
Abstract
A recombinant strain producing a complex of extracellular enzymes including chitinase from Myceliophtora thermophila was created based on the fungus Penicillium verruculosum. The activity of the enzyme preparations obtained from the cultural fluid of the producer strain was 0.55, 0.53, and 0.66 U/mg protein with chitin and chitosans with the molecular weight of 200 and 1000 kDa, respectively. The temperature optimum for the recombinant chitinase was 52-65°C; the pH optimum was 4.5-6.2, which corresponded to the published data for this class of the enzymes. The content of heterologous chitinase in the obtained enzyme preparations was 47% of total protein content in the cultural fluid. Enzyme preparations produced by the recombinant P. verruculosum XT403 strain and containing heterologous chitinase were able to degrade the mycelium of micromycetes, including phytopathogenic ones, and were very efficient in the bioconversion of microbiological industry waste.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
Chitinase (endo-1,4-β-poly-N-acetyl-glucosaminidase) is a key enzyme responsible for the destruction of chitin and chitosan (linear homopolymers of 1,4-β-N-acetyl-D-glucosamine). Chitinase efficiently cleaves glycosidic bonds separated by one carbohydrate residue from the N-acetylglucosamine residues in chitin and chitosan; its activity strongly depends on the degree of substrate deacetylation (DD) [1].
Chitinolytic enzymes are widely used in biotechnology. New approaches have been intensively developed for the application of these enzymes in biochemical transformation of chitin into biologically active chitooligosaccharides possessing antimicrobial (antifungal and antibacterial), elicitor, growth-stimulating, antitumor, and anti-inflammatory activities [1-4]. Moreover, chitin monomers N-acetylglucosamine and D-glucosamine are physiologically active substances [5-7]. Another current application of chitinase is conversion of chitin-containing raw waste into microbial and yeast biomass, protein, and biological fuel [8-11]. Chitinases play a key role in the complex of hydrolases used for the lysis of fungal cell wall and are efficient tools in the isolation of fungal cell protoplasts [12, 13].
Chitinolytic enzymes possess fungicidal, insecticidal, and antiparasitic (nematocidal) activities; they are attractive as biopesticides that could replace chemical pesticides currently widely used in open field and greenhouses [4]. In all these cases, the enzymes have to destroy chitin of the microbial cell walls or structural elements, exoskeletons of insects, and envelopes of parasites’ eggs. Chitinolytic enzymes play an important role as antimicrobial agents in the fight against fungal and bacterial contamination of food and agricultural products [14].
Mycelial fungi occupy a leading position in biotechnological industry due to high productivity upon cultivation on cheap industrial media and ability to synthesize many enzymes and metabolites, such as antibiotics, vitamins, and organic acids. However, in the course of fermentation, fungal mycelium forms large amounts of waste. For example, worldwide production of citric acid using Aspergillus niger leaves 0.34 million tons of mycelium every year [15]. Fungal biomass cannot be stored for a long time and is a source of environment contamination; most frequently, it is destroyed by burning [3, 16]. Chitin is a structurally important component of the fungal cell wall; its content is different in fungi from different taxons and, depending on the systematic position of a fungus and cultivation conditions, varies from 0.2 to 26% of dry mass [17]. Enzymatic destruction of polysaccharides, first of all, chitin, in the cell wall of mycelial fungi is a very promising approach because it represents ecologically safe and economically reasonable way for the processing of fungal mycelium waste in microbiological industry and allows to obtain biologically valuable compounds in easily digestible form [18].
All the above said shows the necessity for creating highly active producer strains for the industrial production of chitinolytic enzymes. The purpose of this work was to create a producer of chitinase based on the recipient Penicillium verruculosum 537 (ΔniaD) strain with a high secretory ability (up to 50-60 g extracellular protein per liter) and reduced repression of glucose catabolism [19-21] and to test the resulting enzyme preparation (EP) in the degradation of the cell wall of microscopic fungi. We chose chitinase from Myceliophtora thermophyla (earlier Chrysosporium lucknowense) for cloning into P. verruculosum 537 (ΔniaD., because enzymes from this ascomycete are known to have wide pH and temperature activity ranges, which makes them more suitable for practical application [22, 23].
MATERIALS AND METHODS
Reagents. Polyacrylamide gels (PAGs) and buffer solutions were prepared using reagents from Bio-Rad (USA), MP Biochemicals (France), Ferak (Germany), and Reakhim (Russia). PAGs were stained with Coomassie Brilliant Blue R-250B, and molecular masses of proteins were determined using protein markers from MW-SDS 200 kit (10-200 kDa; ThermoScientific, USA). Protein concentration was determined by the Lowry method using bovine serum albumin (BSA) as a calibration standard (BelNIIEM, Republic of Belarus).
Substrates. Substrates for the enzymatic activity assays were carboxymethyl cellulose (CMC) sodium salt, birch xylan (Sigma-Aldrich, USA), and microcrystalline cellulose (MCC; MCC Center, Russia). Chitosan with a molecular mass of 1000 kDa and DD of 85%, chitosan with a molecular mass of 200 kDa and DD of 87%, and crab colloidal chitin were kindly provided by Prof. V. P. Varlamov (The Federal Research Centre “Fundamentals of Biotechnology”, Russian Academy of Sciences).
Creation of chitinase-producing P. verruculosum strain. As a recipient strain, we used P. verruculosum B1-537 (ΔniaD) deficient by the niaD gene. Escherichia coli Mach-1 (ΔrecA1398 endA1 tonA Φ80ΔlacM15 ΔlacX74 hsdR(r–Km+K) cells (Invitrogen, USA) were used for gene cloning and plasmid isolation.
Plasmid. The chi403 gene (GenBank: XM_0036663496) coding for M. thermophila chitinase was expressed in the pChi403 plasmid under control of the promoter of homologous cbh1 gene encoding cellobiohydrolase 1 (CBH1) in P. verruculosum [23].
To produce the pChi403 plasmid, the chi403 gene was amplified by PCR using M. thermophila genomic DNA as a template with the following oligonucleotide primers [24]:
(Chi403-LIC) 5′-CAAACAGAAGCAACCGACACAATGGGCGGCGGACCTACGGA,
(Chi403-LIC) 3′-GAGGAGAAGCCCGGTCTAATTGTTCGGGAATCCCTCCCTCA.
Cloning of the resulting PCR product into the pUC-CBH vector ensured expression of the chi403 gene due to its stable integration in the P. verruculosum chromosome using independent ligation sites [25].
Transformation of the recipient strain. Recombinant strains of the XT403 series were obtained by the transformation of the recipient P. verruculosum B1-537 (ΔniaD) strain with the pChi403 plasmid [23].
Isolation of genomic DNA, purification of PCR products from agarose gel, and plasmid DNA isolation were performed using corresponding Qiagen kits (USA). PCR was performed with a Long Polymerase Mix kit (ThermoScientific) in a MyCycler (Bio-Rad) in the following regime: 1 cycle of 94°C for 3.5 min; 30 cycles of 94°C for 30 s, 50°C for 2 min, and 72°C for 1 min; 72°for 10 min; and 4°C for 25 min.
Cultivation of producer strains. P. verruculosum XT403 strains were grown on a shaker in 750-ml Erlenmeyer flasks in 100 ml of the fermentation medium of the following composition (%): KH2PO4 – 1.5; (NH4)2SO4 × 7H2O – 0.5; MgSO4 × 7H2O – 0.03; CaCl2 × 2H2O – 0.03; glucose – 1.0; yeast extract – 1.0; wheat bran – 1.0; MCC – 4.0. After cultivation for six days on a shaker at 220 rpm at 30°C, the cultural fluid (CF) was separated from the mycelium by centrifugation for 10 min at 10,700g and assayed for the protein concentration and activities against colloidal chitin, chitosans, CMC, and xylan.
Preparation of mycelial fungal biomass for hydrolysis by chitinase. Aspergillus awamori M2002 (BKM F-3771D) strain was cultivated in flasks for six days at 35°C in the fermentation medium containing 24% wheat flour treated with thermostable α-amylase. Penicillium canescens Pep-4 (BKM F-4677D) strain was cultivated in flasks for seven days at 30°C in the medium containing (%): soy husk – 4.5, maize extract – 5.0, and KH2PO4 – 2.5.
The biomass of mycelial fungi Fusarium culmorum, Fusarium sambucinum, and Fusarium graminearum was obtained by deep cultivation in the Czapek’s medium; Stagonospora nodorum and Septoria tritici were grown in the potato-glucose medium; Aspergillus flavus was grown in the Payne–Hagler medium. The fungi were cultivated at 220 rpm at 25-26°C for seven days (Fusarium spp., A. flavus) or 10-12 days (St. nodorum, S. tritici). All strains were from the State Collection of phytopathogenic microorganisms and plant identifiers of pathogenic strains of microorganisms, Russian Institute of Phytopathology (Bolshye Vyazemy, Moscow Region).
At the end of fermentation, the fungal biomass was separated by filtration through a nonwoven material.
Enzyme preparations. Selected recombinant strains and the recipient P. verruculosum strain were cultivated in the fermentation KF-108 tanks with a working volume of 1 liter at the Institute of Biochemistry and Physiology of Microorganisms, Russian Academy of Sciences (Pushchino, Moscow Region) for six-seven days at 32.0 ± 0.5°C with aeration at 1 liter/liter medium/min in the medium of the following composition (%): KH2PO4 – 0.7; (NH4)2SO4 × 7H2O – 0.5; MgSO4 × 7H2O – 0.03; CaCl2 × 2H2O – 0.023; glucose – 4.0; yeast extract – 1.0; wheat bran – 1.0; MCC – 4.0 (pH > 4.5). After cultivation, the CF was separated from the mycelium by centrifugation and freeze-dried with a Mini Spray Dryer B-290 (Buchi, Switzerland).
Enzymatic activity assays. Enzymatic activity toward polysaccharide substrates (chitosans, CMC, MCC, xylan) was determined from the initial rate of reducing sugar (RS) formation at 50°C (0.1 M Na-acetate buffer, pH 5.0) at the substrate initial concentration of 5 g/liter. RSs were determined by the modified Nelson–Somogyi method [26-28] or by the bicinchoninate method (in chromatographic fractions) [29]. The amount of the enzyme producing 1 µmol of RS per min was taken as the activity unit.
The activity against colloidal chitin was determined by the modified ferricyanide method [30] or from a decrease in the absorbance of chitin suspension at 700 nm [31]. When using the ferricyanide method, the initial rate of RS (glucosamine) formation was determined at 50°C (Na-acetate buffer, pH 5.0); the amount of the enzyme producing 1 µmol of RS (glucosamine) per min was taken as the enzyme activity unit. In the second method, the reaction was performed at 37°C (Na-acetate buffer pH 5.0); the decrease in the absorbance of chitin suspension by 1% of the initial value at 700 nm was taken as the unit of chitinase activity.
Protein concentration in the CF and enzyme preparations was determined by the modified Lowry method [32] using BSA as a standard. Protein content in the chromatographic fractions was determined from the absorbance at 280 nm, using the coefficient of protein extinction of 2.0.
Electrophoresis in PAG. SDS-electrophoresis in 12% PAG was performed using a MiniProtein device (Bio-Rad) according to the manual.
Chromatographic fractionation of enzyme preparations. The composition of the enzyme preparations was analyzed in three stages: preliminary purification, anion-exchange chromatography, and hydrophobic chromatography. Enzyme preparations were precipitated with (NH4)2SO4 (80% saturation at 25°C), resuspended in 0.02 M bis-Tris-HCl buffer (pH 6.8), and desalted on a P-4 biogel column equilibrated with the same buffer. The following stages were performed on an NGC Chromatography System (Bio-Rad). Anion-exchange chromatography of desalted enzyme preparations was performed on a Source 15Q column (Pharmacia, Sweden) equilibrated with 0.02 M bis-Tris-HCl (pH 6.8). Proteins were eluted with a 0-0.4 M linear gradient of NaCl concentration, and pH of the obtained fractions was adjusted to 5.0 with 1.0 M Na-acetate buffer (pH 5.0). Fractions containing the target enzymes were subjected to hydrophobic chromatography on a 1-ml Source 15 Isopropyl column (Pharmacia) equilibrated with 1.7 M (NH4)2SO4 in 0.05 M Na-acetate buffer (pH 5.0). The proteins were eluted with a 1.7-0 M linear gradient of (NH4)2SO4 concentrations.
The resulting fractions were assayed for the protein content and enzymatic activities toward various substrates. All obtained fractions were analyzed by SDS-electrophoresis in PAG.
The composition of the enzyme preparations was estimated by corresponding the determined enzymatic activities in the chromatographic fractions with the results of electrophoretic analysis. The quantitative composition of the enzyme preparations was calculated as the ratio of the enzyme amount in the homogenous fraction to the total amount of protein in the specimen under study. The content of individual enzymes in the enzyme preparations was determined by densitometric analysis of SDS-PAGs using the Gel Analyser software.
Destruction of fungal biomass. Hydrolysis of deep mycelium biomass of A. awamori, P. canescens, F. culmorum, F. sambucinum, F. graminearum, St. nodorum, S. tritici, and A. flavus was performed in 10-ml graduated glass tubes with ground glass stoppers for 24, 36, or 48 h at a 1 : 10 ratio (1 g wet mycelium per 10 ml distilled water) at 25-27°C without mixing. After addition of the enzyme preparation (10 mg protein per 1 g wet mycelium), its effect on the fungal biomass was assessed visually by estimating the volume of the precipitate in the graduated tube, as well as by microscopy with an Eclipse Ci microscope (Nikon Corporation, Japan).
RESULTS AND DISCUSSION
Obtaining chitinase producer strain. The chi403 gene (GenBank AN: NC_016476), encoding chitinase of M. thermophila belonging to the family of GH18 glycosyl hydrolases was cloned in the pUC-CBHI vector which provided expression of the target gene in the recipient P. verruculosum B1-537 (ΔniaD) strain under control of the inducible promoter of the cbh1 gene encoding CBH1.
The resulting pChi403 plasmid carried the expression cassette containing the promoter and the terminator of the homologous cbh1 gene from P. verruculosum and heterologous chi403 gene fused with the sequence coding for the CBH1 signaling peptide for the enzyme secretion (Fig. 1).

Expression cassette with the chi403 gene encoding GH18 chitinase from M. thermophila in the pChi403 plasmid.
The recipient P. verruculosum B1-537 (ΔniaD) strain was co-transformed with 10 µg of the pChi403 plasmid and 1 µg of the pSTA10 (niaD) plasmid. As a result, ~250 transformants of the XT403 series were obtained, which corresponded to the average transformation efficiency of mycelial fungi [33]. Ten transformants were selected at random, transferred onto the minimal selective medium [23], and analyzed for the presence of the cloned gene by sequencing with oligonucleotides used for its PCR amplification. All analyzed transformants contained the chi403 gene.
The selected transformants were cultivated in flasks on a shaker. After cultivation, the CF was assayed for the protein concentration and enzymatic activity toward colloidal chitin, chitosans with molecular masses of 200 and 1000 kDa, CMC, and xylan. The CF of the recipient strain P. verruculosum B1-537 (ΔniaD) was used as a control. The transformant Chi403-4 with the highest chitinase activity (data not presented) was selected for further cultivation in 1-liter fermenters. The resulting enzyme preparation XT403-4 was lyophilized CF.
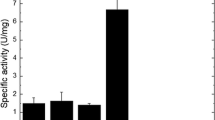
Enzymatic activity of recombinant enzyme preparation. The obtained enzyme preparation XT403-4 was assayed for the protein content and activity toward various substrates (Table 1). The enzyme preparation (lyophilized CF) of the recipient P. verruculosum B1-537 (ΔniaD) strain was used as a control. The specific activity of XT403-4 toward chitosans with the molecular masses of 200 and 1000 kDa was 0.53 and 0.66 U/mg protein, respectively, vs. 0.04 and 0.04 U/mg protein in the control. The activity toward colloidal chitin was 0.55 U/mg protein vs. 0.05 U/mg protein in the control.
The cellobiohydrolase activity of the recombinant enzyme preparation XT403-4 by MCC was 2.7 times lower than in the control, which could be associated with the deficit of positive transcription factors in the recombinant strain [34].
The endoglucanase activity by CMC and xylanase activity by birch xylan of XT403-4 were 6.2 and 9.4 times lower, respectively, in comparison to the control.
Composition of recombinant enzyme preparation. Figure 2 shows the results of the recombinant enzyme preparation analysis by SDS-electrophoresis in PAG. The electrophoregram of the recombinant enzyme differed from that of the control preparation in the presence of a well pronounced new band in the region of 43 kDa (which corresponded to the molecular weight of heterologous chitinase, CHT) and decreased intensities of bands corresponding to cellobiohydrolase 1 (CBHI, 66 kDa), endoglucanase 2 (EG, 39 kDa), and xylanase (XYL, 32 kDa).

SDS-PAGE of XT403-4 (lane 1) and control enzyme preparation from the recipient P. verruculosum B1-537 (ΔniaD) strain (lane 2).
The composition of the recombinant enzyme preparation XT403-4 was studied by anion-exchange chromatography on a Source 15Q column at pH 6.8 after purification from low-molecular-weight compounds by gel-filtration. Fractions containing chitinase were subjected to hydrophobic chromatography on a Source 15 Isopropyl column. All eluted fractions were assayed for the protein content and enzymatic activity against chitosan, chitin, CMC, MCC, and xylan, as well as analyzed by SDS-PAGE. The obtained results were used for the calculation of the content of main enzyme preparation components (Table 2). The amount of heterologous chitinase in XT403-4 was 47% of total protein; the content of CBH1 was ~2.3 times lower than in the control preparation, thus corresponding to the decrease in the specific activity toward MCC (Table 1). The contents of endoglucanase and xylanase were also reduced, which also corresponded to the decrease in the specific activities toward their substrates.
Thermal stability, pH optimum, and temperature optimum of chitinase activity. We also determined pH and temperature optima for the chitinase activity in XT403-4, as well as the enzyme stability at different temperatures (Table 3). The maximal chitinase activity was observed at 60°C and pH 4.7-5.5; the temperature and pH range in which the enzyme displayed 80% of its maximal activity were 52-65°C and pH 4.5-6.2, respectively, which was in a good agreement with the earlier published data for this class of enzymes [1]. Aqueous solution of chitinase remained active when subjected to high temperatures: after 3-h incubation at 40 and 50°C (pH 5), the enzyme retained 90 and 83% of its initial activity, respectively (Table 3).
Degradation of fungal mycelium with chitinase from the enzyme preparation XT403-4. A. awamori (commercial producer of glucoamylase) and P. canescens (commercial producer of proteases) were used as the sources of fungal mycelium. The mycelium was treated with XT403-4 at a dose of 10 mg protein per 1 g wet fungal biomass. The volume of A. awamori micelium decreased 2.2 times after the treatment with XT403-4 for 24 h; the volume of P. canescens decreased ~2 times (Fig. 3). The enzyme preparation from the recipient B1-537 (ΔniaD) strain possessed only the cellulolytic activity (chitinase activity was very low; see Table 1) and did not affect the volume of the fungal biomass from both fungi (Fig. 3).

Residual mycelium volume after hydrolysis of A. awamori (1) and P. canescens (2) fungal biomass for 24 h: I, control (without the enzyme preparation); II, with the XT403-4 enzyme preparation; III, with enzyme preparation from the recipient B1-537 (ΔniaD) strain.
The results of experiments on the hydrolysis of fungal mycelium were confirmed by microscopy (Fig. 4), which revealed destruction of the cell wall in A. awamori and P. canescens fungi after the treatment with XT403-4.

Microscopy images of A. awamori (a) and P. canescens (b) mycelia after hydrolysis for 24 h: 1, control (without enzyme preparation); 2, with the enzyme preparation XT403-4; 3, with the enzyme preparation from the recipient B1-537 (ΔniaD) strain.
The XT403-4 enzyme preparation was used for the hydrolysis of mycelia of the pathogenic fungi F. culmorum, F. sambucinum, F. graminearum, St. nodorum, S. tritici, and A. flavus under conditions described above for the hydrolysis of A. awamori and P. canescens fungi. After incubation with XT403-4, the mycelium volume reduced 2.3, 2.0, 2.3, 2.1, 2.2, and 2.4 times, respectively, in comparison with the control [enzyme preparation from the recipient strain B1-537 (ΔniaD)] (Fig. 5). The results of mycelium hydrolysis were confirmed by microscopy (data not presented).

Residual volume of mycelium after 24-h hydrolysis with XT403-4 (1) and enzyme preparation from the recipient B1-537 (ΔniaD) strain (2). Mycelia of the following phytopathogenic fungi were used: I, F. culmorum; II, F. sambucinum; III, F. graminearum; IV, St. nodorum; V, S. tritici; and VI, A. flavus.
Our results show that the enzyme preparation obtained by expression of chitinase in P. verruculosum is a promising agent for efficient bioconversion of microbiological industry waste, as well as for the degradation of mycelia of phytopathogenic fungi.
Abbreviations
- CBHI:
-
cellobiohydrolase I
- CF:
-
cultural fluid
- CHT:
-
chitinase
- DD:
-
degree of deacetylation
- EG:
-
endoglucanase
- EP:
-
enzyme preparation
- XYL:
-
xylanase
REFERENCES
Melent’ev, A. I., and Aktuganov, G. E. (2013) in Enzymes of Degradation of Chitin and Chitosan. Chitosan [in Russian] (Skryabin, G. K., Mikhailov, C. N., and Varlamov, V. P., eds.) Center “Bioengineering”, Russian Academy of Sciences, Moscow, pp. 71-114.
Chitin, Chitosan, Oligosaccharides and Their Derivatives: Biological Activity and Applications (2011) (Kim, S.-K., ed.) CRC Press, Taylor and Francis Group, Boca Raton, London, N. Y., doi: 10.1201/EBK1439816035.
Berini, F., Katz, C., Gruzdev, N., Casartelli, M., Tettamanti, G., and Marinelli, F. (2018) Microbial and viral chitinases: attractive biopesticides for integrated pest management, Biotechnol. Adv., 36, 818-828, doi: 10.1016/j.biotechadv.2018.01.002.
Vijayakumar, N., and Alagar, S. (2017) Consequence of chitinase from Trichoderma viride integrated feed on digestive enzymes in Corcyra cephalonica (Stainton) and antimicrobial potential, Biosci. Biotech. Res. Asia, 14, 513-519, doi: 10.13005/bbra/2473.
Sakai, K., Uchiyama, T., Matahira, Y., and Nanjo, F. (1991) Immobilization of chitinolytic enzymes and continuous production of N-acetyl D-glucosamine with the immobilized enzymes, J. Ferment. Bioeng., 72, 168-172, doi: 10.1016/0922-338X(91)90211-X.
Saschiwa, H., Fujishima, S., and Yamano, N. (2002) Production of N-acetyl D-glucosamine from alpha-chitin by crude enzymes from Aeromonas hydrophila H-2330, Carbohydr. Res., 337, 761-763, doi: 10.1016/s0008-6215(02)00034-4.
Jain, T., Kumar, H., and Dutta, P. K. (2016) in Chitin and Chitosan for Regenerative Medicine (Dutta, P. K., ed.) Springer India, Bangalore, Mumbai, New Delhi, India, pp. 279-295.
Wang, S.-L., Liang, T.-W., and Yen, Y.-H. (2011) Bioconversion of chitin-containing wastes for the production of enzymes and bioactive materials, Carbohydr. Pol., 84, 732-742, doi: 10.1016/j.carbpol.2010.06.022.
Thadathil, N., and Velappan, S. P. (2014) Recent developments in chitosanase research and its biotechnological applications: a review, Food Chem., 150, 392-399, doi: 10.1016/j.foodchem.2013.10.083.
Sakai, K., Yokota, A., Kurokawa, H., Wakayama, M., and Moriguchi, M. (1998) Purification and characterization of three thermostable endochitinases of a noble Bacillus strain, MH-1, isolated from chitin-containing compost, Appl. Environ. Microbiol., 64, 3397-3402, doi: 10.1128/AEM.64.9.3397-3402.1998.
Revah-Moiseev, S., and Carroad, P. A. (1981) Conversion of the enzymatic hydrolysate of shellfish waste chitin to single-cell protein, Biotechnol. Bioeng., 23, 1067-1078, doi: 10.1002/bit.260230514.
Dahia, N., Tiwari, R., Tiwari, R. P., and Hoondal, G. S. (2005) Production of an antifungal chitinase from Enterobacter sp. NRG4 and its application in protoplast production, World J. Microbiol. Biotechnol., 21, 1611-1615, doi: 10.1007/s11274-005-8343-6.
Johnson, E. A., Villa, T. G., Lewis, M. J., and Phaff, H. J. (1979) Lysis of the cell wall of yeast Phaffia rhodozyme by lytic complex from Bacillus circulans WL-12, J. Appl. Biochem., 1, 273-282.
Biotransformation of Waste Biomass into High Value Biochemicals (2014) (Brar, S. K., Dhillon, G. S., and Soccol, C., eds.) Springer Verlag, N. Y., p. 504.
Kamzolkina, O. V., and Dunaevsky, Y. E. (2017) Biology of a Fungal Cell. A Handbook [in Russian], 2nd Edn., Fellowship of Scientific Publications KMK, Moscow.
Kuranda, M. J., and Robbins, P. W. (1991) Chitinase is required for cell separation during growth of Saccharomyces cerevisiae, J. Biol. Chem., 266, 19707-19758.
Kurchenko, V. P., Buga, S. V., Petrashkevich, H. V., Butkevich, T. V., Vetoshkin, A. A., Demchenkov, E. L., Lodygin, A. D., Zueva, O. Y., Varlamov, V. P., and Borodin, O. I. (2016) Technological bases of obtaining Chitin and chitosan from insects, Trudy BGU, 11, 110-126.
Sereda, A. S., Velikortskaya, I. A., Osipov, D. O., Matys, V. Y., Bubnova, T. V., Nemashkalov, V. A., Sinitsyna, O. A., Rozhkova, A. M., Tsurikova, N. V., and Sinitsyn, A. P. (2018) Enzyme complexes for destruction of the cell wall of mycelial fungi – producers industrial enzymes, Izv. of Res. Center of Ufa, Russian Academy of Sciences, 3, 31-35.
Sinitsyn, A. P., Okunev, O. N., Chernoglazov, V. M., Sinitsyna, O. A., and Popov, V. O. The RF Patent No. 2361918, published 20.07.2009.
Sinitsyn, A. P., Rozhkova , A. M., Sinitsyna, O. A., Fedorova, E. A., Okunev, O. N., Bekkarevich, A. O., Sokolova, L. M., Matys, V. Yu., Koshelev, A. V., Vinetskiy, Yu. P., Chernoglazov, V. M., and Zorov, I. N. The RF Patent No. 2378372, published 10.01.2010.
Sinitsyn, A. P., Korotkova, O. G., Rubtsova, E. A., Sinitsyna, O. A., Kondrat’eva, E. G., Sereda, A. S., Zorov, I. N., and Rozhkova, A. M. (2019) Constructing recombinant producers of enzyme preparations for production of fodder with the expression system based on the fungus Penicillium verruculosum, Biotechnologiya, 35, 6-14, doi: 10.21519/0234-2758-2019-35-4-6-14.
Bukhtoyarov, F. E., Ustinov, B. B., Salanovich, T. N., Antonov, A. I., Gusakov, A. V., Okunev, O. N., and Sinitsyn, A. P. (2004) Cellulase complex of the fungus Chrysosporium lucknowense: isolation and characteristics of endoglucanases and cellobiohydrolases, Biochemistry (Moscow), 69, 542-551.
Merzlov, D. A., Zorov, I. N., Dotsenko, G. S., Denisenko, Y. A., Rozhkova, A. M., Satrutdinov, A. D., Rubtsova, E. A., Kondrat’eva, E. G., and Sinitsyn, A. P. (2015) Properties of enzyme preparations and homogenous enzymes – endoglucanase EG2 Penicillium verruculosum and endoglucanase LAM Myceliophtora thermophila, Biochemistry (Moscow), 80, 473-482, doi: 10/1134/S006297915040112.
Semenova, M. V., Gusakov, A. V., Volkov, P. V., Matys, V. Yu., Nemashkalov, V. A., Telitsyn, V. D., Rozhkova, A. M., and Sinitsyn, A. P. (2019) Enhancement of the enzymatic cellulose saccharification by Penicillium verruculosum multienzyme cocktails containing homologously overexpressed lytic polysaccharide monooxygenase, Mol. Biol. Rep., 46, 2363-2370, doi: 10.1007/s11033-019-04693-y.
Aslanidis, C., and de Jong, J. P. (1990) Ligation-independent cloning of PCR products (LIC-PCR), Nucleic Acids Res., 18, 6069-6075, doi: 10.1093/nar/18.20.6069.
Nelson, N. A. (1944) Photometric adoption of the Somogyi method for the determination of glucose, J. Biol. Chem., 153, 375-379.
Somogyi, M. (1952) Notes on sugar determination, J. Biol. Chem., 195, 19-23.
Sinitsyna, O. A., Bukhtoyarov, F. E., Gusakov, A. V., Okunev, O. N., Bekarevich, A. O., Vinetsky, Y. P., and Sinitsyn, A. P. (2003) Isolation and properties of major components of Penicillium canescens extracellular enzyme complex, Biochemistry (Moscow), 68, 1200-1209, doi: 10.1023/b:biry.0000009134.48246.7e.
Zorov, I. N., Dubasova, M. Y., Sinitsyn, A. P., Gusakov, A. V., Mytchenko, A. A., Baraznenok, V. A., Gutierrez, B., and Popova, N. N. (1997) Application of the bicinchininic method of assay for the reducing sugars to determine carboxymethylcellulase activity of cellulases using a microplate reader, Biochemistry (Moscow), 62, 704-709, doi: 10.1134/S0006297910010062.
Sinitsyn, A. P., Chernoglazov, V. M., and Gusakov, A. V. (1993) Methods of Study and Properties of Cellulolytic Enzymes. Series Biotechnologiya, 25 (Summation of Science and Technique, Academy of Sciences of USSR), VINITI, Moscow, [in Russian].
Decleire, M., De Cat, W., and Tang, V. H. (1996) in Chitin Enzymology (Muzzarelli, R. A. A., ed.) Atec Edizioni, Glottamare, Italy, pp. 165-169.
Peterson, G. L. (1977) Review of the Folin phenol protein quantitation method of Lowry, Rosebrough, Farr and Randall, Anal. Biochem., 100, 201-220, doi: 10.1016/0003-2697(79)90222-7.
Aleksenko, A. Y., Makarova, N. A., Nikolaev, I. V., and Clutterbuck, A. J. (1995) Integrative and replicative transformation of Penicillium canescens with a heterologous nitrate-reductase gene, Curr. Genet., 28, 474-478, doi: 10.1007/BF00310818.
Chulkin, A. M., Kislitsin, V. Y., Zorov, I. N., Sinitsyn, A. P., and Rozhkova, A. M. (2019) Determination of the copy number of the purpose genes of carbohydrases in recombinant strain of the fungus Penicillium verruculosum, Biotekhnologiya, 35, 51-57, doi: 10.21519/0234-2758-2019-35-5-51-57.
Acknowledgements
We used the equipment of the Industrial Biotechnologies Center for Collective Use of the Federal Research Center of Biotechnology, Russian Academy of Sciences.
Funding
This work was supported by the Ministry of Science and Higher Education (unique project identifier RFMEFI61620X0128).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
This article does not contain description of studies with the involvement of humans or animals performed by any of the authors. The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Sinitsyna, O., Rubtsova, E., Sinelnikov, I. et al. Creation of Chitinase Producer and Disruption of Micromycete Cell Wall with the Obtained Enzyme Preparation. Biochemistry Moscow 85, 717–724 (2020). https://doi.org/10.1134/S0006297920060097
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0006297920060097