
Abstract
Pseudoenzymes are homologs of active enzymes that have amino acid substitutions in the active center and, therefore, usually do not possess enzymatic activity. In this work, a recombinant proprotein (proSerPH122) of the homolog of serine peptidases of the S1 family from the yellow mealworm (Tenebrio molitor) was obtained in the yeast producer strain Komagataella kurtzmanii. The target His6-tagged protein was produced in a glycosylated form during secretion in yeast. The properties of both glycosylated and deglycosylated forms were studied. The proSerPH122 homolog with the replacement of active site Ser with Thr was pretreated with trypsin to study the enzymatic properties. The processed, mature homolog SerPH122 was shown to have low, but reliably detectable activity on the chromogenic substrate Suc-Ala-Ala-Pro-Phe-pNA, and this activity did not depend on the glycosylation level.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
According to the MEROPS classification [1], serine peptidases of the S1 family of chymotrypsin represent a large group of proteolytic enzymes. The ancestor of the family is chymotrypsin, the active site of which is a catalytic triad of His57-Asp102-Ser195 amino acid residues (the numbering in the introduction is given according to chymotrypsin A Bos taurus, NCBI ID XP_003587247) [2]). The substrate specificity of serine peptidases is due to the amino acid residues of the S1 binding subsite formed by residues 189, 216, and 226 [3, 4]. Chymotrypsins A/B (Ser-Gly-Gly), chymotrypsins C (Ser-Gly-Val), trypsins (Asp-Gly-Gly), serine elastases (Gly-Val-Ser/Thr) and collagenase (Gly-Gly-Asp) are distinguished depending on the composition of the S1 subsite.
Detailed bioinformatic sequence analysis, which has been widely used in recent years, showed that, in addition to active enzymes that are capable of catalyzing, there are also a number of pseudoenzymes, or pseudopeptidases, that are structural homologs of enzymes. However, they contain substitutions of amino acid residues of the active site, which leads in most cases to the complete absence of catalytic activity [5] or a significant decrease [6]. Currently, there are limited data on the functions of a large group of pseudoenzymes, and they relate mainly to their bioinformatic characteristics [7, 8]. Pseudoenzymes often function as components of complex systems. It is believed that they can take part in the regulation of their catalytically active homologs, acting as partner proteins, and participate in the regulation of metabolic and signaling pathways often associated with pathological conditions. Thus, along with active enzymes, they are potential targets for the development of therapeutic agents [9].
The growing importance and relevance of the study of pseudoenzymes is emphasized both by the wide range of processes in which they are involved and their multiplicity. As more pseudoenzymes are identified, the need to understand their structure, phylogenetic relationships, and functional role increases.
Recent studies indicate that insects are characterized by a large number of pseudoenzymes. Thus, in particular, for Anopheles gambiae and Drosophila melanogaster, as well as many other related species, a large number of both inactive and active trypsin-like serine peptidases were found, and the evolution of inactive peptidases and their new functions were presumably beneficial for insects [7]. An example of such an interaction is the CASPS18 protein found in the mosquito Aedes aegypti. It is a regulator of its active paralog caspase CASPS19 [10]. This assumption can be extended to other organisms, including humans. In this regard, insects of the Tenebrionidae family, in particular, Tenebrio molitor, a well-studied biochemical model due to its large size, may be of great interest for the study of the spectrum and properties of pseudopeptidases.
An extensive transcriptome was previously obtained in the laboratory from the gut of T. molitor larvae [11]. Analysis detected SerPH122 (Serine Peptidase Homolog), a protein homolog of serine peptidases from the S1 family of chymotrypsin. For a detailed characterization of the properties of the SerPH122 homolog, as well as the determination of natural molecules or substrates capable of specific interaction with SerPH122, it is necessary to obtain recombinant preparation of the studied protein. It was previously shown [12, 13] that the methylotrophic yeast Komagataella kurtzmanii is the basis of the expression system, an alternative K. phaffii (formerly Pichia pastoris), and can carry out efficient biosynthesis and secretion of recombinant enzymes.
The goals of this work are to construct a producing strain of the recombinant homolog proprotein proSerPH122 of T. molitor, to produce the proSerPH122 protein, to purify and deglycosylate the proenzyme, to process the proprotein, and to test for the presence of proteolytic activity in the mature protein.
METHOD
Construction of plasmids and strains. A DNA fragment encoding a recombinant homolog proSerPH122 was obtained via amplification with polymerase chain reaction (PCR). The amplification matrix was the plasmid pAL-TA-31-1 (Evrogen, Russia), which contains the cDNA of proSerPH122 homolog (GenBank MW882981). The PCR was performed with Phusion DNA polymerase (Thermo Fisher Scientific, United States) and pNcoI primers (5'-tataccatggaaaagagatctaagcctggagctcgtataatt-3') and pXhoI (5'- taactcgagttaatggtgatggtgatgatggggattgatgatggttctgat-3'). In addition to the restriction sites NcoI and XhoI (underlined in the composition of the corresponding primers), additional sequences were introduced during PCR into the amplified DNA fragment; this extended the N-terminus of the recombinant proSerPH122 protein by a serine residue and the C-terminus by a His6-tag peptide.
The amplified DNA fragment was cloned into the pPH727-AOX727 vector [14] at the NcoI/XhoI restriction sites as described earlier [13], which resulted in the plasmid pPH727-proSerPH122. The reliability of the structural sequence of the proSerPH122 gene in the pPH727-proSerPH122 plasmid was confirmed via sequencing. The regulation of the target-gene expression was under the control of the methanol-induced AOX1 gene promoter of the strain K. kurtzmanii VKPM Y-727 (pAOX727) [14], and the secretion of the target protein was guided by the leader area artHH as described earlier [13]. Top10 Escherichia coli cells (Invitrogen, United States) were used during the cloning and amplification of the pPH727-proSerPH122 plasmid. They were cultivated at a temperature of 37°C on LB medium supplemented with ampicillin at a concentration of 50 μg/mL.
The cells of the recipient yeast strain K. kurtzmanii Y727his4Δ were transformed according to the method described in [14]. Prior to transformation, the integrating DNA fragment was linearized via hydrolysis of the pPH727-proSerPH122 plasmid at the MluI restriction sites.
The resulting transformants were cultivated on YPGM medium of the following composition (%): yeast extract, 1.0 (0207; BioSpringer, France); soy peptone, 2.0 (P140; Amresco, United States); glycerin, 0.5 (Panreac, Spain); and methanol, 1.0 (technical grade A GOST 2222-95, Russia). Methanol was added to the medium to a final concentration of 1% during yeast inoculation and then at 24-h intervals. The production of samples of culture medium (CM) containing the proSerPH122 protein was carried out for 72 h at a temperature of 29°C with a rotary shaker with a rotation speed of 250 rpm.
Protein electrophoresis. Electrophoresis of proteins in the CM of pPH727-proSerPH122 transformants was carried out in 15% PAGE under reducing conditions (SDS-PAGE) as described earlier [13]. Prestained protein markers (Thermo Fisher Scientific, United States) were used. During the preparation of samples, proteins were concentrated prior to their application to the gel as described earlier [15]. Preparations concentrated from 100 μL of CM were applied to the lanes.
Purification of the recombinant T. molitor proSerPH122. Purification of the recombinant protein from the yeast CM was performed via metal chelate affinity chromatography as described earlier [13], with the difference that the transfer of the purified protein from buffer A into mQ water was carried out via ultrafiltration with an ultrafiltration cell (Amicon, United States) and membranes with a cut-off threshold of 3 kDa (Millipore, USA). Immediately after the desalting of the protein solution, the preparation purity was assessed via SDS-PAGE. The concentration was measured photometrically at a wavelength of 280 nm on a NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific, United States). The preparation was frozen in liquid nitrogen and lyophilized on a FreeZone device (Labconco, United States).
Deglycosylation of the recombinant T. molitor proSerPH122 with endoglycosidase-H. The lyophilized preparation obtained in the previous step was used for the deglycosylation reaction. Deglycosylation was performed with Endo H endoglycosidase (New England Biolabs, United States) according to the manufacturer’s recommendations. The protein was desalted and transferred into mQ water via ultrafiltration as described above. The concentration was measured photometrically at a wavelength of 280 nm on a NanoDrop 1000 spectrophotometer. The preparation was frozen in liquid nitrogen and lyophilized on a FreeZone device (Labconco, United States).
Mass-spectrometric analysis. Mass-spectrometric analysis of the purified protein was performed on an UltrafleXtreme MALDI time-of-flight mass spectrometer (Bruker Daltonics, Germany) equipped with a UV laser (Nd). The mass spectra were obtained in the linear, positive-ion mode with a reflectron. The accuracy of the measured average masses was 10 Da. The Vector NTI software package (Thermo Fisher Scientific, United States) was used to calculate the protein masses.
Bioinformatic analysis of the SerPH122 sequence of T. molitor. The complete amino acid sequence of the T. molitor SerPH122 protein, which was obtained as a result of translation of the coding sequence GenBank MW882981, was aligned with an ancestor of the S1 family—the bovine chymotrypsin NCBI ID XP_003587247 with the use of the Clustal Omega server for multiple sequence alignment [16]. Analysis and visualization of the active site and substrate-binding subsite were performed in GeneDoc [17]. The signal peptide was predicted on the SignalP-5.0 server [18].
Processing of the T. molitor proSerPH122 protein. To activate proSerPH122, lyophilized glycosylated and deglycosylated proprotein preparations were dissolved to a concentration of 15 μg/mL in 0.1 M acetate–phosphate–borate universal buffer at a pH of 7.9; a trypsin solution was added to a final concentration of 0.25 μg/mL, and the mixture was incubated for 60 min at 37°C in a thermostat (Binder, Germany).
Determination of the enzymatic activity of the recombinant homolog SerPH122 of T. molitor. To test the enzymatic activity of the original proprotein and processed homolog SerPH122, N-succinyl-Ala-Ala-Pro-Phe- (p-nitroanilide) - Suc-Ala-Ala-Pro-Phe-pNA (Bachem, Switzerland) was used as a chromogenic substrate. The reaction and the calculation of the enzymatic activity were carried out according to the previously published procedure [19]. The activity was measured at pH levels of 7.9 and 5.6 in 0.1 M acetate–phosphate–borate universal buffer [20].
RESULTS AND DISCUSSION
Preparation, purification, and structural study of proSerPH122. Biosynthesis of the recombinant proSerPH122 protein was performed with the K. kurtzmanii expression system, which has proven itself as an effective alternative to the widespread expression system based on K. phaffii (previously, P. pastoris) (Invitrogen, United States) [13, 14].
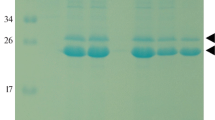
In order to obtain a producer strain of the recombinant proSerPH122 protein, cells of the recipient yeast strain K. kurtzmanii Y727his4Δ were transformed with an expression construct pPH727-proSerPH122. The transformants were grown in glass flasks under methanol induction at 20 and 30°C for 4 and 3 days, respectively. The obtained CM samples were analyzed via SDS-PAGE. The results are shown in Fig. 1. The protein products found in the CM samples are represented on the electrophoregram in the form of a separate band located in the region of 36 kDa and in the form of a diffuse sludge distributed in a range of molecular weights of 40 to 80 kDa. The staining of these products was significantly more intense in the sample with induction at 30°C; such products were absent in a sample of the recipient strain K. kurtzmanii Y727his4Δ (negative control) (Fig. 1). The discrepancy between the obtained molecular weights and the calculated one (26.6 kDa) is probably due to glycosylation.

Electrophoresis of proteins in the CM of transformants pPH727-proSerPH122. M, molecular mass markers; K–, negative control (Y727his4Δ); 20 and 30, samples of the target proSerPH122 protein obtained via culturing of the transformants pPH727-proSerPH122 at 20 and 30°C respectively. The arrow shows the location of the major band of the target glycosylated protein (P122). *The area of the distribution of hyperglycosylated forms of the target protein is indicated.
The recombinant protein sequence proSerPH122 contained two potential N-glycosylation sites: N48AT and N118ET (the residue numbering corresponds to the mature, activated enzyme). Protein treatment with endoglycosidase Endo H and subsequent analysis of the reaction products via SDS-PAGE (Fig. 2) and mass spectrometry (Fig. 3 and Table 1) showed that the target protein was actually secreted in the form of an N-glycosylated product. In particular, according to electrophoresis data, as a result of treatment of the secretion product with Endo H, the molecular weight of the major protein band decreased from 36 to 28 kDa (Fig. 2). At the same time, the intensity of the major band increased significantly, and the staining practically disappeared in the region of hyperglycosylation.

Electrophoretic control of deglycosylation of the purified preparation proSerPH122 with endoglycosidase Endo H. M, molecular mass markers; 1, protein preparation proSerPH122 after purification via metal chelate affinity chromatography; 2, purified protein preparation proSerPH122 after deglycosylation. Arrows show the location of the major bands of glycosylated (P122) and deglycosylated (P122dg) proteins.

Mass spectra of samples of purified proSerPH122 protein before deglycosylation (a) and after deglycosylation with Endo H endoglycosidase (b).
According to mass spectrometry data, the average molecular mass of the spectrum of protein products decreased from approximately 31 to 27 kDa (Fig. 3). Analysis of the derivatives of the target protein (Table 1) suggested that the major deglycosylated product is a protein containing two N-acetyglucosamine residues (MM = 27003, Table 1). The appearance of such a product was expected as a result of the complete deglycosylation of the protein at two sites containing carbohydrate components linked to asparagine residues by an N-glycosidic bond. The other variants were most likely derivatives of the major deglycosylated product, which contained one or two mannose residues: Man and Man2 (Table 1). They are the potential products of protein O-glycosylation and one more protein degradation product that lacks the terminal histidine (desHisCend).
Enzymatic properties of SerPH122. The complete sequence of protein homolog SerPH122 (GenBank MW882981) contains a signal peptide (Fig. 4, highlighted with blue bracket). According to modern concepts, the signal peptide is cleaved during secretion, and the propeptide (Fig. 4, highlighted with green bracket), which ends with an arginine residue (Fig. 4, marked with a red arrow), is usually further processed with trypsin [21, 22]. As compared to the ancestor of the S1 family—bovine chymotrypsin (NCBI ID XP_003587247), which has the His-Asp-Ser catalytic triad, the SerPH122 homolog contains His-Asp-Thr amino acid residues in the active center (Fig. 4, marked in red). Replacement of the Ser amino acid residue, which is essential for catalysis, with a synonymous Thr residue in the homolog made it possible to assume the presence of proteolytic activity in it.

Multiple sequence alignment of T. molitor SerPH122 and the ancestor of the S1 family—bovine chymotrypsin (chtr_Btaur, NCBI ID XP_003587247). The signal peptide of SerPH122 is highlited with a blue bracket, the propeptide with a green bracket, and the putative trypsin-processing site is marked with a red arrow. Amino acid residues of the active site are highlighted in red, and amino acid residues of the substrate binding subsite S1 are highlighted in yellow.
The proSerPH122 homolog obtained as a proprotein was pretreated with trypsin to search for possible enzymatic properties. It turned out that the mature, processed homolog SerPH122 was proteolytically active with the chromogenic peptide substrate Suc-Ala-Ala-Pro-Phe-pNA, which is widely used to test the activity of chymotrypsin. Both processed forms (glycosylated, SerPH122g and deglycosylated, SerPH122dg) were proteolytically active and hydrolyzed the Suc-Ala-Ala-Pro-Phe-pNA substrate (Fig. 5). Interestingly, both unprocessed forms of proSerPH122 (glycosylated and deglycosylated) also had proteolytic activity, but it was 1.5 times lower than that of processed mature forms (Fig. 5). This result may indicate that glycosylation does not affect the proteolytic activity of homolog. Compared to SerP38 peptidase of T. molitor from the S1 family (NCBI ID QRE01764) [13], the specific activity of SerPH122 with the same substrate Suc-Ala-Ala-Pro-Phe-pNA was 600 times lower. Proteolytic activity has recently been detected in a homolog of serine peptidase from an ectoparasitoid venom Scleroderma guani. The active site of the protein contained Ser-Asp-Ser amino acid residues instead of the typical catalytic triad of serine peptidases His-Asp-Ser. The homolog exhibited trypsin-like activity during hydrolysis of the substrate Bz-Arg-pNA (Nα-benzoyl-Arg- (p-nitroanilide)) [6].

Proteolytic activity of the homolog SerPH122 in glycosylated (SerPH122g) and deglycosylated (SerPH122dg) forms with chromogenic substrate Suc-Ala-Ala-Pro-Phe-pNA. 1, Original protein (proSerPH122); 2, processed mature protein after trypsin treatment (SerPH122).
The pH value optimal for the functioning of the SerPH122 homolog was determined in accordance with the physiological acidity levels in the gut of T. molitor larvae: a pH of 5.6 in the anterior part and a pH of 7.9 in the posterior part of the midgut [23]. Figure 6 shows that the SerPH122 activity is higher at a pH of 7.9.

Dependence of the specific activity (nmol/min mg) of the processed homolog SerPH122 on the pH value: 1, pH 5.6; 2, pH 7.9.
CONCLUSIONS
Thus, a method to obtain a recombinant protein proSerPH122—serine peptidase homolog of T. molitor was developed, the conditions for its purification via metal-chelate affinity chromatography were selected, and deglycosylation with Endo H endoglycosidase and proprotein processing to a mature form were carried out. It was shown that (1) the resulting homolog was processed under the action of trypsin. (2) It exhibited chymotrypsin-like activity with the substrate Suc-Ala-Pro-Phe-pNA. The activity of SerPH122 at a pH of 7.9 under slightly alkaline conditions characteristic of the contents of the posterior midgut of T. molitor larvae is significantly higher, than that at a pH of 5.6 under conditions of the contents of the anterior part of the midgut. (3) It was also shown that glycosylation did not affect the studied properties. This work contributes to the research on the poorly studied field of pseudopeptidase functioning and can serve as a basis for the clarification of the relationship between the structure and function of serine peptidases and their homologs.
REFERENCES
Rawlings, N.D., Barrett, A.J., Thomas, P.D., Huang, X., Bateman, A., and Finn, R.D., Nucleic Acids Res., 2018, vol. 46, no. D1, pp. D624–D632. https://doi.org/10.1093/nar/gkx1134
Brown, J.R. and Hartley, B.S., Biochem. J., 1966, vol. 101, no. 1, pp. 214–228. https://doi.org/10.1042/bj1010214
Perona, J.J., Hedstrom, L., Rutter, W.J., and Fletterick, R.J., Biochemistry, 1995, vol. 34, no. 5, pp. 1489–1499. https://doi.org/10.1021/bi00005a004
Perona, J.J. and Craik, C.S., Protein Sci., 1995, vol. 4, no. 3, pp. 337–360. https://doi.org/10.1002/pro.5560040301
Wex, T., Lipyansky, A., Bromme, N.C., Wex, H., Guan, X.Q., and Bromme, D., Biochemistry, 2001, vol. 40, no. 5, pp. 1350–1357. https://doi.org/10.1021/bi0022660
Wu, C.Y., Huang, J.M., Zhao, Y.J., Xu, Z.W., and Zhu, J.Y., Toxicon, 2020, vol. 183, pp. 29–35. https://doi.org/10.1016/j.toxicon.2020.05.011
Pils, B. and Schultz, J., J. Mol. Biol., 2004, vol. 340, no. 3, pp. 399–404. https://doi.org/10.1016/j.jmb.2004.04.063
Yang, L., Lin, Z., Fang, Q., Wang, J., Yan, Z., Zou, Z., Song, Q., and Ye, G., Dev. Comp. Immunol., 2017, vol. 77, pp. 56–68. https://doi.org/10.1016/j.dci.2017.07.014
Reynolds, S.L. and Fischer, K., Biochem. J., 2015, vol. 468, no. 1, pp. 17–24. https://doi.org/10.1042/BJ20141506
Bryant, B., Ungerer, M.C., Liu, Q., Waterhouse, R.M., and Clem, R.J., Insect Biochem. Mol. Biol., 2010, vol. 40, no. 7, pp. 516–523. https://doi.org/10.1016/j.ibmb.2010.04.011
Martynov, A.G., Elpidina, E.N., Perkin, L., and Oppert, B., BMC Genomics, 2015, vol. 16, no. 75. https://doi.org/10.1186/s12864-015-1306-x
Naumov, G.I., Naumova, E.S., Tyurin, O.V., and Kozlov, D.G., Antonie van Leeuwenhoek, 2013, vol. 104, no. 3, pp. 339–347. https://doi.org/10.1007/s10482-013-9956-7
Gorbunov, A.A., Akent’ev, F.I., Gubaidullin, I.I., Zhiganov, N.I., Tereshchenkova, V.F., Elpidina, E.N., and Kozlov, D.G., Biotekhnologiya, 2020, vol. 36, no. 6, pp. 78–86. https://doi.org/10.21519/0234-2758-2020-36-6-78-86
Matveeva, A.Yu., Gubaidullin, I.I., Fedorov, A.S., and Kozlov, D.G., Biotekhnologiya, 2019, vol. 35, no. 5, pp. 3–11. https://doi.org/10.21519/0234-2758-2019-35-5-3-11
Kozlov, D.G. and Yagudin, T.A., Biotechnol. Lett., 2008, vol. 30, no. 9, pp. 1661–1663. https://doi.org/10.1007/s10529-008-9739-9
Sievers, F., Wilm, A., Dineen, D., Gibson, T.J., Karplus, K., Li, W., Lopez, R., McWilliam, H., Remmert, M., Soding, J., Thompson, J.D., and Higgins, D.G., Mol. Syst. Biol., 2011, vol. 7, p. 539. https://doi.org/10.1038/msb.2011.75
Nicholas, K.B., Nicholas, H.B.Jr., and Deerfield, D.W., Embnew. News, 1997, vol. 4, p. 14. http://www.psc.edu/biomed/genedoc
Armenteros, J.J., Tsirigos, K.D., Sonderby, C.K., Petersen, T.N., Winther, O., Brunak, S., von Heijne, G., and Nielsen, H., Nat. Biotechnol., 2019, vol. 37, no. 4, pp. 420–423. https://doi.org/10.1038/s41587-019-0036-z
Tereshchenkova, V.F., Klyachko, E.V., Benevolenskii, S.V., Belozerskii, M.A., Dunaevskii, Ya.E., Filippova, I.Yu., and Elpidina, E.N., Appl. Biochem. Microbiol., 2019, vol. 55, no. 3, pp. 218–223.
Frugoni, J.A.C., Gazz. Chem. Ital., 1957, vol. 87, pp. 403–407.
Parry, M.A., Fernandez-Catalan, C., Bergner, A., Huber, R., Hopfner, K.P., Schlott, B., Guhrs, K.H., and Bode, W., Nat. Struct. Biol., 1998, vol. 5, no. 10, pp. 917–923. https://doi.org/10.1038/2359
Hedstrom, L., Chem. Rev., 2002, vol. 102, no. 12, pp. 4501–4524. https://doi.org/10.1021/cr000033x
Vinokurov, K.S., Elpidina, E.N., Oppert, B., Prabhakar, S., Zhuzhikov, D.P., Dunaevsky, Y.E., and Belozersky, M.A., Comp. Biochem. Physiol. B. Biochem. Mol. Biol., 2006, vol. 145, no. 2, pp. 126–137. https://doi.org/10.1016/j.cbpb.2006.05.005
ACKNOWLEDGMENTS
The authors are grateful to M.V. Serebryakova for the mass-spectrometric analysis of the preparation of the recombinant prohomolog proSerPH122.
Funding
The study was carried out with the financial support of a grant from the Russian Science Foundation (project no. 19-74-00080).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflict of interest. This article does not contain any studies involving animals or human participants performed by any of the authors.
Rights and permissions
About this article

Cite this article
Tereshchenkova, V.F., Zhiganov, N.I., Akentyev, P.I. et al. Preparation and Properties of the Recombinant Tenebrio molitor SerPH122—Proteolytically Active Homolog of Serine Peptidase. Appl Biochem Microbiol 57, 579–585 (2021). https://doi.org/10.1134/S0003683821050161
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0003683821050161