
Abstract
The kinetics of free radical peroxidation of different classes of blood plasma lipoproteins (nanoparticles involved in lipid transport in the body) was studied. The susceptibility of atherogenic low-density lipoproteins (LDLs) to the Cu2+-initiated free radical peroxidation in vitro was found to be more than ten times higher than that of antiatherogenic high density lipoproteins (HDLs). The baseline content of acyl hydroperoxy derivatives of phospholipids (primary products of free radical peroxidation) in the outer layer of LDL particles in vivo measured per particle exceeded the baseline content of these compounds in HDL particles by more than an order of magnitude. The susceptibility to oxidation of the HDL2 subfraction of HDLs was higher than the susceptibility of total HDL fraction and HDL3 subfraction. The data obtained confirm an important role of free radical peroxidation of LDLs in the molecular mechanisms of vascular wall damage in atherosclerosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
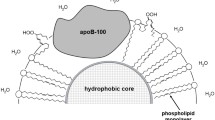
The transport of lipids in the body involves the two major classes of lipid-protein nanostructures – low-density lipoproteins (LDLs) and high-density lipoproteins (HDLs) – differing in their metabolic functions, size, and chemical composition [1-5]. HDL particles contain apolipoproteins A1, C2, and E (ApoA1, ApoC2, and ApoE, respectively); larger LDL particles contain solely apolipoprotein B-100 (ApoB100) [1-4]. Liver-produced LDLs transport lipids to peripheral body tissues, whereas HDLs enable reverse transport of lipids to the liver for their utilization [5-8]. ApoB100 in the LDLs can be modified by low-molecular-weight dicarbonyls formed as secondary products in the reactions of free radical oxidation of polyene lipids [4-hydroxy-2-nonenal and malondialdehyde (MDA)] or glucose and other six-atom carbohydrates (glyoxal and methylglyoxal) [9]. MDA is an isomer of methylglyoxal generated enzymatically from triose phosphates accumulated during glycolysis in the case of hyperglycemia [10], whereas glyoxal (MDA homolog) is formed in the reactions of glucose autooxidation [11] or co-oxidation with polyene lipids [11, 12]. Previously, we demonstrated that methylglyoxal is efficiently formed from glucose phosphates attacked by lipid hydroperoxide radicals [13], which implies accumulation of methylglyoxal as a result of non-enzymatic free radical co-oxidation of six-atom sugars and unsaturated lipids during oxidative stress. Oxidatively modified LDL particles are actively captured by the scavenger receptors in the vascular wall cells resulting in the emergence of pre-atherogenic lipoid vascular lesions in atherosclerosis and diabetes mellitus [9, 12]. The molecular mechanisms of modification of LDL apolipoproteins by carbonyls in atherosclerosis (MDA) and diabetes (glyoxal and methylglyoxal) are similar [12, 14], although the efficiency of protein modification by different dicarbonyls may differ [15, 16]. Moreover, it was shown that oxidized LDL particles form complexes with the surface endothelial cell receptor LOX-1, followed by the stimulation of apoptosis and dysfunction of the endothelium [17]. We found that the vascular scavenger receptors preferentially bind dicarbonyl-modified LDL particles and not enzymatically oxidized LDLs (obtained by oxidation of LDL by animal C-15 lipoxygenase) [18]. Therefore, the effects related to the atherogenic activity of oxidized LDLs described in numerous studies are due not so much to the action of oxidized LDL particles containing acyl hydroperoxy derivatives of phospholipids in the outer layer, but rather result from the modification of apolipoproteins by accumulated low-molecular-weight dicarbonyls [9, 14]. Hence, oxidative transformation of lipoproteins may play a critical role in the etiology and pathogenesis of atherosclerosis and diabetes mellitus [9, 14]. However, there is no consensus opinion on the comparative susceptibility of LDL and HDL particles to oxidation [19-27], since the studies on the assessment of oxidation rates of these compounds have used different analytical assays, methods for the oxidation initiation, and approaches for data analysis (the reaction rate is typically calculated per mg protein, which complicates data interpretation due to a large difference in the apolipoprotein content in the LDL and HDL particles). Based on the above, we investigated the ability of LDL and HDL particles, as well as the HDL2 and HDL3 subfractions, to undergo Cu2+-initiated free radical peroxidation in vitro and assessed the baseline content of hydroperoxy derivatives of phospholipids in the outer layers of LDL and HDL particles in vivo.
MATERIALS AND METHODS
Isolation of LDLs, HDLs, and HDL subfractions (HDL2, HDL3) by preparative ultracentrifugation. LDLs, total HDLs, and HDL2 and HDL3 subfractions were isolated from the peripheral blood serum of three apparently healthy volunteers by preparative ultracentrifugation in a NaBr density gradient using a Beckman Optima XPN-80 ultracentrifuge (Beckman Coulter, USA) [28]. After adding 1 mM EDTA, the plasma samples were transferred into centrifuge tubes, carefully overlaid with NaBr solution (density, 1.006 g/ml), and centrifuged at 105,000g for 18 h at 4°C. After centrifugation, the top fraction containing very low-density lipoproteins (VLDLs) was decanted and the tube content was mixed with the calculated amount of fine NaBr powder to produce a solution with a density of 1.065 g/ml. More NaBr solution with the same density was added to the tubes, and the tubes were centrifuged again at 105,000g for 18 h at 4°C. The top fraction containing floating LDLs was carefully collected. The density of the remaining solution in the centrifuge tube was adjusted to 1.125 mg/ml with NaBr powder. After centrifugation at 150,000g for 24 h at 4°C, the top fraction containing HDL2 was collected, and the density of the solution was adjusted to 1.21 g/ml with NaBr. The solution was centrifuged again as 150,000g for 24 h at 4°C, and the top fraction containing HDL3 was collected. To obtain the total HDL fraction, the density of the solution remaining after collecting LDLs was adjusted to 1.21 g/liter with NaBr, and the solution was centrifuged at 150,000g for 24 h at 4°C. Finally, all isolated lipoprotein fractions were dialyzed for 18 h at 4°C against 2000 volumes of 0.145 M NaCl in 50 mM K,Na-phosphate buffer (pH 7.4).
Kinetics of Cu2+-initiated free radical oxidation of LDLs, HDLs, and subfractions HDL2 and HDL3. After dialysis, the protein content in the lipoprotein samples was measured by the Lowry method followed by diluting the samples to 50 µg protein/ml with 0.154 M NaCl in 50 mM K,Na-phosphate buffer (pH 7.4). Oxidation of LDLs, HDLs, and HDL2 and HDL3 subfractions at 37°C was induced by adding 30 µM CuSO4 to the incubation medium followed by the assessment of lipid hydroperoxide (LOOH) accumulation at fixed time points at 233 nm using a UV-2600 Shimadzu spectrophotometer (Shimadzu, Japan) [12, 29, 30]. The content of LOOHs (conjugated dienes, ΔD233) in the LDL and HDL particles was calculated using the molar extinction coefficient 22,000 M−1 cm−1. The content of LOOHs per one LDL or HDL particle was estimated based on levels of ApoB100 in LDLs and ApoA1 in HDLs (LDL and HDL particles contain one molecule of the corresponding apolipoprotein per particle). The levels of ApoB100 and ApoA1 were assessed using an Abbott Architect C8000 chemistry analyzer (Abbott, USA) and commercial test kits (Abbott). The kinetics of in vitro free radical Cu2+-initiated lipoprotein oxidation was analyzed using lipoprotein particles isolated from the plasma samples of three volunteers (plasma lipoproteins isolated from each volunteer were used in independent experiments).
Statistical data processing was conducted using STATISTICA 10 (Statsoft, USA), MedCalc v. 12.7.0.0 (MedCalc Software, Belgium), and Microsoft Excel 2010 v. 14.0.7263.5000 software. Because data analysis revealed that the parameter distribution differed from normal, the difference between the groups was evaluated using the non-parametric Mann–Whitney U test. The difference was considered significant at p < 0.05.
RESULTS AND DISCUSSION
Comparative assessment of the susceptibility of different classes lipoproteins to oxidation. The mechanism of Cu-initiated free radical peroxidation of lipoproteins has been thoroughly investigated [29, 30]. Although the kinetics of in vitro lipoprotein oxidation significantly depends on the particle composition, chemical nature (unsaturation) of oxidation substrates (polyene phospholipids of the particle outer layer), and their quantity, it is mostly strongly determined by the presence of primary oxidation products (LOOHs) accumulated in the particles during their circulation in the blood [12]. The decomposition of these unstable LOOHs may occur spontaneously:
LOOH → LOO• + H+.
However, the Cu-dependent LOOH decomposition resulting in the formation of peroxyl (LOO•) and alkoxyl (LO•) radicals occurs more efficiently:
LOOH + Cu2+ → LOO• + H+ + Cu+
LOOH + Cu+ → LO• + OH− + Cu2+ [12, 28, 29].
The above equations show that the initiation rate of the lipoprotein free radical oxidation is determined by the baseline content of LOOHs accumulated in the particles in vivo. Further polyene lipid (LH) oxidation in lipoprotein particles occurs via a chain mechanism with the formation of lipid radicals (L•) as intermediates:
LH + LOO• → LOOH + L•
LH + LO• → LOH + L•
The comparison of the oxidation kinetics for the LDL particles and total HDL fraction is shown in Fig. 1.

Kinetics of the Cu2+-induced free radical oxidation of LDLs (curve 1) and HDLs (curve 2); significant difference between curve 1 and curve 2 (p < 0.05) at all time points starting from 20 min after the onset.
Under identical in vitro conditions, the rate of the free radical oxidation per LDL particle exceeded by more than two orders of magnitude the oxidation rate per HDL particle (Fig. 1). After 90 min of Cu2+-initiated oxidation, the content of accumulated LOOHs in LDL particles was (66.9 ± 3.26) × 10−11 pmol per particle, whereas the content of LOOHs in the total HDL fraction was as little as (1.85 ± 0.09) × 10−11 pmol per particle (Fig. 2).

LOOH accumulation in LDL particles, total HDL fraction, and HDL2 and HDL3 subfractions 90-min after initiation of the Cu2+-dependent free radical oxidation of phospholipids in the particle outer layer; * significant difference with all HDL fractions (p < 0.05); ** significant difference between HDL2 and total HDL, HDL3 (p < 0.05).
These data unambiguously show that the LDL particles were much more sensitive to the induced free radical oxidation of phospholipids in the outer particle layer than the HDL particles. The baseline LOOH concentration in the isolated LDLs was (4.2 ± 0.11) × 10−11 pmol LOOH per particle vs. (0.20 ± 0.04) × 10−11 pmol LOOH per particle in the total HDL fraction (Fig. 3).

The baseline LOOH concentration (in vivo LOOH level) in the LDL particles, total HDL fraction, and HDL2 and HDL3 subfractions assessed immediately after lipoprotein isolation by ultracentrifugation in the presence of EDTA; * significant difference with all HDL fractions (p < 0.05); ** significant difference between HDL2 and total HDL, HDL3 (p < 0.05).
The baseline level of lipoprotein oxidation in vivo was more than ten times higher in the LDL particles than in the HDL particles. This suggests that the rate of the in vivo free radical oxidation of LDL particles is substantially higher than that of the HDL particles, because LOOH degradation generates active alkoxy radicals LO• capable of initiating further free radical oxidation of lipid substrates via a chain mechanism. Therefore, an increased sensitivity of LDL particles to the initiation of the free radical oxidation should markedly depend on the rate of initiation [12]. An elevated content of lipid peroxides in LDLs in vivo [25, 26] may be accounted for by the initiation of oxidation of LDLs by reactive oxygen species (ROS) during the development of oxidative stress [9, 14, 26], in particular, in the atherogenesis-associated hyperlipidemia [9, 12, 26].
Studying the kinetics of induced free radical oxidation in the most anti-atherogenic HDL subfractions (HDL2 and HDL3) revealed that the lipid oxidation rate in the HDL2 particles markedly (severalfold) exceeded that in the HDL3 particles, as well as in the total HDL fraction (Fig. 4).

Comparative kinetics of the free radical Cu2+-induced oxidation for total HDL fraction (curve 1), HDL2 subfraction (curve 2), and HDL3 subfraction (curve 3); significant difference (p < 0.05) between curve 2 and curve 1 and between curve 2 and curve 3 at all-time points starting from 20 min after the onset.
We found that after 90 min of oxidation, the amount of accumulated LOOHs in HDL2 particles was 3.5 times higher than in the total HDL fraction (6.49 ± 0.66× 10–11 vs. 1.85 ± 0.09 × 10–11 pmol LOOH per particle, respectively). The difference between the LOOH accumulation in the HDL3 particles and in total HDL fraction was insignificant (Fig. 4). The baseline LOOH level in HDL2 particles was more than 2.5 times higher than in the total HDL fraction (0.52 ± 0.087 × 10–11 vs. 0.20 ± 0.04 × 10–11 pmol LOOH per particle, respectively), while no significant difference was observed between the HDL3 particles and total HDL fraction (Fig. 3).
These data convincingly demonstrate that LDL particles are much more prone to the in vitro free radical oxidation and in vivo spontaneous oxidation. LDL particles are a preferential substrate for the free radical oxidation both in in vitro and in vivo, which is in line with the concept on the crucial role of oxidatively modified LDLs in molecular mechanisms involved in the formation of vascular wall lesions in atherosclerosis [9, 27] and diabetes mellitus [9, 12, 14-16]. LDL particles enriched in hydroperoxy derivatives of phospholipids readily undergo modification by the secondary products of LOOH degradation, such as 4-hydroxy-2-nonenal and MDA [14-16]. Modification of ApoB100 in LDL particles by carbonyls might play a key role in the triggering of the mechanisms of atherogenesis [9, 14] and diabetes mellitus [9, 12, 14, 15]. Some authors have also reported higher susceptibility of LDLs vs. HDLs [24, 26, 27], which is in agreement with our data (Figs. 1-3), but there are also studies that have drawn the opposite conclusions [19-23, 25]. This controversy may be related to various factors, such as the use of different inducers for the initiation of oxidation in vitro, methods for the detection of oxidation products, and approaches for data interpretation [19-27]. In particular, we believe that presenting the data on the accumulation of oxidation products per mg total protein (as typically done in such studies) for comparing the susceptibility of LDL and HDL particles to oxidation is incorrect because of a great difference in the apolipoprotein levels in these particles [31]. Based on our calculations, the baseline content of LDL and HDL lipid peroxides, as well as their accumulation in the free radical oxidation reaction in vitro, could be more properly compared by presenting the data as LOOH content per particle. In our opinion, a higher susceptibility to oxidation of HDLs vs. LDLs is very unlikely even in theory due to a higher percentage content of apolipoproteins in HDLs and, even to a greater extent, due to a significantly lower content of oxidation substrates (phospholipids) in HDLs vs. LDLs [31]. Indeed, HDL2 and HDL3 particles contain 5 to 13 times less phospholipid molecules than LDL particles [31]. At the same time, the protein content in the HDL2 and HDL3 subfractions is 2 to 2.6 times higher than in LDLs [31]. As we demonstrated earlier by comparing the oxidation kinetics of lipoprotein (a) [Lp(a)] and LDL particles containing the same amount of oxidation substrates, a markedly lower susceptibility of Lp(a) particles to oxidation may be related to a higher apolipoprotein (a) content in them [32]. It is possible that the less pronounced oxidation of Lp(a) particles may be due to the presence of a long glycoprotein tail that distinguishes apolipoprotein (a) from ApoB100, because it can mask phospholipid polyene acyl moieties in the outer layer of Lp(a) particles, thereby reducing their accessibility to the free radical oxidation (i.e., acting as a “structural antioxidant”) [32]. This hypothesis is corroborated by the observation that carbonyl modification of Lp(a) resulting in the abolishment or attenuation of the phospholipid masking by apolipoprotein (a) increased the rate of Lp(a) free radical oxidation [32].
At present, the reasons for the observed discrepancies in the susceptibility of HDL2 and HDL3 particles to oxidation remain unclear (Figs. 2-4); however, it should be noted that the free radical oxidation of HDL particles results in the modification of their apolipoproteins so that HDLs lose their capacity to facilitate reverse cholesterol transport [33-35]. An elevated susceptibility of HDL2 particles to oxidative modification (Figs. 2-4) might affect their transformation to HDL3 [36] and contributes to the negative effect of the free radical oxidation on reverse cholesterol transport [33-35]. Our data on the increased susceptibility of LDL particles to the free radical oxidation in vitro and in vivo are in good agreement with the earlier data [37] evidencing that the elevated susceptibility of LDLs to oxidation serves as a predictor of developing coronary atherosclerosis [37].
CONCLUSION
Here, we used classical kinetics methods to obtain the data on the content of primary products of free radical oxidation in the LDL and HDL particles and HDL subfractions (HDL2 and HDL3). We found that calculating LOOH content per mg protein (as in previous publications) produces ambiguous results and should be replaced by assessing the LOOH content per LDL or HDL particle. Even in healthy people, the baseline LOOH content and the rate of induced LOOH accumulation in the atherogenic LDLs markedly exceeded those in the anti-atherogenic HDL particles. Our data convincingly show that LDL particles accumulate the major amount of the free radical oxidation products in the blood plasma upon atherogenesis-associated oxidative stress.
Abbreviations
- HDL:
-
high-density lipoprotein
- LDL:
-
low-density lipoprotein
- Lp(a):
-
lipoprotein (a)
- LOOH:
-
lipid hydroperoxide
References
Tomkin, G. H. (2010) Atherosclerosis, diabetes and lipoproteins, Expert Rev. Cardiovasc. Ther., 8, 1015-1029, https://doi.org/10.1586/erc.10.45.
Arnao, V., Tuttolomondo, A., Daidone, M., and Pinto, A. (2019) Lipoproteins in atherosclerosis process, Curr. Med. Chem., 26, 1525-1543, https://doi.org/10.2174/0929867326666190516103953.
Carr, S. S., Hooper, A. J., and Sullivan, D. R. (2019) Non-HDL-cholesterol and apolipoprotein B compared with LDL-cholesterol in atherosclerotic cardiovascular disease risk assessment, Pathology, 51, 148-154, https://doi.org/10.1016/j.pathol.2018.11.006.
Getz, G. S., and Reardon, C. A. (2020) Atherosclerosis: cell biology and lipoproteins, Curr. Opin. Lipidol., 31, 286-290, https://doi.org/10.1097/MOL.0000000000000704.
Wang, H. H., Garruti, G., Liu, M., Portincasa, P., and Wang, D. H. (2017) Cholesterol and lipoprotein metabolism and atherosclerosis: recent advances in reverse cholesterol transport, Ann. Hepatol., 16 (Suppl. 1), S27-S42, https://doi.org/10.5604/01.3001.0010.5495.
Lee, J. M. S., and Choudhury, R. P. (2010) Atherosclerosis regression and high-density lipoproteins, Expert Rev. Cardiovasc. Ther., 8, 1325-1334, https://doi.org/10.1586/erc.10.108.
Bryan, H., and Brewer, Jr. (2011) Clinical review: the evolving role of HDL in the treatment of high-risk patients with cardiovascular disease, J. Clin. Endocrinol. Metab., 96, 1246-1257, https://doi.org/10.1210/jc.2010-0163.
Hernáez, Á., Soria-Florido, M., Schröder, H., Ros, E., Pintó, X., et al. (2019) Role of HDL function and LDL atherogenicity on cardiovascular risk: A comprehensive examination, PLoS One, 14, e0218533, https://doi.org/10.1371/journal.pone.0218533.
Lankin, V. Z., and Tikhaze, A. K. (2017) Role of oxidative stress in the genesis of atherosclerosis and diabetes mellitus: a personal look back on 50 years of research, Curr. Aging Sci., 10, 18-25, https://doi.org/10.2174/1874609809666160926142640.
Schalkwijk, C. G., and Stehouwer, C. D. A. (2020) Methylglyoxal, a highly reactive dicarbonyl compound, in diabetes, its vascular complications, and other age-related diseases, Physiol. Rev., 100, 407-461, https://doi.org/10.1152/physrev.00001.2019.
Spiteller, G. (2008) Peroxyl radicals are essential reagents in the oxidation steps of the maillard reaction leading to generation of advanced glycation end products, Ann. NY Acad. Sci., 1126, 128-133, https://doi.org/10.1196/annals.1433.031.
Lankin, V. Z., Konovalova, G. G., Tikhaze, A. K., Shumaev, K. B., Kumskova, E. M., et al. (2014) The initiation of the free radical peroxidation of low-density lipoproteins by glucose and its metabolite methylglyoxal: a common molecular mechanism of vascular wall injures in atherosclerosis and diabetes, Mol. Cell. Biochem., 395, 241-252, https://doi.org/10.1007/s11010-014-2131-2.
Lankin, V. Z., Shadyro, O. I., Shumaev, K. B., Tikhaze, A. K., and Sladkova, A. A. (2019) Non-enzymatic methylglyoxal formation from glucose metabolites and generation of superoxide anion radical during methylglyoxal-dependent cross-links reaction, J. Antioxidant Activity, 1, 33-45, https://doi.org/10.14302/issn.2471-2140.jaa-19-2997.
Lankin, V. Z., Tikhaze, A. K., and Melkumyants, A. M. (2022) Dicarbonyl-dependent modification of LDL as a key factor of endothelial dysfunction and atherosclerotic vascular wall damage, Antioxidants, 11, 1565, https://doi.org/10.3390/antiox11081565.
Lankin, V. Z., Tikhaze, A. K., Kapel’ko, V.I., Shepel’kova, G. S., Shumaev, K. B., et al. (2007) Mechanisms of oxidative modification of low density lipoproteins under conditions of oxidative and carbonyl stress, Biochemistry (Moscow), 72, 1081-1090, https://doi.org/10.1134/s0006297907100069.
Lankin, V. Z., Tikhaze, A. K., Konovalova, G. G., Kumskova, E. M., and Shumaev, K. B. (2010) Aldehyde-dependent modification of low density lipoproteins, in Handbook of Lipoprotein Research, NY., pp. 85-107.
Sun, Y., and Chen, X. (2011) Ox-LDL-induced LOX-1 expression in vascular smooth muscle cells: role of reactive oxygen species, Fundam. Clin. Pharmacol., 25, 572-579, https://doi.org/10.1111/j.1472-8206.2010.00885.x.
Lankin, V. Z., Tikhaze, A. K., and Kumskova, E. M. (2012) Macrophages actively accumulate malonyldialdehyde-modified but not enzymatically oxidized low density lipoprotein, Mol. Cell. Biochem., 365, 93-98, https://doi.org/10.1007/s11010-012-1247-5.
Bowry, V. W., Stanley, K. K., and Stocker, R. (1992) High density lipoprotein is the major carrier of lipid hydroperoxides in human blood plasma from fasting donors, Proc. Natl. Acad. Sci. USA, 89, 10316-10320, https://doi.org/10.1073/pnas.89.21.10316.
Suzukawa, M., Ishikawa, T., Yoshida, H., and Nakamura, H. J. (1995) Effect of in-vivo supplementation with low-dose vitamin E on susceptibility of low-density lipoprotein and high-density lipoprotein to oxidative modification, J. Am. Coll. Nutr., 14, 46-52, https://doi.org/10.1080/07315724.1995.10718472.
Garner, B., Witting, P. K., Waldeck, A. R., Christison, J. K., Raftery, M., et al. (1998) Oxidation of high density lipoproteins. I. Formation of methionine sulfoxide in apolipoproteins AI and AII is an early event that accompanies lipid peroxidation and can be enhanced by alpha-tocopherol, J. Biol. Chem., 273, 6080-6087, https://doi.org/10.1074/jbc.273.11.6080.
Ohmura, H., Watanabe, Y., Hastumi, C., Sato, H., Daida, H., et al. (1999) Possible role of high susceptibility of high-density lipoprotein to lipid peroxidative modification and oxidized high-density lipotein in genesis of coronary artery spasm, Atherosclerosis, 142, 179-184, https://doi.org/10.1016/s0021-9150(98)00235-4.
Raveh, O., Pinchuk, I., Fainaru, M., and Lichtenberg, D. (2001) Kinetics of lipid peroxidation in mixtures of HDL and LDL, mutual effects, Free Radic. Biol. Med., 31, 1486-1497, https://doi.org/10.1016/s0891-5849(01)00730-4.
Parthasarathy, S., Barnett, J., and Fong, L. G. (1990) High-density lipoprotein inhibits the oxidative modification of low-density lipoprotein, Biochim. Biophys. Acta, 1044, 275-283, https://doi.org/10.1016/0005-2760(90)90314-n.
Raveh, O., Pinchuk, I., Schnitzer, E., Fainaru, M., Schaffer, Z., et al. (2000) Kinetic analysis of copper-induced peroxidation of HDL, autoaccelerated and tocopherol-mediated peroxidation, Free Radic. Biol. Med., 29, 131-146, https://doi.org/10.1016/s0891-5849(00)00332-4.
Nourooz-Zadeh, J., Tajaddini-Sarmad, J., Ling, K. L., and Wolff, S. P. (1996) Low-density lipoprotein is the major carrier of lipid hydroperoxides in plasma. Relevance to determination of total plasma lipid hydroperoxide concentrations, Biochem. J., 313 (Pt 3), 781-786, https://doi.org/10.1042/bj3130781.
Lankin, V. Z., and Tikhaze, A. K. (2003) in Free Radicals, Nitric Oxide, and Inflammation: Molecular, Biochemical, and Clinical Aspects (Tomasi, A., Ozben, T., Skulachev, V. P., eds) IOS Press, NATO Science Series, Amsterdam, 344, pp. 218-231.
Lindgren, F. T. (1975) in Analysis of Lipids and Lipoproteins (Perkins, E. G., ed) Champaign: Amer. Oil. Chemists Soc., pp. 204-224.
Mark, J., and Burkitt, A. (2001) Critical overview of the chemistry of copper-dependent low density lipoprotein oxidation: roles of lipid hydroperoxides, a-tocopherol, thiols, and ceruloplasmin, Arch. Biochem. Biophys., 394, 117-135, https://doi.org/10.1006/abbi.2001.2509.
Patel, R. P., and Darley-Usmar, V. (1999) Molecular mechanisms of the copper dependent oxidation of low-density lipoprotein, Free Rad. Res., 30, 1-9, https://doi.org/10.1080/10715769900300011.
Shen, B.W., Scanu, A. M., and Kezdy, F. J. (1977) Structure of human serum lipoproteins inferred from compositional analysis, Proc. Natl. Acad. Sci. USA, 74, 837-841, https://doi.org/10.1073/pnas.74.3.837.
Lankin, V. Z., Afanasieva, O. I., Konovalova, G. G., Utkina, E. A., Dmitrieva, O. A., et al. (2011) Modification of lipoprotein(a) by natural dicarbonyls induced their following free radical peroxidation, Dokl. Biochem. Biophys., 441, 287-289, https://doi.org/10.1134/S1607672911060159.
Nagano, Y., Arai, H., and Kita, T. (1991) High density lipoprotein loses its effect to stimulate efflux of cholesterol from foam cells after oxidative modification, Proc. Natl. Acad. Sci. USA, 88, 6457-6461, https://doi.org/10.1073/pnas.88.15.6457.
Salmon, S., Maziere, C., Auclair, M., Theron, L., Santus, R., et al. (1992) Malondialdehyde modification and copper-induced autooxidation of high-density lipoprotein decrease cholesterol efflux from human cultured fibroblasts, Biochim. Biophys. Acta, 1125, 230-235, https://doi.org/10.1016/0005-2760(92)90050-6.
Gao, D., and Podrez, E. A. (2018) Characterization of covalent modifications of HDL apoproteins by endogenous oxidized phospholipids, Free Radic. Biol. Med., 115, 57-67, https://doi.org/10.1016/j.freeradbiomed.2017.11.012.
Nestel, P. J. (1987) High-density lipoprotein turnover, Am. Heart J., 113 (Pt. 2), 518-521, https://doi.org/10.1016/0002-8703(87)90624-7.
Aoki, T., Abe, T., Yamada, E., Matsuto, T., and Okada, M. (2012) LDL susceptibility to oxidation accelerates future carotid artery atherosclerosis, Lipids Health Dis., 11, 4, https://doi.org/10.1186/1476-511X-11-4.
Acknowledgments
We are grateful to A. A. Panferova and Dr. G. G. Konovalova, for their assistance in lipoprotein isolation and conduction of experiments and to Dr. K. B. Shumaev for valuable discussion of the study results.
Funding
The study was supported by the Russian Science Foundation (project no. 22-15-00013).
Author information
Authors and Affiliations
Contributions
V. Z. Lankin conceived and supervised the research, discussed experimental data, and wrote the manuscript; A. K. Tikhaze discussed experimental data, wrote and edited the manuscript; V. Ya. Kosach conducted experiments, discussed the results, and edited the manuscript.
Corresponding author
Ethics declarations
The authors declare no conflict of interests. All procedures performed in the study with human subjects were conducted in compliance with the ethics standards approved by the National Research Ethics Committee, as well as the Declaration of Helsinki 1964 and its subsequent revisions or comparable ethical standards. All persons enrolled in the study provided informed voluntary consent.
Rights and permissions
About this article

Cite this article
Lankin, V.Z., Tikhaze, A.K. & Kosach, V.Y. Comparative Susceptibility to Oxidation of Different Classes of Blood Plasma Lipoproteins. Biochemistry Moscow 87, 1335–1341 (2022). https://doi.org/10.1134/S0006297922110128
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0006297922110128