
Abstract
It has been established that acylhydroperoxy derivatives of phospholipids from oxidized rat liver mitochondria are captured predominantly by LDL particles but not by HDL during co-incubation with blood plasma lipoproteins, which refutes the previously suggested hypothesis about the involvement of HDL in the reverse transport of oxidized phospholipids and confirms the possibility of different mechanisms of lipohydroperoxide accumulation in LDL during oxidative stress.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
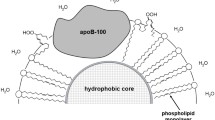
Lipid transport in vivo is executed by blood plasma lipoproteins present as two major classes referred to as low-density lipoproteins (LDL) and high-density lipoproteins (HDL). Particles (nanoscale size) of such lipoprotein classes profoundly differ in terms of related metabolic functions as well as particle size and chemical composition [1-4]. HDL particles bear apoproteins A1, C2, and E [1-4], whereas larger-sized LDL particles contain only apoprotein B-100 [1-4]. LDL generated in the liver transport lipids to peripheral tissues, whereas HDL are involved in lipid reverse transport back to the liver [5-8]. It has been demonstrated that accumulation of oxidized LDL is the main event causing development and progression of atherosclerosis [9-11]. In contrast, high level of HDL protects from atherosclerosis development [12, 13].
The only protein in the LDL particles (apoprotein B-100) could be subjected to chemical modification with participation of low molecular weight dicarbonyl agents formed during free radical peroxidation of polyene lipids (such as malonic dialdehyde – MDA) as well as enzymatic oxidation or autooxidation of glucose and other 6-atom carbohydrates (e.g., glyoxal and methylglyoxal) [14-16]. The dicarbonyl-modified LDL particles are recognized by scavenger receptors and are captured by vascular wall cells, which are subsequently converted into the lipid-rich “foam cells” [11, 14-17]. The foam cells create primary pre-atherogenic lipoid lesions in the vascular wall in atherosclerosis and diabetes [11, 14-17]. The oxidized LDL particles can also bind to endotheliocyte LOX-1 scavenger receptor inducing NADPH oxidase expression and generation of superoxide anion radicals [15, 16, 18]. Overproduction of reactive oxygen species (ROS) ultimately promotes apoptosis and leads to endothelial dysfunction [15, 16, 18]. Thus, oxidative transformations of lipoproteins could play a crucial role in the molecular mechanisms of vascular wall damage during atherogenesis and diabetogenesis [14, 15]. Based on our own data and available publications, we hypothesized that vascular wall damage in atherosclerosis and diabetes mellitus occur via the same mechanism in which accumulation of the dicarbonyl-modified LDL plays an essential role [15, 19]. LDL particles are highly susceptible to initiation of free radical peroxidation in the outer phospholipid monolayer, whereas HDL particles remain quite resistant to lipid peroxidation [20]. Nevertheless, it has been shown that HDL could not only inhibit oxidation of LDL upon in vitro co-incubation [21], but also exert an antioxidant effect (suppress LDL oxidation) in the bloodstream in vivo [22, 23]. Moreover, it was hypothesized that HDL particles are capable of capturing oxidized phospholipids from the supramolecular lipid–protein complexes (lipoproteins and biomembranes) to transport them to the liver, where enzymatic detoxification of lipid peroxides occurs [24]. Unfortunately, this hypothesis was not rigorously validated experimentally by using proper approaches and methods allowing to obtain irrefutable and unambiguous results [25, 26]. Based on the aforementioned, here we assessed ability of the LDL and HDL particles to absorb hydroperoxy-derivatives of phospholipids from the surface of oxidized biomembranes.
MATERIALS AND METHODS
Isolation of various lipoprotein classes (LDL, HDL) and subfractions (HDL2, HDL3) with preparative ultracentrifugation. Preparative isolation of blood plasma LDL, total HDL fraction, as well as HDL2 and HDL3 subfractions from apparently healthy donors was carried out by using NaBr density gradient centrifugation with an Optima XPN-80 ultracentrifuge (Beckman, USA) as described earlier [20, 27]. Lipoproteins of various classes were isolated from three blood plasma samples obtained from three individual healthy donors. Blood samples were collected into test tubes containing EDTA (1 mM) as an anticoagulant and antioxidant. Protein level in the isolated lipoprotein samples purified further by dialysis [20] was assessed by Lowry method.
Isolation and free radical oxidation of rat liver mitochondria. To isolate mitochondria, rat liver (WKY strain, males, 250-300 g) was perfused with a cooled isotonic KCl solution followed by homogenization (1 : 10, w/v) in a Potter homogenizer with Teflon pestle in a medium containing 0.25 M sucrose solution, 10 mM MOPS and 1 mM EDTA [28]. Nuclei and debris were precipitated by centrifugation at 700g for 10 min in an Eppendorf 5804 R refrigerated centrifuge (Eppendorf, Germany) followed by repeated centrifugation of the final supernatant at 8000g for 10 min to precipitate heavy mitochondria [28]. The pellet was resuspended in an EDTA-free isolation medium, and mitochondria were precipitated twice followed by resuspension (1 mg protein/ml) in an isotonic K/Na-phosphate buffer (pH 7.4). Free radical peroxidation of polyene phospholipids of mitochondrial membranes was induced by adding 0.5 mM ascorbate to reduce endogenous iron ions. Kinetics of mitochondrial free radical peroxidation under aerobic conditions was measured by assessing accumulation of conjugated double bond-bearing lipid hydroperoxides (LOOH) based on absorption at 233 nm wavelength using a UV-2600 spectrophotometer (Shimadzu, Japan) with an ISR-2600 integrating sphere attachment for measurements in a turbid medium. After a 4-hour incubation (reaching a plateau on kinetic curves), mitochondrial oxidation was inhibited by adding EDTA to a final concentration of 1 mM.
Assessing transfer of acylhydroperoxy phospholipid derivatives from oxidized mitochondrial membranes into lipoprotein particles under conditions of co-incubation. A pellet of oxidized mitochondria was resuspended in a medium containing isotonic K/Na-phosphate buffer (pH 7.4) and 1 mM EDTA, precipitated/resuspended twice at 8000g for 10 min (1 mg protein/ml) in an isotonic K/Na-phosphate buffer (pH 7.4) containing 1 mM EDTA. Next, LDL, HDL, or HDL2 and HDL3 subfractions (200 μg protein/ml) were added to the medium with oxidized mitochondria and incubated for 6 h. At specific time points, aliquots of the incubation medium were collected, centrifuged at 10,000g for 15 min to precipitate mitochondria and fragments of their membranes followed by evaluating LOOH level in the supernatant containing only lipoprotein particles based on absorption of the conjugated dienes at 233 nm. The experiments were repeated three times by using lipoprotein samples obtained from individual donors in each experiment. The methodology we developed allowed quick separation of the oxidized membranes from lipoprotein particles, which facilitated monitoring kinetics of LOOH accumulation in LDL and HDL. The amount of LOOH per LDL and HDL particle was calculated based on the determined levels of apoprotein B-100 and apoprotein A-1, respectively, by using molar extinction coefficient of 22,000 M–1 cm–1. Concentration of LOOH per one LDL and HDL particles was calculated based on the level of apoprotein B-100 and apoprotein A1 in LDL and HDL particles, respectively (each of these apoproteins is found at the level of 1 molecule per LDL and HDL particle, respectively). The level of apoprotein B-100 and apoprotein A1 was measured using an Architect C8000 chemical analyzer (Abbott, USA) and relevant test kits (Abbott) [20].
Statistical analysis of the data was carried out using the software packages STATISTICA 10 (Statsoft, USA), MedCalc version 12.7.0.0 (MedCalc Software, Belgium), and Microsoft Excel 2010, version 14.0.7263.5000. The data were presented as a mean ± standard error of the mean. Considering that in all cases the analyzed parameters did not exhibit normal distribution, nonparametric statistical methods were applied. Analysis of intergroup differences for quantitative parameters was performed by using the nonparametric Mann–Whitney U-test. Differences were considered significant at p < 0.05.
RESULTS
Free radical oxidation of outer membranes of rat liver mitochondria. Kinetics of free radical oxidation occurring in rat mitochondria outer membranes is shown in Fig. 1. As can be seen from the kinetic curve no further oxidation of polyene phospholipids within the mitochondrial membranes occurs after 3-hour incubation (Fig. 1).

Kinetics curve of free radical oxidation in the rat liver mitochondria outer membranes. For details of experimental settings see “Materials and Methods” section.
Based on this fact, 3 h after the onset of incubation, main portion of the medium containing mitochondria was collected followed by suppression of further mitochondrial oxidation by adding EDTA to a final concentration of 1 mM for binding variable valency metal ions that catalyze oxidation. After three precipitation–resuspension cycles to remove membrane fragments and soluble low molecular weight oxidation products, the final preparation of oxidized heavy mitochondria was used to assess absorption of acylhydroperoxy-containing phospholipids of mitochondrial membranes by the isolated unoxidized LDL and HDL particles.
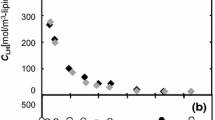
Assessing absorption kinetics of acylhydroperoxy-containing phospholipids of mitochondrial membranes by LDL and HDL particles. The results of investigation of the possibility for transfer of acylhydroperoxy phospholipid derivatives from oxidized biomembranes to LDL and HDL particles are shown in Fig. 2.

Kinetic curve depicting incorporation of acylhydroperoxy phospholipid derivatives from the rat hepatic mitochondrial membranes subjected to free radical peroxidation into LDL (curve 1) and HDL (curve 2) particles. For details of experimental settings see “Materials and Methods” section.
The obtained data indicate that the native LDL particles co-incubated with oxidized mitochondria predominantly capture acylhydroperoxy phospholipid derivatives (Fig. 2, curve 1), whereas the rate of oxidized phospholipid transfer from the biomembranes to HDL particles is insignificant (Fig. 2, curve 2). Kinetics of oxidized phospholipid absorption by the HDL subfractions – HDL2 and HDL3 – is presented in Fig. 3.

Kinetics curve of incorporation of acylhydroperoxy phospholipid derivatives from the rat hepatic mitochondrial membranes subjected to free radical oxidation into total HDL fraction (curve 1), HDL2 (curve 2), and HDL3 (curve 3) particles. Differences in the levels of incorporated LOOH into HDL2 and HDL3 particles found insignificant at all time points; * significant differences in the level of incorporated LOOH for total HDL fraction as well as HDL2 or HDL3 particles (p < 0.05). For details of experimental settings see “Materials and Methods” section.
Hence, the obtained data allow concluding that the difference in the levels of acylhydroperoxy phospholipid derivatives absorbed from oxidized mitochondrial membranes by HDL2 and HDL3 particles were insignificant (Fig. 3, curves 2 and 3), whereas they were significantly lower than the level of oxidized phospholipids absorbed by the total HDL fraction (Fig. 3, curve 1) at all time points examined.
DISCUSSION
The data obtained in our study (Fig. 2) contradict the previously proposed hypothesis suggesting the possibility of the HDL-mediated reverse transport of oxidized lipids to the liver [24]. It should be noted that the possibility of the transfer of phospholipid acylhydroperoxy derivatives from the oxidized erythrocyte membranes (shadows of erythrocytes) to co-incubated LDL has been demonstrated earlier [28] and is not considered as unusual. It was shown in other similar studies [29] that the multi-hour oxidation of erythrocyte shadows with the free radical oxidation initiators resulted in generation of a large number of secondary products of aldehyde nature [16] presence of which could be a source of artifacts [16]. It is worth mentioning that the erythrocyte membranes are not particularly suitable for such experiments, because, unlike liver microsomes and mitochondria, erythrocytes are highly resistant even to enzymatic lipid peroxidation [30]. In accordance with our data, LDL particles, unlike HDL, can not only be easily oxidized via the free radical mechanism [20], but also can capture oxidized phospholipids from other lipid–protein supramolecular complexes when they interact (Fig. 2). Destruction of the hydroperoxy derivatives of phospholipids accumulated in the LDL should undoubtedly elicit atherogenic modifications of LDL particles by the secondary products of oxidation such as low molecular weight dicarbonyls, e.g., 4-hydroxy-2-nonenal and MDA [14, 16, 17] contributing to atherogenesis [14-17]. Nevertheless, there are data available showing that the oxidized lipids can be actively transferred from the outer phospholipid layer of LDL particles to HDL particles during their co-incubation [31]. Moreover, oxidized lipids of the hydrophobic core of LDL are not transferred to HDL and oxidized HDL lose their capability to adsorb lipoperoxides from oxidized LDL [31]. Indeed, the acylhydroperoxy derivatives of phospholipids of the outer layer of LDL particles are significantly more polar than the non-oxidized phospholipids [32]. As a result, the phospholipid hydroperoxy acyls should enter the water phase [32] and, probably, could be more efficiently exchanged with the other lipid–protein supramolecular complexes upon contact. Despite that it is HDL subfractions HDL2 and HDL3, which are credited with a cardioprotective effect [33-35], our data (Fig. 3) indicate that if such effect exists, it probably has nothing to do with their presumed ability to participate in detoxification of lipid hydroperoxides [24]. It is evident that the overall LOOH absorption by HDL2 and HDL3 subfractions is lower (Fig. 3, curves 2 and 3) than that by the total HDL fraction (Fig. 3, curve 1). This apparent contradiction may be due to the fact that the number of total HDL subfractions is not limited solely to HDL2 and HDL3 [34]. Moreover, preparative isolation of HDL2 and HDL3 may be associated with the loss of relevant oxidized species present in the total HDL fraction [35].
CONCLUSION
The data obtained in our study imply existence of diverse mechanisms causing emergence of atherogenic LDL enriched in free radical peroxidation products, which further highlights the threat posed by the pathogenetically-important oxidative transformation of LDL in vivo [14-16]. Antioxidant action of HDL particles [36] may be related to the presence of paraoxonase-1, which is capable of reducing lipid peroxides within LDL particles upon co-incubation [29, 37]. Obviously, convincing data are necessary to validate this putative mechanism for blood plasma lipid peroxides utilization in vivo involving an effective exchange between the LDL-derived oxidized phospholipids and HDL particles. Unfortunately, no such data are currently available. What is clear that enrichment of the LDL particles with the oxidized lipids, as shown by our data, may occur both during intense lipid free radical peroxidation as well as by the capture of oxidized lipids from the membranes of blood cell (erythrocytes, etc.). Both these processes should contribute to the increased atherogenicity of the oxidatively modified LDL, i.e., facilitate their involvement in the vascular wall damage [14-16].
Abbreviations
- HDL:
-
high density lipoproteins
- LDL:
-
low density lipoproteins
- LOOH:
-
lipid hydroperoxides
References
Tomkin, G. H. (2010) Atherosclerosis, diabetes and lipoproteins, Expert Rev. Cardiovasc. Ther., 8, 1015-1029, https://doi.org/10.1586/erc.10.45.
Arnao, V., Tuttolomondo, A., Daidone, M., and Pinto, A. (2019) Lipoproteins in atherosclerosis process, Curr. Med. Chem., 26, 1525-1543, https://doi.org/10.2174/0929867326666190516103953.
Getz, G. S., and Reardon, C. A. (2020) Atherosclerosis: cell biology and lipoproteins, Curr. Opin. Lipidol., 31, 286-290, https://doi.org/10.1097/MOL.0000000000000704.
Wang, H. H., Garruti, G., Liu, M., Portincasa, P., Wang, D. H. (2017) Cholesterol and lipoprotein metabolism and atherosclerosis: recent advances in reverse cholesterol transport, Ann. Hepatol., 16 (Suppl. 1), s27-s42, https://doi.org/10.5604/01.3001.0010.5495.
Lee, J. M. S., and Choudhury, R. P. (2010) Atherosclerosis regression and high-density lipoproteins, Expert Rev. Cardiovasc. Ther., 8, 1325-1334, https://doi.org/10.1586/erc.10.108.
Brewer, H. B. Jr. (2011) Clinical review: the evolving role of HDL in the treatment of high-risk patients with cardiovascular disease, J. Clin. Endocrinol. Metab., 96, 1246-1257, https://doi.org/10.1210/jc.2010-0163.
Hernáez, Á., Soria-Florido, M. T., Schröder, H., Ros, E., Pintó, X., Estruch, R., Salas-Salvadó, J., Corella, D., Arós, F., Serra-Majem, L., Martínez-González, A. M., Fiol, M., Lapetra, J., Elosua, R., Lamuela-Raventós, R. M., and Fitó, M. (2019) Role of HDL function and LDL atherogenicity on cardiovascular risk: a comprehensive examination, PLoS One, 14, e0218533, https://doi.org/10.1371/journal.pone.0218533.
Carr, S. S., Hooper, A. J., and Sullivan, D. R. (2019) Non-HDL-cholesterol and apolipoprotein B compared with LDL-cholesterol in atherosclerotic cardiovascular disease risk assessment, Pathology, 51, 148-154, https://doi.org/10.1016/j.pathol.2018.11.006.
Steinberg, D., and Witztum, J. L. (2002) Is the oxidative modifications hypothesis relevant to human atherosclerosis? Do the antioxidant trials conducted to date reflect the hypothesis?, Circulation, 105, 2107-2111, https://doi.org/10.1161/01.CIR.0000014762.06201.06.
Parthasarathy, S., Santanam, N., and Auge, N. (1998) Oxidised low-density lipoprotein: a two-faced Janus in coronary artery disease?, Biochem. Pharmacol., 56, 279-284, https://doi.org/10.1016/S0006-2952(98)00074-4.
Khatana, C., Saini, N. K., Chakrabarti, S., Saini, V., Sharma, A., Saini, R.V., and Saini, A. K. (2020) Mechanistic insights into the oxidized low-density lipoprotein induced atherosclerosis, Oxid. Med. Cell. Longev., 2020, 1-14, https://doi.org/10.1155/2020/5245308.
Barter, P. J., and Rye, K. A. (1996) High-density lipoproteins and coronary heart disease, Atherosclerosis, 121, 1-12, https://doi.org/10.1016/0021-9150(95)05675-0.
Rubins, H. B., Robins, S. J., Collins, D., Fye, C. L., and Anderson, J. W. (1999) Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. Veterans affairs high-density lipoprotein cholesterol intervention trial study group, N. Engl. J. Med., 341, 410-418, https://doi.org/10.1056/NEJM199908053410604.
Lankin, V. Z., and Tikhaze, A. K. (2017) Role of oxidative stress in the genesis of atherosclerosis and diabetes mellitus: a personal look back on 50 years of research, Curr. Aging Sci., 10, 18-25, https://doi.org/10.2174/1874609809666160926142640.
Lankin, V. Z., Tikhaze, A. K., and Melkumyants, A. M. (2022) Dicarbonyl-dependent modification of LDL as a key factor of endothelial dysfunction and atherosclerotic vascular wall damage, Antioxidants, 11, 1565, https://doi.org/10.3390/antiox11081565.
Lankin, V. Z., Tikhaze, A. K., and Melkumyants, A. M. (2023) Malondialdehyde as a important key factor of molecular mechanisms of vascular wall damage under heart diseases development, J. Int. Mol. Sci., 24, 128, https://doi.org/10.3390/ijms24010128.
Lankin, V. Z., Tikhaze, A. K., and Kumskova, E. M. (2012) Macrophages actively accumulate malonyldialdehyde-modified but not enzymatically oxidized low density lipoprotein, Mol. Cell Biochem., 365, 93-98, https://doi.org/10.1007/s11010-012-1247-5.
Sun, Y., and Chen, X. (2011) Ox-LDL-induced LOX-1 expression in vascular smooth muscle cells: role of reactive oxygen species, Fundam. Clin. Pharmacol., 25, 572-579, https://doi.org/10.1111/j.1472-8206.2010.00885.x.
Lankin, V. Z., Konovalova, G. G., Tikhaze, A. K., Shumaev, K. B., Kumskova, E. M., and Viigimaa, M. (2014) The initiation of the free radical peroxidation of low-density lipoproteins by glucose and its metabolite methylglyoxal: a common molecular mechanism of vascular wall injure in atherosclerosis and diabetes, Mol. Cell. Biochem., 395, 241-252, https://doi.org/10.1007/s11010-014-2131-2.
Lankin, V. Z., Tikhaze, A. K., and Kosach, V. Ya. (2022) Comparative susceptibility to oxidation of different classes of blood plasma lipoproteins, Biochemistry (Moscow), 87, 1335-1341, https://doi.org/10.1134/S0006297922110128.
Raveh, O., Pinchuk, I., Fainaru, M., and Lichtenberg, D. (2001) Kinetics of lipid peroxidation in mixtures of HDL and LDL, mutual effects, Free Radic. Biol. Med., 31, 1486-1497, https://doi.org/10.1016/s0891-5849(01)00730-4.
Fumiaki, I., and Tomoyuki, I. (2020) High-density lipoprotein (HDL) triglyceride and oxidized HDL: new lipid biomarkers of lipoprotein-related atherosclerotic cardiovascular disease, Antioxidants (Basel), 9, 362, https://doi.org/10.3390/antiox9050362.
Bowry, V. W., Stanley, K. K., and Stocker, R. (1992) High density lipoprotein is the major carrier of lipid hydroperoxides in human blood plasma from fasting donors, Proc. Natl. Acad. Sci. USA, 89, 10316-10320, https://doi.org/10.1073/pnas.89.21.10316.
Klimov, A. N., Nikiforova, A. A., Kuzmin, A. A., Kuznetsov, A. S., and Mackness, M. I. (1998) Is high density lipoprotein a scavenger for oxidized phospholipids of low density lipoprotein? In Advances in Lipoprotein and Atherosclerosis Research, Diagnostics and Treatment, Jena, Gustav Fisher Verlag, pp. 78-82.
Klimov, A. N., Kozhemyakin, L. A., Pleskov, V. M., and Andreeva, L. I. (1987) Antioxidative effect of high density lipoproteins in the oxidation of low density lipoproteins, Bull. Expt. Biol. Med., 103, 550-556, https://doi.org/10.1007/BF00841817.
Klimov, A. N., Gurevich, V. S., Nikiforova, A. A., Shatilina, L. V., Kuzmin, A. A., Plavinsky, S. L., and Teryukova, N. P. (1993) Antioxidative activity of high density lipoproteins in vivo, Atherosclerosis, 100, 13-18, https://doi.org/10.1016/0021-9150(93)90063-z.
Lindgren, F. T. (1975) Preparative ultracentrifugal laboratory procedures and suggestions for lipoprotein analysis, in Analysis of Lipids and Lipoproteins (Perkins, E. G., ed) Champaign: Amer. Oil. Chemists Soc., pp. 204-224.
Vila, A., Korytowski, W., and Girotti, A. W. (2002) Spontaneous transfer of phospholipid and cholesterol hydroperoxides between cell membranes and low-density lipoprotein: assessment of reaction kinetics and prooxidant effects, Biochemistry, 41, 13705-13716, https://doi.org/10.1021/bi026467z.
Mastorikou, M., Mackness, B., Liu, Y., and Mackness, M. (2008) Glycation of paraoxonase-1 inhibits its activity and impairs the ability of high-density lipoprotein to metabolize membrane lipid hydroperoxides, Diabetic Med., 25, 1049-1055, https://doi.org/10.1111/j.1464-5491.2008.02546.x.
Lankin, V. (2003) The enzymatic systems in the regulation of free radical lipid peroxidation, in “Free Radicals, Nitric Oxide, and Inflammation: Molecular, Biochemical, and Clinical Aspects, Amsterdam etc.: IOS Press, 2003, NATO Science Series, 344, pp. 8-23.
Rasmiena, A. A., Barlow, C. K., Ng, T. W., Tull, D., and Meikle, P. J. (2016) High density lipoprotein efficiently accepts surface but not internal oxidised lipids from oxidised low density lipoprotein, Biochim. Biophys. Acta, 1861, 69-77, https://doi.org/10.1016/j.bbalip.2015.11.002.
Lankin, V. Z., Tikhaze, A. K., and Osis, Yu. G. (2002) Modeling the cascade of enzymatic reactions in liposomes including successive free radical peroxidation, reduction, and hydrolysis of phospholipid polyenoic acyls for studying the effect of these processes on the structural-dynamic parameters of the membranes, Biochemistry (Moscow), 67, 566-574, https://doi.org/10.1023/a:1015502429453.
Superko, H. R., Pendyala, L., Williams, P. T., Momary, K. M., King, S. B., and Garrett, B. C. (2012) High-density lipoprotein subclasses and their relationship to cardiovascular disease, J. Clin. Lipidol., 6, 496-523, https://doi.org/10.1016/j.jacl.2012.03.001.
Williams, P. T., and Feldma, D. E. (2011) Prospective study of coronary heart disease vs. HDL2, HDL3, and other lipoproteins in Gofman’s Livermore Cohort, Atherosclerosis, 214, 196-202, https://doi.org/10.1016/j.atherosclerosis.2010.10.024.
Honda, H., Hirano, T., Ueda, M., Kojima, S., Mashiba, S., Hayase, Y., Michihata, T., and Shibata, T. (2016) High-density lipoprotein subfractions and their oxidized subfraction particles in patients with chronic kidney disease, J. Atheroscler. Thromb., 23, 81-94, https://doi.org/10.5551/jat.30015.
Mackness, B., and Mackness, M. (2012) The antioxidant properties of high-density lipoproteins in atherosclerosis, Panminerva Med., 54, 83-90.
Mackness, M., and Mackness, B. (2013) Targeting paraoxonase-1 in atherosclerosis, Expert Opin. Ther. Targets., 17, 829-837, https://doi.org/10.1517/14728222.2013.790367.
Acknowledgments
We are grateful to Dr. A.V. Doroshchuk for help in performing individual experiments.
Funding
The study was financially supported by the Russian Science Foundation (grant no. 22-15-00013).
Author information
Authors and Affiliations
Contributions
V.Z.L. – supervised the study, discussed the data; A.K.T. – wrote and edited the manuscript; V.Y.K., G.G.K. – performed experiments, prepared the manuscript.
Corresponding author
Ethics declarations
The authors declare no conflict of interests in financial or any other sphere. All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Rights and permissions
About this article

Cite this article
Lankin, V.Z., Tikhaze, A.K., Kosach, V.Y. et al. Adsorption of Acylhydroperoxy-Derivatives of Phospholipids from Biomembranes by Blood Plasma Lipoproteins. Biochemistry Moscow 88, 698–703 (2023). https://doi.org/10.1134/S0006297923050127
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0006297923050127