenius’ J. Anal. Chem. 350, 474–480 (1994).
M. Blumer, “Removal of elemental sulfur from hydrocarbon fractions,” Anal. Chem. 29, 1039–1041 (1957).
I. S. Castañeda and S. Schouten, “A review of molecular organic proxies for examining modern and ancient lacustrine environments,” Quat. Sci. Rev. 30, 2851–2891 (2011).
I. S. Castañeda, J. P. Werne, T. C. Johnson, and T. R. Filley, “Late Quaternary vegetation history of southeast Africa: The molecular isotopic record from Lake Malawi,” Palaeogeogr., Palaeoclimatol., Palaeoecol. 275, 100–112 (2009).
J. A. Collins, E. Schefuß, S. Mulitza, M. Prange, M. Werner, T. Tharammal, A. Paul, and G. Wefer, “Estimating the hydrogen isotopic composition of past precipitation using leaf-waxes from western Africa,” Quat. Sci. Rev. 65, 88–101 (2013).
J. W. Collister, G. Rieley, B. Stern, G. Eglinton, and B. Fry, “Compound-specific δ13C of leaf lipids from plants with differing carbon dioxide metabolisms,” Org. Geochem. 21, 617–627 (1994).
T. B. Coplen, J. K. Böhlke, P. D. Bièvre, T. Ding, N. E. Holden, J. A. Hopple, H. R. Krouse, A. Lamberty, H. S. Peiser, K. Revesz, S. E. Rieder, K. J. R. Rosman, E. Roth, P. D. P. Taylor, R. D. Vocke, Jr., and Y. K. Xiao, “Isotope-abundance variations of selected elements (IUPAC Technical Report),” Pure Appl. Chem. 74, 1987–2017 (2002).
H. Craig, “Isotopic standards for carbon and oxygen and correction factors for mass-spectrometric analysis of carbon dioxide,” Geochim. Cosmochim. Acta. 12, 133–149 (1957).
H. Craig and C. D. Keeling, “The effects of atmospheric NO2 on the measured isotopic composition of atmospheric CO2,” Geochim. Cosmochim. Acta. 27, 549–551 (1963).
P. A. Cranwell, “Diagenesis of free and bound lipids in terrestrial detritus deposited in a lacustrine 554 sediment,” Org. Geochem. 3, 79–89 (1981).
A. F. Diefendorf and E. J. Freimuth, “Extracting the most from terrestrial plant-derived n-alkyl lipids and their carbon isotopes from the sedimentary record: a review,” Org. Geochem. 103, 1–21 (2017).
S. Doddipatla, C. He, R. I. Kaiser, Y. Luo, R. Sun, G. R. Galimova, A. M. Mebel, and T. J. Millar, “A chemical dynamics study on the gas phase formation of thioformaldehyde (H2CS) and its thiohydroxycarbene isomer (HCSH),” Proc. Natl. Acad. Sci. U. S. A. 117, 22712–22719 (2020).
B. Hattendorf and D. Günther, “Strategies for method development for an inductively coupled plasma mass spectrometer with bandpass reaction cell. Approaches with different reaction gases for the determination of selenium,” Spectrochim. Acta, Part B 58, 1–13 (2003).
D. Hausler, “Trace element analysis of organic solutions using inductively coupled plasma-mass spectrometry,” Spectrochim. Acta. 428, 63–73 (1987).
Y. Huang, G. Eglinton, P. Ineson, P. M. Latter, R. Bol, D. D. Harkenss, “Absence of carbon isotope fractionation of individual n-alkanes in a 23-year field decomposition experiment with Cufluna vulgaris,” Org. Geochem. 26, 497–501 (1997).
M. Jin, Y. Xia, T. Lei, S. Ma, J. Qiu, R. Zhang, “Note on sulfur removal with metallic copper,” Geochem. Int. 45, 620–623 (2007).
J. Kaiser, “Reformulated 17O correction of mass spectrometric stable isotope measurements in carbon dioxide and a critical appraisal of historic ‘absolute’ carbon and oxygen isotope ratios,” Geochim. Cosmochim. Acta. 72, 1312–1334 (2008).
D. B. Kemp and A. L. Coe, “A nonmarine record of eccentricity forcing through the Upper Triassic of southwest England and its correlation with the Newark Basin astronomically calibrated geomagnetic polarity time scale from North America,” Geology 35, 991 (2009).
S. Killops and V. Killops, “Production, preservation and degradation of organic matter,” in Introduction to Organic Geochemistry, Ed. by S. Killops and V. Killops (Blackwell, 2013), pp. 71–116.
M. Kohler and J. A. Leary, “Gas phase reactions of doubly charged alkaline earth and transition metal(II)-ligand complexes generated by electrospray ionization,” Int. J. Mass Spectrom. Ion Processes. 162, 17–34 (1997).
H. Kylin, S. Atuma, L. Hovander, and S. Jensen, “Elemental sulphur (S8) in higher plants - biogenie or anthropogenic origin?,” Experientia 50, 80–85 (1994).
G. A. Logan, M. C. Hinman, M. R. Walter, and R. E. Summons, “Biogeochemistry of the 1640 Ma McArthur River (HYC) lead-zinc ore and host sediments, Northern Territory, Australia,” Geochim. Cosmochim. Acta. 65, 2317–2336 (2001).
W. Meier-Augenstein, “GC and IRMS technology for 13C and 15N analysis on organic compounds and related gases,” in Handbook of Stable Isotope Analytical Techniques (Elsevier, Amsterdam, 2004), pp. 153–176.
W. Meier-Augenstein, H. F. Kemp, and C. M. Lock, “N2: a potential pitfall for bulk 2H isotope analysis of explosives and other nitrogen-rich compounds by continuous-flow isotope-ratio mass spectrometry,” Rapid Commun. Mass Spectrom. 23, 2011–2016 (2009).
M. Morosini, J. Schreitmueller, U. Reuter, and K. Ballschmiter, “Correlation between C-6/C-14 chlorinated hydrocarbons levels in the vegetation and in the boundary layer of the troposphere,” Environ. Sci. Technol. 27, 1517–1523 (1993).
D. Pick, M. Leiterer, and J. W. Einax, “Reduction of polyatomic interferences in biological material using dynamic reaction cell ICP-MS,” Microchem. J. 95, 315–319 (2010).
S. Reinnicke, D. Juchelka, S. Steinbeiss, A. Meyer, A. Hilkert, and M. Elsner, “Gas chromatography/isotope ratio mass spectrometry of recalcitrant target compounds: performance of different combustion reactors and strategies for standardization,” Rapid Commun. Mass Spectrom. 26, 1053–1060 (2012).
G. Rieley, R. J. Collier, D. M. Jones, G. Eglinton, P. A. Eakin, and A. E. Fallick, “Sources of sedimentary lipids deduced from stable carbon-isotope analyses of individual compounds,” Nature 352, 425–427 (1991).
D. Sachse, J. Radke, and G. Gleixner, “δD values of individual n-alkanes from terrestrial plants along a climatic gradient—Implications for the sedimentary biomarker record,” Org. Geochem. 37, 469–483 (2006).
J. Santrock, S. A. Studley, and J. M. Hayes, “Isotopic analyses based on the mass spectrum of carbon dioxide,” Anal. Chem. 57, 1444–1448 (1985).
E. Schefuß, V. Ratmeyer, J. B. W. Stuut, J. H. F. Jansen, and J. S. Sinninghe Damsté, “Carbon isotope analyses of n-alkanes in dust from the lower atmosphere over the central eastern Atlantic,” Geochim. Cosmochim. Acta. 67, 1757–1767 (2003).
J. Schmitt, B. Seth, M. Bock, C. Veen, L. Möller, C. J. Sapart, M. Prokopiou, T. Sowers, T. Röckmann, and H. Fischer, “On the interference of 86Kr2+ during carbon isotope analysis of atmospheric methane using continuous flow combustion - isotope ratio mass spectrometry,” Atmos Meas Tech. 6, 1409–1460 (2013).
S. Schouten, M. Woltering, W.I.C. Rijpstra, A. Sluijs, H. Brinkhuis, J.S. Sinninghe Damsté, “The Paleocene–Eocene carbon isotope excursion in higher plant organic matter: Differential fractionation of angiosperms and conifers in the Arctic,” Earth Planet. Sci. Lett. 258, 581–592 (2007).
V. F. Schwab, Y. Garcin, D. Sachse, G. Todou, O. Séné, J. M. Onana, G. Achoundong, and G. Gleixner, “Effect of aridity on δ13C and δD values of C3 plant- and C4 graminoid-derived leaf wax lipids from soils along an environmental gradient in Cameroon (Western Central Africa),” Org. Geochem. 78, 99–109 (2015).
J. P. Severinghaus, A. Grachev, B. Luz, and N. Caillon, “A method for precise measurement of argon 40/36 and krypton/argon ratios in trapped air in polar ice with applications to past firn thickness and abrupt climate change in Greenland and at Siple Dome, Antarctica,” Geochim. Cosmochim. Acta. 67, 325–343 (2003).
J. S. Sinninghe Damsté, D. Verschuren, J. Ossebaar, J. Blokker, R. Houten, M. T. J. Meer, B. Plessen, and S. Schouten, “A 25,000-year record of climate-induced changes in lowland vegetation of eastern equatorial Africa revealed by the stable carbon-isotopic composition of fossil plant leaf waxes,” Earth Planet. Sci. Lett. 302, 236–246 (2011).
F. Smith, S. Wing, and K. Freeman, “Magnitude of the carbon isotope excursion at the Paleocene–Eocene thermal maximum: the role of plant community change,” Earth Planet. Sci. Lett. 262, 50–65 (2007).
R. E. Summons, P. Metzger, C. Largeau, A. P. Murray, and J. M. Hope, “Polymethylsqualanes from Botryococcus braunii in lacustrine sediments and crude oils,” Org. Geochem. 33, 99–109 (2002).
J. E. Tierney, J. M. Russell, J. S. Sinninghe Damsté, Y. Huang, and D. Verschuren, “Late Quaternary behavior of the East African monsoon and the importance of the Congo Air Boundary,” Quat. Sci. Rev. 30, 798–807 (2011).
B. J. Tipple and M. Pagani, “A 35Myr North American leaf-wax compound-specific carbon and hydrogen isotope record: Implications for C4 grasslands and hydrologic cycle dynamics,” Earth Planet. Sci. Lett. 299, 250–262 (2010).
V. S. Wadi, K. Halique, and S. M. Alhassan, “Polypropylene–elemental sulfur (S8) composites: effect of sulfur on morphological, thermal, and mechanical properties,” Ind. Eng. Chem. Res. 59, 13079–13087 (2020).
J. Wu, Y. Lin, J. Lu, and C. Wilson, “Copper clean-up procedure for ultrasonic extraction and analysis of pyrethroid and phenylpyrazole pesticides in sediments by gas chromatography-electron capture detection,” Sci. Total Environ. 409, 3482–3491 (2011).
E. D. Young, A. Galy, and H. Nagahara, “Kinetic and equilibrium mass-dependent isotope fractionation laws in nature and their geochemical and cosmochemical significance,” Geochim. Cosmochim. Acta. 66, 1095–1104 (2002).
D. Zhu, Y. Huang, J. Cao, S. C. Lee, M. Chen, and Z. Shen, “Cobalt nanoparticles encapsulated in porous nitrogen-doped carbon: Oxygen activation and efficient catalytic removal of formaldehyde at room temperature,” Appl. Catal. B. 258, 117981 (2019).
ACKNOWLEDGMENTS
This study was financially supported by the National Natural Science Foundation of China (41888101 and 41672168), the Strategic Priority Research Program of Chinese Academy of Sciences (XDA2007020202 and XDA2007020205), and the Beijing Geological survey project (11000022T000000440137 and 11000022T000000440175). In addition, we are very grateful to the associated editor Prof. Mikhail Spasennykh and two anonymous reviewers for their valuable comments on our article, as well as the time and effort of Dr. Tatiana Shishkina in editing and contacting.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of interest.
Rights and permissions
About this article

Cite this article
Mengmeng Cao, Cao, Y., Sun, J. et al. Effect of Elemental Sulfur (S8) on Carbon Isotope Analysis of n-Alkanes. Geochem. Int. 61, 1155–1163 (2023). https://doi.org/10.1134/S0016702923080049
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0016702923080049