
Abstract
Stable carbon (C) and nitrogen (N) isotope ratios of sedimentary organic matter (OM) can reflect the biogeochemical history of aquatic ecosystems. However, diagenetic processes in sediments may alter isotope records of OM via microbial activity and preferential degradation of isotopically distinct organic components. This study investigated the isotope alteration caused by preferential degradation in surface sediments sampled from a eutrophic reservoir in Germany. Sediments were treated sequentially with hot water extraction, hydrochloric acid hydrolysis, hydrogen peroxide oxidation and di-sodium peroxodisulfate oxidation to chemically simulate preferential degradation pathways of sedimentary OM. Residue and extracts from each extraction step were analyzed using elemental analyzer-isotope ratio mass spectrometry and solid-state 13C nuclear magnetic resonance spectroscopy. Our results show that stable C and N isotope ratios reacted differently to changes in the biochemical composition of sedimentary OM. Preferential degradation of proteins and carbohydrates resulted in a 1.2‰ depletion of 13C, while the isotope composition of 15N remained nearly the same. Sedimentary δ15N values were notably altered when lignins and lipids were oxidized from residual sediments. Throughout the sequential fractionation procedure, δ13C was linearly correlated with the C:N of residual sediments. This finding demonstrates that changes in biochemical composition caused by preferential degradation altered δ13C values of sedimentary OM, while this trend was not observed for δ15N values. Our study identifies the influence of preferential degradation on stable C isotope ratios and provide additional insight into the isotope alteration caused by post-depositional processes.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The biogeochemical cycling of organic carbon (OC), inorganic carbon and nutrients in aquatic ecosystems is affected by the properties of sedimentary organic matter (OM), which determine the degradation and burial of OM (Herczeg et al. 2001). Stable carbon (C) and nitrogen (N) isotope ratios, along with the C:N of sedimentary OM, serve as proxies for inferring the biogeochemical dynamics and for reconstructing paleo-environments in lakes and reservoirs (Meyers 1997). Because photosynthesis has multiple inorganic sources and pathways, aquatic and terrestrial sedimentary OM are different in original isotope composition (Meyers and Ishiwatari 1993; Ogrinc et al. 2005; Teodoru et al. 2013). Changes in environmental properties in sediments, such as redox conditions, microbiology and thermodynamics, can lead to a shift of the original isotope composition of sedimentary OM (Arndt et al. 2013). By interpreting depth profiles of stable C and N isotope ratios of OM in sediment cores, changes in lake productivity, trophic state, external discharge or water levels in freshwater or coastal ecosystems can be tracked (Brenner et al. 1999; Gu et al. 1996; Lamb et al. 2006; Woodward et al. 2012).
These isotope signals must be interpreted with caution, however, since early diagenesis may cause additional alterations in stable isotope ratios of sedimentary OM that could modify the original isotope signal (Meyers and Lallier-Vergès 1999; Schelske and Hodell 1995). Previous studies indicate that both aerobic and anaerobic degradation of OM can alter stable C and N isotope ratios in sediments (Freudenthal et al. 2001; McArthur et al. 1992). Similarly, enrichment of 15N in residual sedimentary OM has been observed after microbial degradation of algae-derived OM (Altabet et al. 1995; Saino and Hattori 1980). Organic components with distinct stable isotope ratios exhibit different degradation kinetics in sediments (Fabiano et al. 1995; Guillemette et al. 2016; Hatcher et al. 2014; Hayes 1993; Hayes et al. 1990; Mahmoudi et al. 2017). Therefore, changes in their biochemical composition are often accompanied by alterations in stable isotope ratios during early diagenesis of sedimentary OM (Hatcher et al. 1983).
Organic geochemists have long studied mechanisms of early diagenesis-induced alterations in stable C and N isotope ratios of sedimentary OM. Macko and Estep (1984) examined the influence of microbial activity on the distribution of sedimentary stable C and N isotopes, testing microbial isotopic fractionation factors in different substrates. Their results demonstrated that changes in the biochemical environment of the sediment could mediate microbial isotope alterations. Lehmann et al. (2002) investigated diagenetic alterations in stable isotope ratios in anoxic incubation experiments and also hypothesized systematic effects of microbial activity and preferential degradation on stable C and N isotope ratios. Sun et al. (2016) argued that changes in the isotope composition of sedimentary OM represented the rate of OM decomposition rather than the consequence of early diagenesis in a high elevation alpine lake. In contrast, a 12-week in-site incubation experiment on the biochemistry of estuarine plants showed no significant stable C and N isotope alterations during decomposition (Lanari et al. 2018), thus indicating that isotope alterations may require more time.
It remains difficult to distinguish microbially mediated isotope fractionation from preferential degradation of individual organic components when isotope compositions of bulk sedimentary OM are altered during early diagenesis. Inconsistent results for the influence of microbial activity on stable isotope ratios are likely related to varying post-depositional environments (Freudenthal et al. 2001; Holmes et al. 1999; Lehmann et al. 2002). These unresolved issues make it difficult to assess the influence of preferential degradation on the bulk stable isotope ratios during early diagenesis. Researchers usually consider two or more mechanisms to explain alterations in stable isotopes during early diagenesis (Freudenthal et al. 2001; McArthur et al. 1992). Hence, studies of changes in biochemical composition associated with preferential degradation but without bacterial effects would advance mechanistic understanding of the diagenetic alteration of sedimentary stable isotope ratios.
New insights into correlations between biochemical compositions and stable isotope ratios of sedimentary OM could provide information to support studies of diagenetic alteration of stable isotope ratios. In previous studies, early diagenesis of OM was described using chemical analysis such as solid-state 13C nuclear magnetic resonance (13C-NMR) spectroscopy (Hatcher et al. 2014; Mao et al. 2011), or by determining stable isotope composition (McArthur et al. 1992). Combined 13C-NMR and stable isotope analyses can provide information to explain mechanisms involved in early diagenesis of OM in lacustrine and estuarine sediments (Krull et al. 2009; Longbottom and Hockaday 2019). It is widely accepted that components of sedimentary OM are preferentially degraded in the following order: proteins, carbohydrates, carbonyls, lipids, lignins and chars (Arndt et al. 2013; Burdige 2007; Last and Smol 2006; Middelburg 2018; Wakeham et al. 1997).
We aimed to distinguish the effects of preferential degradation caused by microbial metabolism during early diagenesis of OM on sedimentary stable isotopes as indicators of the paleo-environment and paleo-limnology. We performed a sequential extraction procedure and joint isotope and 13C-NMR analyses of the sedimentary OM to simulate and clarify effects of preferential degradation. We hypothesized that preferential degradation can change the biochemical composition of OM and thus cause post-depositional alteration of stable C and N isotopes. Because of preferential degradation, stable C and N isotopes were expected to change asymmetrically, since differing degrees of microbial activity may be involved in the early diagenesis of OM.
Materials and methods
Field site and sediment sampling
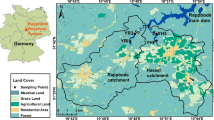
This study was conducted in the Hassel and Rappbode pre-dams of the Rappbode Reservoir System in the Harz Mountains, Germany (51.74° N, 10.89° E, Fig. 1), which came into service in the 1960s to avoid heavy sediment and nutrient loads in the main reservoir. The Hassel pre-dam is a eutrophic water impoundment (Rinke et al. 2013) with a surface area, water capacity, maximum depth and water retention time of 0.288 km2, 1.64 million m3, 14 m and 65 days, respectively (Dadi et al. 2015). The Rappbode pre-dam is an oligotrophic impoundment with a surface area, water capacity, maximum depth and water retention time of 0.218 km2, 1.66 million m3, 17 m and 52 days, respectively (Friese et al. 2014). These systems have been the subject of previous studies of isotopes in the water column (Barth et al. 2017).

Map of the Rappbode Reservoir System with sampling sites (red points) and location in Germany (top left). The geological data come from ATKIS1 DGM50 M745; Geobasisdaten© Vermessungsverwaltungen der Bundesländer und BKG (www.bkg.bund.de); The map was created using ArcMap (ESRI 2014). ArcGIS Release 10.2. Redlands, California USA). (Color figure online)
Sediments were collected with a simple grab sampler in April 2011 in the Hassel and Rappbode pre-dam reservoirs. The pH of sediments was 7–8. To simplify the simulated degradation system, sediments used for sequential fractionation in this study were an equal mixture of the top 20 cm material from 10 sampling points (5 from each pre-dam, Fig. 1). Aliquots of freeze-dried samples from the 10 sampling points were mixed by continuously rotating the material in a closed barrel. The homogeneity of the sample was confirmed by repeated analysis until the coefficient of variation in total C or N dry weight was less than 5%.
Sequential fractionation of sediments
The fractionation procedure consisted of four steps: (i) hot water extraction, (ii) hydrochloric acid (HCl) hydrolysis, (iii) hydrogen peroxide (H2O2) oxidation, and (iv) disodium peroxodisulfate (Na2S2O8) oxidation (Fig. 2). In principle, hot water extracts proteins and carbohydrates that have high bioavailability (Haynes 2005; Leinweber et al. 1995; Sparling et al. 1998), while HCl preferentially hydrolyzes most amides, nucleic acids, polysaccharides and certain carboxyl compounds (Paul et al. 1997; Paul et al. 2006). Subsequently, both H2O2 and Na2S2O8 oxidize aliphatic and aromatic organic compounds (Jagadamma et al. 2010; Mikutta et al. 2005; von Lützow et al. 2007). However, H2O2 is more effective in removal of alkyl compounds, while Na2S2O8 tends to remove O/N-alkyl compounds (Helfrich et al. 2007). The C and N stable isotope ratios and biochemical compositions of all residue generated in this fractionation procedure were analyzed.

Diagram of the sequential fractionation procedure. Three replicates were fractionated with successive extraction with hot water, hydrolysis with 6 M HCl, and oxidization with 10% H2O2 and Na2S2O8. After each fractionation step, extracts and a portion of residual sediments were collected for analysis, and the other portion was maintained for additional fractionation until the end of the procedure. In the diagram, IRMS-EA and CP/MAS 13C NMR are abbreviations for isotope ratio mass spectrometer coupled with elemental analyzer and cross polarization/magic angle spinning 13C nuclear magnetic resonance, respectively
Extraction with hot water
Hot water extraction of sediments was modified following the method of Ghani et al. (2003). Three replicates were prepared for the extraction of sedimentary OM. Aliquots of dry and ground sediments (3 g) were suspended in 30 mL of distilled water in a 50 mL polypropylene centrifuge tube and mixed on a shaker for 30 min at 200 rotations per minute (rpm). The suspension was centrifuged at 3500 rpm at 20 °C for 20 min (Heraeus Labofuge 400R, Heraeus Quarzglas GmbH & Co. KG, Bitterfeld-Wolfen, Germany). The supernatant was collected, and the residue was suspended in 30 mL of distilled water. The residue and water were mixed on a vortex shaker (Multifunction Vortex Mixer Set VM-10, Witeg Labortechnik GmbH, Wertheim, Germany) for 30 s and then left in a water bath at 80 °C for 20 h. To release the hot water extractable organic compounds completely, centrifuge tubes were shaken for 60 s on a vortex shaker, followed by re-centrifuging at 3500 rpm at 20 °C for 20 min. The supernatant was filtered through a 0.45 µm cellulose nitrate membrane filters. The filtrate was mixed with the supernatant, and the filter residue was combined with the corresponding centrifugal residue. All residue was dried at 60 °C and ground manually with an agate mortar. The hot water-resistant residue was weighted and kept in a vacuum desiccator for analysis or additional treatments.
Acid hydrolysis with HCl
Three replicates (2 g each) of the hot water-resistant residue were placed in three 50 mL centrifuge tubes for hydrolysis with 6 M HCl (25 mL, Fig. 2). All tubes with suspension were placed on a shaker (TitroWiCo Orbital Shaker, Bochum, Germany), shaken at 200 rpm for 2 h, and then placed in a water bath at 80 °C for 20 h (Silveira et al. 2008). Next, 6 M HCl resistant residue was separated from the supernatant by centrifuging at 3500 rpm at 20 °C for 25 min. The residue was repeatedly rinsed with 10 mL of distilled water and centrifuged (3000 rpm at 20 °C for 15 min). The last step was repeated three times. Centrifugation supernatant and rinses were combined in brown glass bottles and dried at 60 °C. The residue was ground into homogeneous powder and weighed before the next treatment. A portion of the residue (ca. 1 g) was collected for H2O2 oxidation, while the remaining material was kept in a vacuum desiccator for subsequent analysis.
Oxidation with H2O2
The H2O2 oxidation procedure was modified from the methods of Helfrich et al. (2007) and Jagadamma et al. (2010) to consider differences between soils and sediments. One gram of 6 M HCl resistant residue was suspended in 10 mL of distilled water in a 50 mL centrifuge tube. The first two doses of 10 mL 10% H2O2 were added to the suspension at an interval of 3 h. The mixed solution was kept in an ultrasonic bath to ensure that the reactants responded adequately. After thoroughly mixing the H2O2- and HCl-resistant residue, and the centrifuge tubes were placed in a water bath at 50 °C overnight. The last dose of H2O2 (10%, 10 mL) was added, and the tubes were left in the water bath (50 °C) until the suspensions stopped effervescing. The suspensions were shaken on a vortex shaker for 60 s to separate the oxidized OM completely from the residue. The residue was isolated by centrifuging (3500 rpm at 20 °C, 20 min), washed with 10 mL of 10% H2O2 and placed in a water bath at 50 °C for 2 h to react. After re-centrifugation of the suspension, the residue was washed three times with 10 mL of distilled water. The centrifugation supernatant, and the H2O2 and distilled water rinses were collected for the analysis of stable C isotope ratio. The H2O2-resistant residue was dried in an oven at 45 °C and ground for analysis or was retained for the fourth step of the fractionation procedure.
Oxidation with Na2S2O8
The residue that had been treated with hot water, HCl and H2O2 was then oxidized by Na2S2O8. Specifically, 0.5 g of H2O2-resistant residue was suspended in 40 mL of Milli-Q water in an ultrasonic bath for 20 min. The suspension was mixed with 4 g of Na2S2O8, and the reaction was simultaneously buffered with 4.4 g NaHCO3 (Helfrich et al. 2007; Lorenz et al. 2008). H2O2-resistant residue and chemical reagents were mixed completely on a vortex shaker before being placed in a water bath at 80 °C for 18–24 h, until the effervescence stopped. The centrifuge tubes were again placed on a vortex shaker and then centrifuged at 3500 rpm at 20 °C for 20 min. The supernatant was collected, and the residue was rinsed three times with 10 mL of MilliQ water. Subsequently, rinses were centrifuged (3500 rpm at 20 °C for 10 min) to further separate and purify the residue. To avoid retaining carbonate in the moist residue, the residue was dried and homogenized and then acidized with 20 mL of 1 M HCl for 18 h while being agitated at 180 rpm at 25 °C. The suspension was re-centrifuged at 3500 rpm at 20 °C for 20 min, and the residue was rinsed with Milli-Q water until the pH exceed 6. All supernatants and rinses were collected and combined. Residue was dried at 40 °C and then homogenized for isotope ratio and 13C-NMR analyses.
Analysis of sedimentary organic matter in residue and extracts
Total OC (TOC), total N (TN) and stable C and N isotope ratios of chemically resistant residue were measured. In addition, extracts from the fractionation procedure were analyzed for TOC and stable C isotope composition.
Analysis of elemental and stable isotope ratios
Extracts were analyzed with an isotope ratio mass spectrometer (IRMS, Thermo Fisher Delta V Plus, Bremen, Germany) coupled in continuous-flow mode with an OI Analytical Aurora 1030W TOC analyzer (College Station, Texas, USA). Elemental (C, N) and stable isotope 13C/12C and 15N/14N ratios of the original sediments and chemically resistant residue were analyzed using a Flash 2000 elemental analyzer coupled with a Delta V Advantage IRMS (Thermo Fisher Scientific, Bremen, Germany). The original mixed sediments (“Field site and sediment sampling” section) with known elemental and isotope compositions were used as reference materials to test the stability of the instruments. Reference materials run after the measurements in this study were interspersed with analyses of samples from other studies. For this procedure, 4 mg of residual sediments were packed into a tin capsule, which was then closed and crimped. Contents of C and N were measured as percentages (by weight), with a standard deviation of ± 0.08% and 0.01% for C (n = 6) and N (n = 6) in standards, respectively. These percentages were converted into absolute C and N mass per gram of dry sediments (mg g− 1), and the corresponding atomic C:N was calculated.
Stable isotope composition was converted into δ13C and δ15N using the following equation:
where R is 13C/12C or 15N/14Nand δ is the isotope ratio of 13C or 15N relative to the international reference materials: Vienna PeeDee Belemnite for δ13C and atmospheric N2 for δ15N. Standard deviations of isotope measurements of sediment standards were less than ± 0.1‰ for C (n = 6) and ± 0.2‰ for N (n = 6).
Analysis of biochemical compositions
The compositions of original and residual sediments were analyzed using solid-state 13C-NMR spectroscopy (Bruker Avance 300, Bruker Biospin, Bremen, Germany). The cross polarization/magic angle spinning (CP/MAS) 13C-NMR spectra were obtained at a 13C frequency of 75.47 MHz. The spectroscope was equipped with a 4 mm MAS probe head (product number: Bruker BL4). The 90° proton pulse was set at 3.3 µs, and the decoupling strength during acquisition was 69 kHz. The following conditions were applied: spinning rate of 15 kHz, recycle delay of 3 s and contact time of 2 ms. The samples were then introduced into 4 mm zirconium oxide rotors.
A line-broadening function (LB = 1 Hz) was used for Fourier transformation. Chemical shifts were externally referenced to the glycine resonance at 176.03 ppm. Chemical shift regions of − 10 to 45, 45 to 60, 60 to 95, 95 to 110, 110 to 145, 145 to 160 and 160 to 210 ppm were assigned to alkyl compounds; N-alkyl-methoxy compounds; O-alkyl compounds; O2-alkyl compounds; aromatic compounds; O-aromatic structures with phenols; and carboxyl or carbonyl compounds, respectively (Pane et al. 2013; Rodríguez-Murillo et al. 2011). The 13C-NMR spectral areas were integrated according to the chemical shift regions to assess the relative contributions of these functional groups. The main resonance peaks were referred to their typical spectroscopic signals of chemical compounds (Hatcher 1987; KoÈgel-Knabner 2002).
Using a molecular mixing model to estimate biochemical composition
The mixing model of Nelson and Baldock (2005) was used to infer the biochemical composition of sedimentary OM. It categorized OM into six components: carbohydrates, proteins, lignins, lipids, carbonyls and char. The model assumed that these six components were linearly combined and then tested for the best fit to the area-integration results of 13C-NMR spectra. For example, the acquired percentages of several functional groups (Table 2) and the C:N of residual sediments (Fig. 3) were used to constrain the model. This model was used to estimate the percentages of these six organic components in the residual sediments at each step of the sequential extraction procedure. The biochemical compositions of two consecutive extraction steps derived from the 13C-NMR spectra were compared to estimate the removal of organic components by the latter step.

a Carbon (C) and b nitrogen (N) contents in residual sediments during the sequential fractionation procedure. Error bars represent one standard deviation of three replicates samples
Statistical analysis
The effectiveness of each extraction step was estimated as the percentage of TOC and TN contents extracted by each extraction step relative to the TOC and TN contents in the original sediments. The effectiveness of this procedure was also estimated by changes in δ13C and δ15N values and the percentages of various components between two consecutive extraction steps. A Student’s t-test (unpaired) was used to compare the extraction effectiveness between two extraction steps using R software (RCore Team 2013). Differences were considered significant at p < 0.05. The correlation between C:N and δ13C or δ15N values was tested by a Pearson correlation analysis using the correlation test package of R Studio. Correlations were considered significant at p < 0.05. All results were calculated as a mean ± standard deviation (1σ) of three replicate samples.
Results
Variations in stable isotopic compositions of sedimentary organic matter
As the fractionation procedure progressed, OC and N contents of the residual sediments decreased gradually (Fig. 4). The OC content in hot water-resistant residue decreased by 10.8 mg g− 1 after 6 M HCl hydrolysis. During H2O2 oxidation OC was oxidized to CO2 rather than dissolved in solvents, thus C concentration in the H2O2 extracts was low (Table 1). The oxidation with H2O2 decreased OC content by an additional 16.3 mg g− 1. Similarly, Na2S2O8 further oxidized the OC in the H2O2-resistant residue, which resulted in an extremely low C concentration in the Na2S2O8 extracts (Table 1). Overall, hot water, HCl, H2O2 and Na2S2O8 extraction removed ca. 5%, 18%, 27% and 44% of TOC, respectively.

a The atomic carbon-to-nitrogen ratio (C:N), b stable C isotope ratio and c stable N isotope ratio of residual sediments at each step of the sequential fractionation procedure. Error bars represent one standard deviations of three replicate samples
Reaction with 6 M HCl removed organic N effectively, decreasing the 5.4 mg N g− 1 in hot water-resistant to 2.8 mg N g− 1, which accounted for about half of the TN in original sediments. In further steps, H2O2 and Na2S2O8 oxidation decreased N content by 1.0 and 1.2 mg g− 1, respectively. Hot water, HCl, H2O2 and Na2S2O8 extraction removed 8%, 45%, 16% and 17% of TN from original bulk sediments, thus indicating that hot water extraction and 6 M HCl hydrolysis removed larger proportions of N than OC. Consequently, the C:N of residual sediments increased with each extraction step until OC was extensively oxidized by Na2S2O8 (Fig. 3a).
Compared to the original bulk sediments, the δ13C value of residual sediments that were treated by hot water decreased by 0.2‰ while the δ15N value increased by 0.5‰ (Fig. 3b and c). Both changes associated with hot water extraction in δ13C and δ15N values were significant (p < 0.05), as indicated by a Student’s t-test. The changes in residue δ13C values coincided with the changes in δ13C values between the original bulk sediments and the hot water extracts (Table 1). Hydrolysis with 6 M HCl decreased δ13C and δ15N values significantly (by 1.0 and 0.7‰, respectively, both p < 0.005, Student’s t-test, Fig. 3b and c), which was consistent with the higher δ13C values (− 27.2‰) of HCl extracts (Table 1). The oxidation with H2O2 oxidation decreased δ13C values in residue slightly (Fig. 3b) but decreased δ15N in residue significantly (by 1.3‰, p < 0.05, Student’s t-test, Fig. 3c), which suggests that the δ13C value of H2O2-oxidizable OM was similar to that of 6 M HCl non-hydrolysable OM. Since Na2S2O8 oxidized nearly all OC, the δ13C value of its residue increased from − 29.4‰ to − 27.2‰ (p < 0.001, Student’s t test, Fig. 3b), and its δ15N value also decreased by a similar degree (5.2‰, Fig. 3c).
Solid-state 13C-NMR analysis of sedimentary organic matter
With solid-state CP/MAS 13C-NMR analysis, representative organic compounds in the original and residual sediments were identified by typical signal peaks of chemical shifts (Fig. 5). The signal peak at 22 ppm may have corresponded to long-chain methyl and methylene OC in fatty acids, waxes and resins (Jagadamma et al. 2010; Vane et al. 2005). Based on previous findings, the peak at 65 ppm was in the overlapping regions of chemical shifts, which represented for proteinaceous C or carbohydrates (Baldock et al. 1992).

Solid-state cross-polarization/magic-angle-spinning 13C nuclear magnetic resonance spectra of original bulk sediments and residual sediments obtained from the sequential fractionation procedure. Spectra were analyzed and stacked using Bruker TopSpin4.0.6
The O/O2-alkyl C that peaked at 87 ppm and 97 ppm were the most prominent polysaccharides or carbohydrates with anomeric C (Baldock et al. 2004; Vane et al. 2005). Resonance peaks at 121 ppm with shoulders were interpreted as a signal of lignins (Hatcher 1987), while a peak at 166 ppm was interpreted as to carboxyl, ester and amide compounds (Monteil-Rivera et al. 2000).
After the 6 M HCl hydrolysis and H2O2 oxidation, the peak at 22 ppm in the 13C-NMR spectra of residual sediments remained and became sequentially sharper (Fig. 5), due to a change in biochemical composition or the removal of paramagnetic impurities. Although the peak at 121 ppm strengthened after the 6 M HCl hydrolysis, it decreased after H2O2 oxidation (Fig. 5). Sedimentary OM was transformed during the fractionation, since three peaks that occurred after 6 M HCl hydrolysis (in the vicinities of 48, 65 and 79 ppm) were changed to a broad peak at 65 ppm after the H2O2 oxidation (Fig. 5). The oxidation of H2O2-resistant residue with Na2S2O8 resulted in ineffective cross-polarization of the residual sediments, which led to a non-identifiable signal in the 13C-NMR spectrum (Fig. 5).
The chemical shift regions in the 13C-NMR spectra were integrated to quantify functional groups in the original and residual sediments (Table 2). About 40%, 30% and 21% of OC in the original bulk sediments was alkyl C; substituted alkyl C; and aromatic and O-aromatic C, respectively. Hot water extraction changed the chemical structure of sedimentary OM slightly (Fig. 5), decreasing the proportions of N-alkyl, O-alkyl and O-aromatic C by 1.4, 1.8 and 1.5 percentage points, respectively. In the 13C-NMR spectra of 6 M HCl-resistant residue, the percentage of alkyl C increased by ca. 10 percentage points, while the total percentage of O-alkyl and carboxyl C decreased by the same amount. The alkyl C percentage in the 6 M HCl resistant residue increased considerably (ca. 10 percentage points) after H2O2 oxidation. The percentage of aromatic C steadily increased with the hot water and HCl extraction, but decreased after H2O2 oxidation. Since the noise in the spectrum of Na2S2O8 resistant residue was high, the integrated results were not shown in Table 2.
Changes in the biochemical composition of sedimentary organic matter
Although quantitative characterization of sedimentary OM could not be obtained by solid-state CP/MAS 13C-NMR analysis, the mixing model helped to convert signals in the spectrum of each extraction residue into the biochemical composition of sedimentary OM. The model predicted percentages of six major components of OM in the original and residual sediments (Fig. 6), suggesting that chars and carbonyls were absent in the original sediments. Sedimentary OM consisted mainly of lipids, proteins and lignins, and less than 15% of carbohydrates. Since hot water extracted a small fraction of proteins and carbohydrates, the percentage of lipids in the residual sediments increased to nearly 40%. The biochemical composition of hot water non-extractable OM differed little from the original sedimentary OM. A substantial reduction of proteins and carbohydrates and proteins decreased substantially (from 37 to 16%) after 6 M HCl hydrolysis. After oxidation of lignins and complex proteins using H2O2, the percentage of lipids in residual sediments was twice that in the original bulk sediments.

Estimated percentages of carbon assigned to the main organic components in residual sediments. Data were derived from results of 13C nuclear magnetic resonance analyses, which were calculated using a mixing model of Nelson and Baldock (2005)
Discussion
Using sequential chemical fractionation to remove sedimentary organic matter
Sequential treatment of sediments with hot water, HCl, H2O2 and Na2S2O8 changed the chemical characteristics of residual sediments. After the final step of the chemical fractionation procedure, 95% of TOC and 90% of TN were removed from the original bulk sediments. Labile proteins and carbohydrates in sediments were extracted first using hot water and HCl, while lipids and lignins were sequentially removed using H2O2 and Na2S2O8.
Although we did not analyze hot water-extracted proteins and carbohydrates, previous studies (Heller and Weiss 2015; Haynes and Francis 1993; Leinweber et al. 1995) consistently demonstrated the microbial origin of this OM fraction. Inherent structural complexity may be one explanation for the existence of proteins and carbohydrates in HCl-resistant residue that were hot water-insoluble but HCl-hydrolysable (Fig. 6). This structural complexity makes these molecules more stable than water soluble ones. Silveira et al. (2008) observed a similar difference when comparing hot water-extractable C and HCl-hydrolysable C. However, it is more likely that some of proteins and carbohydrates extracted using HCl have a chemistry similar to that of the OM extracted by hot water. If so, they may have been physically protected by encapsulation within biogenic minerals or attachment to a mineral matrix or surfaces of organic macromolecules (Ingalls et al. 2004; Ingalls et al. 2003). This hypothesis is supported by the nearly unchanged 13C-NMR spectra (Fig. 5) and stable isotope ratios after hot water extraction (Fig. 3).
Lipids extracted using H2O2 and Na2S2O8 are considered to have different structures, as indicated by the shifts of resonance peaks in the 13C-NMR spectra after H2O2 and Na2S2O8 residue oxidations. Moreover, the chemical resistances of H2O2 and Na2S2O8-oxidized lipids can be differentiated, because they counteract H2O2 oxidation differently. These findings are supported by the similar study of Jagadamma et al. (2010) who revealed distinct structures of H2O2- and Na2S2O8-oxidized lipids using 13C-NMR analyses of extracted soil OM.
In the early diagenesis of sedimentary OM, proteins and carbohydrates are susceptible to microbial reformation and decomposition due to their weak chemical bonds and high bioavailability (Arndt et al. 2013; de Leeuw and Largeau 1993). The stability of lipids varies greatly in sediments and is determined by the degree of saturation and stereochemical and structural properties (Middelburg 2019). Nonetheless, most lipids are less degradable in comparison with proteins and carbohydrates. For lignins, primary studies have reported their limited degradability in lacustrine sediments, which is due to their refractory nature in anoxic environments after deposition (Ishsiwatari and Uzaki 1987; Louchouarn et al. 1997; Szklarz and Leonowicz 1986). In general, the sequential removal of sedimentary OM by our fractionation procedure is consistent with the preferential degradation of organic components in sediments as revealed by the stable isotope composition and 13C-NMR analyses. Thus, the fractionation procedure can be interpreted as simplified early in-situ diagenetic processes.
Influence of biochemical composition on stable isotope ratios
Early diagenesis of sedimentary OM involves selective preservation of a succession of compounds as well as a vast range of enzymatic reactions, including microbial degradation of labile components and growth of bacteria (Harvey et al. 1995; Hedges and Keil 1995).
Thus, preferential removal of N- or C-enriched organic compounds will change the biochemical composition and the C:N of OM in residual sediments. Moreover, labile organic components (proteins and carbohydrates) are generally enriched in 15N and 13C, while more stable components (lipids and lignins) are relatively depleted in these isotopes (Böttcher et al. 1998; Hobbie and Werner 2004; Ogrinc et al. 2005; Rieley et al. 1991). Preferential degradation of proteins and carbohydrates during early diagenesis would enrich the lighter isotopes and thus decreased the isotope composition of 15N and 13C in the residual OM.
Pearson correlation test revealed that δ13C values of the original bulk sediments and residual sediments are negatively and linearly correlated to C:N (r2 = 0.95, p < 0.005, Fig. 7a). Theoretically, the gradual depletion of 13C during early in-situ diagenesis of sediments can be attributed to (i) preferential degradation of isotopically heavy OM components, (ii) formation of isotopically light OM components (e.g. for bacterial growth) and (iii) isotopic fractionation related to hydrolysis of reactive OM components (Freudenthal et al. 2001; Meyers and Ishiwatari 1993). Although the influence of each mechanism was difficult to distinguish in our study, the 13C-NMR and isotope ratio analyses of residue indicate the influence of preferential removal of N-enriched proteins and OC-enriched carbohydrates on the stable isotope composition of bulk sediments. For example, during the oxidation of lignins and lipids by H2O2 and Na2S2O8, the δ13C value increased greatly (Fig. 3b) and the biochemical composition of the residual sediments changed (Fig. 6). Therefore, it is reasonable to assume that changes in biochemical composition are closely related to the stable C isotope composition of sedimentary OM.

Correlation between stable carbon (a) or nitrogen (b) isotope ratio and atomic carbon-to-nitrogen ratio (C:N) in original and residual sediments from the fractionation procedure (hot water, HCl, H2O2 and Na2S2O8). Error bars represent one standard deviations of three replicate samples
The correlation between δ15N and C:N was not significant (p > 0.05, Fig. 7b). The lack of correlation between δ15N and C:N is consistent with previous studies of other lacustrine and marine sediment samples (Hassan et al. 1997; Ogrinc et al. 2005; Woodward et al. 2011). No evident shifts in the δ15N value (less than 0.2‰) were observed after hot water extraction and 6 M HCl hydrolysis, despite the substantial change in the C:N of residual sediments (Fig. 7b). The nearly invariable δ15N value in the present study indicates that changes in biochemical composition do not fundamentally alter the stable N isotope composition of bulk sediments. It also agrees well with Lehmann et al. (2002), who attributed the decline of sedimentary δ15N value primarily to the addition of a 15N-depleted substrate caused by bacterial growth rather than the preferential degradation of proteins. Alternatively, the results of the present study help to interpret the increase in δ15N value as being a consequence of isotopic fractionation processes during deamination (Freudenthal et al. 2001). Our findings supplement laboratory studies that indicate alteration of isotope signals associated with microbial growth and metabolism (Freudenthal et al. 2001; Lehmann et al. 2002).
Implications for the application of stable carbon and nitrogen isotope ratios in research
Stable C and N isotope compositions in lacustrine sediments may provide information about past nutrient limitations (Brenner et al. 1999), primary production (Teranes and Bernasconi 2000) and early diagenesis in aquatic ecosystems (Macko et al. 1994). They may also record extreme weather and vegetation successions in watersheds (Leng and Marshall 2004; Meyers 2003). Nonetheless, since the 1980s, organic geochemists have observed that diagenetic processes can modify stable isotope signals in lacustrine and marine sediments (Hatcher et al. 1983; Spiker and Hatcher 1984). The present study demonstrates that changes in biochemical composition (Fig. 6) have a strong influence on bulk C:N and stable C isotope compositions, causing a shift up to 1 per mil point (Fig. 3b). The magnitude of shifts in stable isotope ratios induced by preferential degradation is similar to those of isotope alterations, which are interpreted as changes in paleo-environments. For example, a decrease of 1.3‰ in the δ13C value was observed in a sediment core from the Rochester Basin of Lake Ontario for the period 1980 to 1990. This decrease has been explained exclusively as a decline in productivity in this region (Hodell and Schelske 1998). The present study, however, demonstrates that preferential degradation of isotopically light OM components during early diagenesis can also explain the decrease in the δ13C value in the core.
The δ15N value decreased by only 0.2‰ (Fig. 3c) after more than half of the N-containing organic compounds had been extracted using hot water and HCl (Fig. 4b). The decrease in δ15N increased to 1.6‰ after another 16% of TN from recalcitrant lignins and lipids had been removed using H2O2 under oxic conditions (Figs. 3c, 4b). This degree of removal of this recalcitrant TN fraction during in-situ diagenesis is unusual due to the absence of oxygen in the sediment in nature. Consequently, in-situ diagenesis may not shift the stable N isotope composition appreciably. In principle, our results suggest that preferential degradation of proteins and carbohydrates has a limited influence on the stable N isotope composition of bulk sediments. However, as previous studies demonstrated, microbial metabolism involved in early diagenesis can shift δ15N values by up to 4‰ (Macko and Estep 1984; Meyers and Ishiwatari 1993; Sigman et al. 1999). Thus, it is possible that diagenetic alterations in stable N isotope ratios mask the initial isotope information of sediments and thus complicate application of δ15N in paleo-environmental research.
Conclusions
Diagenetic processes in sediments may alter paleo-environmental signals from stable isotope ratios and thus hinder accurate interpretations. In particular, preferential degradation of isotopically heavy proteins and carbohydrates results in a post-depositional alteration of stable C isotope ratios. This effect might become large enough to interfere with isotope records used for paleo-environmental reconstruction. The strong correlation between δ13C and C:N values of residual sedimentary OM during sequential fractionation processes implies that stable C isotope ratios depend greatly on the biochemical composition of OM in sediments. In contrast, degradation during early diagenesis seemed an unlikely primary process for mediating alterations in stable N isotope ratios after deposition. As alternative explanations, we suggest microbial growth and metabolism.
Our simulation of preferential degradation of sedimentary OM identified the complex mechanisms that alter stable C and N isotope ratios during early diagenesis. Stable isotope measurements combined with chemical structure analyses provide insights into mechanisms that govern diagenetic alteration of sedimentary OM. Overall, we suggest future studies that focus more on paleo-signal formation and its diagenetic alteration.
References
Altabet MA, Francois R, Murray DW, Prell WL (1995) Climate-related variations in denitrification in the Arabian Sea from sediment 15N/14N ratios. Nature 373:506–509
Arndt S, Jørgensen BB, LaRowe DE, Middelburg JJ, Pancost RD, Regnier P (2013) Quantifying the degradation of organic matter in marine sediments: a review and synthesis. Earth Sci Rev 123:53–86
Barth JAC, Mader M, Nenning F, van Geldern R, Friese K (2017) Stable isotope mass balances versus concentration differences of dissolved inorganic carbon–implications for tracing carbon turnover in reservoirs. Isot Environ Health Stud 53:413–426
Bada JL, Schoeninger MJ, Schimmelmann A (1989) Isotopic fractionation during peptide bond hydrolysis. Geochim Cosmochim Acta 53:3337–3341
Baldock J, Oades J, Waters A, Peng X, Vassallo A, Wilson M (1992) Aspects of the chemical structure of soil organic materials as revealed by solid-state 13C-NMR spectroscopy. Biogeochemistry 16:1–42
Baldock JA, Masiello CA, Gélinas Y, Hedges JI (2004) Cycling and composition of organic matter in terrestrial and marine ecosystems. Mar Chem 92:39–64
Böttcher ME, Oelschläger B, Höpner T, Brumsack HJ, Rullkötter J (1998) Sulfate reduction related to the early diagenetic degradation of organic matter and “black spot” formation in tidal sandflats of the German Wadden Sea (southern North Sea): stable isotope (13C, 34S, 18O) and other geochemical results. Org Geochem 29:1517–1530
Brenner M, Whitmore TJ, Curtis JH, Hodell DA, Schelske CL (1999) Stable isotope (δ13C and δ15N) signatures of sedimented organic matter as indicators of historic lake trophic state. J Paleolimnol 22:205–221
Burdige D (2007) Preservation of organic matter in marine sediments: controls, mechanisms, and an imbalance in sediment organic carbon budgets? Chem Rev 107:467–485
Dadi T, Völkner C, Koschorreck M (2015) A sediment core incubation method to measure the flux of dissolved organic carbon between sediment and water. J Soils Sediments 15(12):2350–2358
de Leeuw JW, Largeau C (1993) A review of macromolecular organic compounds that comprise living organisms and their role in kerogen, coal and petroleum formation. In: Engel MH, Macko SA (eds) Organic Geochemistry. Plenum Publishing Group, New York, pp 23–72
Fabiano M, Danovaro R, Fraschetti S (1995) A three-year time series of elemental and biochemical composition of organic matter in subtidal sandy sediments of the Ligurian Sea (northwestern Mediterranean). Cont Shelf Res 15:1453–1469
Freudenthal T, Wagner T, Wenzhöfer F, Zabel M, Wefer G (2001) Early diagenesis of organic matter from sediments of the eastern subtropical Atlantic: evidence from stable nitrogen and carbon isotopes. Geochim Cosmochim Acta 65:1795–1808
Friese K, Schultze M, Boehrer B, Büttner O, Herzsprung P, Koschorreck M, Kuehn B, Rönicke H, Tittel J, Wendt-Potthoff K, Wollschläger U, Dietze M, Rinke K (2014) Ecological response of two hydro-morphological similar pre-dams to contrasting land-use in the Rappbode reservoir system (Germany). Int Rev Hydrobiol 99:335–349
Ghani A, Dexter M, Perrott K (2003) Hot-water extractable carbon in soils: a sensitive measurement for determining impacts of fertilisation, grazing and cultivation. Soil Biol Biochem 35:1231–1243
Gu B, Schelske CL, Brenner M (1996) Relationship between sediment and plankton isotope ratios (δ13C and δ15N) and primary productivity in Florida lakes. Can J Fish Aquat Sci 53:875–883
Guillemette F, McCallister SL, del Giorgio PA (2016) Selective consumption and metabolic allocation of terrestrial and algal carbon determine allochthony in lake bacteria. The ISME Journal 10:1373–1382
Harvey HR, Tuttle JH, Bell JT (1995) Kinetics of phytoplankton decay during simulated sedimentation: changes in biochemical composition and microbial activity under oxic and anoxic conditions. Geochim Cosmochim Acta 59:3367–3377
Hassan KM, Swinehart JB, Spalding RF (1997) Evidence for Holocene environmental change from C/N ratios, and δ13C and δ15N values in Swan Lake sediments, western Sand Hills, Nebraska. J Paleolimnol 18:121–130
Hatcher PG (1987) Chemical structural studies of natural lignin by dipolar dephasing solid-state 13C nuclear magnetic resonance. Org Geochem 11:31–39
Hatcher PG, Ravin A, Behar F, Baudin F (2014) Diagenesis of organic matter in a 400 m organic rich sediment core from offshore Namibia using solid state 13C-NMR and FTIR. Org Geochem 75:8–23
Hatcher PG, Spiker EC, Szeverenyi NM, Maciel GE (1983) Selective preservation and origin of petroleum-forming aquatic kerogen. Nature 305:498
Hayes J (1993) Factors controlling 13C contents of sedimentary organic compounds: principles and evidence. Mar Geol 113:111–125
Hayes J, Freeman KH, Popp BN, Hoham CH (1990) Compound-specific isotopic analyses: a novel tool for reconstruction of ancient biogeochemical processes. Org Geochem 16:1115–1128
Haynes R (2005) Labile organic matter fractions as central components of the quality of agricultural soils: an overview. Adv Agron 85:221–268
Hedges JI, Keil RG (1995) Sedimentary organic matter preservation: an assessment and speculative synthesis. Mar Chem 49:81–115
Helfrich M, Flessa H, Mikutta R, Dreves A, Ludwig B (2007) Comparison of chemical fractionation methods for isolating stable soil organic carbon pools. Eur J Soil Sci 58:1316–1329
Heller C, Weiss K (2015) Approaching a standardized method for the hot-water extraction of peat material to determine labile SOM in organic soils. Commun Soil Sci Plant Anal 46:1044–1060
Herczeg A, Smith A, Dighton J (2001) A 120 year record of changes in nitrogen and carbon cycling in Lake Alexandrina, South Australia: C:N, δ15N and δ13C in sediments. Appl Geochem 16:73–84
Hobbie EA, Werner RA (2004) Intramolecular, compound-specific, and bulk carbon isotope patterns in C3 and C4 plants: a review and synthesis. New Phytol 161:371–385
Hodell DA, Schelske CL (1998) Production, sedimentation, and isotopic composition of organic matter in Lake Ontario. Limnol Oceanogr 43:200–214
Holmes B, Eichner C, Struck U, Wefer G (1999) Reconstructions of surface ocean nitrate utilization using stable nitrogen isotopes in sinking particles and sediments. In: Fischer G, Wefer G (eds) Use of proxies in Paleoceanography: examples from the South Atlantic. Springer-Verlag, Berlin, pp 447–468
Ingalls AE, Lee C, Wakeham SG, Hedges JI (2003) The role of biominerals in the sinking flux and preservation of amino acids in the Southern Ocean along 170 W. Deep Sea Research Part II. Top Stud Oceanogr 50:713–738
Ingalls AE, Aller RC, Lee C, Wakeham SG (2004) Organic matter diagenesis in shallow water carbonate sediments. Geochim Cosmochim Acta 68:4363–4379
Ishiwatari R, Uzaki M (1987) Diagenetic changes of lignin compounds in a more than 0.6 million-year-old lacustrine sediment (Lake Biwa, Japan). Geochim Cosmochim Acta 51:321–328
Jagadamma S, Lal R, Ussiri DA, Trumbore SE, Mestelan S (2010) Evaluation of structural chemistry and isotopic signatures of refractory soil organic carbon fraction isolated by wet oxidation methods. Biogeochemistry 98:29–44
KoÈgel-Knabner I (2002) The biomacromolecular organic composition of plant and microbial residue as inputs to soil organic matter. Soil Biol Biochem 34:139–162
Krull E, Haynes D, Lamontagne S, Gell P, McKirdy D, Hancock G, McGowan J, Smernik R (2009) Changes in the chemistry of sedimentary organic matter within the Coorong over space and time. Biogeochemistry 92:9–25
Lamb AL, Wilson GP, Leng MJ (2006) A review of coastal palaeoclimate and relative sea-level reconstructions using δ13C and C/N ratios in organic material. Earth Sci Rev 75:29–57
Lanari M, Claudino MC, Garcia AM, da Silva Copertino M (2018) Changes in the elemental (C, N) and isotopic (δ13C, δ15N) composition of estuarine plants during diagenesis and implications for ecological studies. J Exp Mar Biol Ecol 500:46–54
Last WM, Smol JP (2006) Tracking environmental change using lake sediments. Volume 2: physical and geochemical methods. Kluwer Academic, Dordrecht
Lehmann MF, Bernasconi SM, Barbieri A, McKenzie JA (2002) Preservation of organic matter and alteration of its carbon and nitrogen isotope composition during simulated and in situ early sedimentary diagenesis. Geochim Cosmochim Acta 66:3573–3584
Leinweber P, Schulten HR, Körschens M (1995) Hot water extracted organic matter: chemical composition and temporal variations in a long-term field experiment. Biol Fertil Soils 20:17–23
Leng MJ, Marshall JD (2004) Palaeoclimate interpretation of stable isotope data from lake sediment archives. Quatern Sci Rev 23:811–831
Longbottom TL, Hockaday WC (2019) Molecular and isotopic composition of modern soils derived from kerogen-rich bedrock and implications for the global C cycle. Biogeochemistry 143:239–255
Louchouarn P, Lucotte M, Canuel R, Gagné JP, Richard LF (1997) Sources and early diagenesis of lignin and bulk organic matter in the sediments of the Lower St. Lawrence Estuary and the Saguenay Fjord. Mar Chem 58:3–26
Lorenz K, Lal R, Shipitalo M (2008) Chemical stabilization of organic carbon pools in particle size fractions in no-till and meadow soils. Biol Fertil Soils 44:1043–1051
Macko SA, Estep MLJOG (1984) Microbial alteration of stable nitrogen and carbon isotopic compositions of organic matter. Org Geochem 6:787–790
Macko SA, Engel MH, Qian Y (1994) Early diagenesis and organic matter preservation: a molecular stable carbon isotope perspective. Chem Geol 114:365–379
Mahmoudi N, Beaupre SR, Steen AD, Pearson A (2017) Sequential bioavailability of sedimentary organic matter to heterotrophic bacteria. Environ Microbiol 19:2629–2644
Mao J, Tremblay L, Gagné JP (2011) Structural changes of humic acids from sinking organic matter and surface sediments investigated by advanced solid-state-NMR: insights into sources, preservation and molecularly uncharacterized components. Geochim Cosmochim Acta 75:7864–7880
McArthur J, Tyson R, Thomson J, Mattey D (1992) Early diagenesis of marine organic matter: alteration of the carbon isotopic composition. Mar Geol 105:51–61
Meyers PA (1997) Organic geochemical proxies of paleoceanographic, paleolimnologic, and paleoclimatic processes. Org Geochem 27:213–250
Meyers PA (2003) Applications of organic geochemistry to paleolimnological reconstructions: a summary of examples from the Laurentian Great Lakes. Org Geochem 34:261–289
Meyers PA, Ishiwatari R (1993) Lacustrine organic geochemistry: an overview of indicators of organic matter sources and diagenesis in lake sediments. Org Geochem 20:867–900
Meyers PA, Lallier-Vergès E (1999) Lacustrine sedimentary organic matter records of Late Quaternary paleoclimates. J Paleolimnol 21:345–372
Middelburg J (2018) Review and syntheses: to the bottom of carbon processing at the seafloor. Biogeosciences 15:413–427
Middelburg JJ (2019) Marine carbon biogeochemistry: a primer for earth system scientists. Springer Nature, Cham
Mikutta R, Kleber M, Kaiser K, Jahn R (2005) Review: organic matter removal from soils using hydrogen peroxide, sodium hypochlorite, and disodium peroxodisulfate. Soil Sci Soc Am J 69:120–135
Monteil-Rivera F, Brouwer EB, Masset S, Deslandes Y, Dumonceau J (2000) Combination of X-ray photoelectron and solid-state 13C nuclear magnetic resonance spectroscopy in the structural characterisation of humic acids. Anal Chim Acta 424:243–255
Nelson PN, Baldock JA (2005) Estimating the molecular composition of a diverse range of natural organic materials from solid-state 13C-NMR and elemental analyses. Biogeochemistry 72:1–34
Ogrinc N, Fontolan G, Faganeli J, Covelli S (2005) Carbon and nitrogen isotopic compositions of organic matter in coastal marine sediments (the Gulf of Trieste, N Adriatic Sea): indicators of sources and preservation. Mar Chem 95:163–181
Pane C, Piccolo A, Spaccini R, Celano G, Villecco D, Zaccardelli M (2013) Agricultural waste-based composts exhibiting suppressivity to diseases caused by the phytopathogenic soil-borne fungi Rhizoctonia solani and Sclerotinia minor. Appl Soil Ecol 65:43–51
Paul E, Follett R, Leavitt S, Halvorson A, Peterson G, Lyon D (1997) Radiocarbon dating for determination of soil organic matter pool sizes and dynamics. Soil Sci Soc Am J 61:1058–1067
Paul EA, Morris SJ, Conant RT, Plante AF (2006) Does the acid hydrolysis–incubation method measure meaningful soil organic carbon pools? Soil Sci Soc Am J 70:1023–1035
R Core Team (2013) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.r-project.org
Rieley G, Collier RJ, Jones DM, Eglinton G, Eakin PA, Fallick AE (1991) Sources of sedimentary lipids deduced from stable carbon-isotope analyses of individual compounds. Nature 352:425–427
Rinke K, Kuehn B, Bocaniov S, Wendt-Potthoff K, Büttner O, Tittel J, Schultze M, Herzsprung P, Rönicke H, Rink K, Rinke K, Dietze M, Matthes M, Paul L, Friese K (2013) Reservoirs as sentinels of catchments: the Rappbode Reservoir Observatory (Harz Mountains, Germany). Environ Earth Sci 69:523–536
Rodríguez-Murillo JC, Almendros G, Knicker H (2011) Wetland soil organic matter composition in a Mediterranean semiarid wetland (Las Tablas de Daimiel, Central Spain): insight into different carbon sequestration pathways. Org Geochem 42:762–773
Saino T, Hattori A (1980) 15N natural abundance in oceanic suspended particulate matter. Nature 283:752–754
Schelske CL, Hodell DA (1995) Using carbon isotopes of bulk sedimentary organic matter to reconstruct the history of nutrient loading and eutrophication in Lake Erie. Limnol Oceanogr 40:918–929
Sigman DM, Altabet MA, Francois R, McCorkle DC, Gaillard JF (1999) The isotopic composition of diatom-bound nitrogen in Southern Ocean sediments. Paleoceanography 14:118–134
Silveira ML, Comerford NB, Reddy KR, Cooper WT, El-Rifai H (2008) Characterization of soil organic carbon pools by acid hydrolysis. Geoderma 144:405–414
Sparling G, Vojvodić-Vuković M, Schipper L (1998) Hot-water-soluble C as a simple measure of labile soil organic matter: the relationship with microbial biomass C. Soil Biol Biochem 30:1469–1472
Spiker EC, Hatcher PG (1984) Carbon isotope fractionation of sapropelic organic matter during early diagenesis. Org Geochem 5:283–290
Sun W, Zhang E, Jones RT, Liu E, Shen J (2016) Biogeochemical processes and response to climate change recorded in the isotopes of lacustrine organic matter, southeastern Qinghai-Tibetan Plateau, China. Palaeogeogr Palaeoclimatol Palaeoecol 453:93–100
Szklarz G, Leonowicz A (1986) Cooperation between fungal laccase and glucose oxidase in the degradation of lignin derivatives. Phytochemistry 25:2537–2539
Teodoru CR, Del Giorgio PA, Prairie YT, St-Pierre A (2013) Depositional fluxes and sources of particulate carbon and nitrogen in natural lakes and a young boreal reservoir in Northern Québec. Biogeochemistry 113:323–339
Teranes JL, Bernasconi SM (2000) The record of nitrate utilization and productivity limitation provided by δ15N values in lake organic matter: a study of sediment trap and core sediments from Baldeggersee, Switzerland. Limnol Oceanogr 45:801–813
Vane CH, Drage TC, Snape CE, Stephenson MH, Foster C (2005) Decay of cultivated apricot wood (Prunus armeniaca) by the ascomycete Hypocrea sulphurea, using solid state 13C-NMR and off-line TMAH thermochemolysis with GC–MS. Int Biodeterior Biodegrad 55:175–185
von Lützow M, Kögel-Knabner I, Ekschmitt K, Flessa H, Guggenberger G, Matzner E, Marschner B (2007) SOM fractionation methods: relevance to functional pools and to stabilization mechanisms. Soil Biol Biochem 39:2183–2207
Wakeham SG, Lee C, Hedges JI, Hernes PJ, Peterson MJ (1997) Molecular indicators of diagenetic status in marine organic matter. Geochim Cosmochim Acta 61:5363–5369
Woodward CA, Potito AP, Beilman DW (2011) Carbon and nitrogen stable isotope ratios in surface sediments from lakes of western Ireland: implications for inferring past lake productivity and nitrogen loading. J Paleolimnol 47:167–184
Woodward CA, Potito AP, Beilman DW (2012) Carbon and nitrogen stable isotope ratios in surface sediments from lakes of western Ireland: implications for inferring past lake productivity and nitrogen loading. J Paleolimnol 47:167–184
Acknowledgements
This study was supported by a grant to X.Q. Liu from the China Scholarship Council. We acknowledge Ines Locker for the elemental and isotopic analyses of all the solid samples, Christian Hanke for measuring isotope ratios in the extraction solutes and Katrin Wendt-Potthoff for critical reading and English editing.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Responsible Editor: Stephen D. Sebestyen.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Liu, X., Hilfert, L., Barth, J.A.C. et al. Isotope alteration caused by changes in biochemical composition of sedimentary organic matter. Biogeochemistry 147, 277–292 (2020). https://doi.org/10.1007/s10533-020-00640-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10533-020-00640-3