
Abstract
A common haplotype of the flavin-containing monooxygenase gene FMO3 is associated with aberrant mRNA splicing, a twofold reduction in in vivo nicotine N-oxidation and reduced nicotine dependence. Tobacco remains the largest cause of preventable mortality worldwide. CYP2A6, the primary hepatic nicotine metabolism gene, is robustly associated with cigarette consumption but other enzymes contribute to nicotine metabolism. We determined the effects of common variants in FMO3 on plasma levels of nicotine-N-oxide in 170 European Americans administered deuterated nicotine. The polymorphism rs2266780 (E308G) was associated with N-oxidation of both orally administered and ad libitum smoked nicotine (P⩽3.3 × 10−5 controlling for CYP2A6 genotype). In vitro, the FMO3 G308 variant was not associated with reduced activity, but rs2266780 was strongly associated with aberrant FMO3 mRNA splicing in both liver and brain (P⩽6.5 × 10−9). Surprisingly, in treatment-seeking European American smokers (n=1558) this allele was associated with reduced nicotine dependence, specifically with a longer time to first cigarette (P=9.0 × 10−4), but not with reduced cigarette consumption. As N-oxidation accounts for only a small percentage of hepatic nicotine metabolism we hypothesized that FMO3 genotype affects nicotine metabolism in the brain (unlike CYP2A6, FMO3 is expressed in human brain) or that nicotine-N-oxide itself has pharmacological activity. We demonstrate for the first time nicotine N-oxidation in human brain, mediated by FMO3 and FMO1, and show that nicotine-N-oxide modulates human α4β2 nicotinic receptor activity in vitro. These results indicate possible mechanisms for associations between FMO3 genotype and smoking behaviors, and suggest nicotine N-oxidation as a novel target to enhance smoking cessation.
Similar content being viewed by others


Introduction
Despite reduced prevalence, tobacco use remains the largest cause of preventable mortality both in the US and worldwide.1, 2 And although more than half of American smokers attempt to quit every year, only 6% succeed.3 Currently, three medications—varenicline, nicotine replacement therapy and bupropion—are prescribed to aid smoking cessation with modest success,4 but the heavy burden of tobacco-related disease warrants pursuing further pharmacotherapy targets.
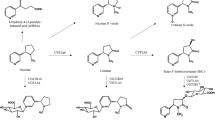
Nicotine is extensively metabolized. In most smokers, the large majority of nicotine (70–80%) is metabolized to cotinine, the product of cytochrome P450 2A6 (CYP2A6)-catalyzed oxidation,5, 6 but two further pathways also contribute to human hepatic nicotine metabolism: N-glucuronidation catalyzed by the UDP-glucuronsyltransferase UGT2B10, and N-oxidation by the flavin monooxygenase FMO3. Variation in in vivo nicotine metabolism is strongly genetically determined and significantly predicts smoking-related behaviors including cessation.7, 8, 9 Specifically, CYP2A6 is among the few loci consistently associated with cigarette consumption and related pulmonary disease in genome-wide association studies.10, 11, 12 But, importantly, CYP2A6’s influence on smoking behavior is primarily related to cigarette consumption among dependent smokers13 unlike functional variation in the CHRNA5 nicotinic acetylcholine receptor subunit gene, which is associated with consumption and dichotomous nicotine dependence.14, 15, 16
On average <7% of total nicotine equivalents (the sum of nicotine and six metabolites) are typically excreted as nicotine-N-oxide.17, 18, 19 However, a notable feature of the FMOs is their expression and activity in the human brain,20, 21 where CYP2A6 is not detected.22 FMO3 is also highly polymorphic, including several common amino acid altering variants with reported substrate specific consequences.23 The FMO3 polymorphisms that determine heritable differences in nicotine N-oxidation are not established; here we describe experiments that evaluated nicotine N-oxidation in vivo to identify common FMO3 alleles associated with altered nicotine metabolism. We also pursued the mechanisms by which these variants alter enzymatic activity, and the mechanisms by which variation in nicotine N-oxidation may influence nicotine dependence.
Materials and methods
Human subjects
The study complies with the Code of Ethics of the World Medical Association. Written informed consent from participants and approval from the appropriate institutional review boards was obtained. Metabolism experiment participants were recruited from the Collaborative Genetic Study of Nicotine Dependence (COGEND).16, 24 Subjects in the clinical phenotypes experiments were European Americans in the University of Wisconsin Transdisciplinary Tobacco Use Research Center (UW-TTURC) cessation trials.25, 26, 27
Clinical nicotine disposition
Nicotine was administered and plasma collected 240 min later, as described.24 We previously demonstrated the validity of single time point measures of nicotine metabolites as correlates of genetic variation.24, 28 Nicotine-N-oxide and d2-nicotine-N-oxide were analyzed by liquid chromatography tandem mass spectrometry as previously.19 Selective reaction monitoring was carried out for nicotine-N-oxide (m/z 179→130 and m/z 179→117), d2-nicotine N-oxide (m/z 181→131 and m/z 181→117) and the internal standard d3-nicotine-N-oxide (m/z 182→130 and m/z 182→117). Percent of total nicotine converted to nicotine-N-oxide was calculated as nicotine-N-oxide/(nicotine+nicotine-N-oxide+cotinine+trans-3′-hydroxycotinine+nicotine-glucuronide). Nicotine, cotinine, trans-3′-hydroxycotinine and nicotine-glucuronide were previously reported.24, 28
Genotyping and haplotype determination
Metabolism experiment subjects were genotyped and a nicotine metabolism metric based on CYP2A6 genotype determined as previously reported.16, 24, 28, 29 FMO3 haplotypes were determined using PHASE version 2.1.1.30 In all other subjects FMO3 SNPs were genotyped by the Center for Inherited Disease Research at Johns Hopkins University using the Illumina Omni2.5 microarray (www.illumina.com), with data cleaning led by the GENEVA Coordinating Center at the University of Washington.29 All SNPs analyzed conformed to Hardy-Weinberg equilibrium.
Gene expression and splicing in human liver and brain
Genomic DNAs and cDNAs were previously prepared from de-identified normal liver biopsy samples and postmortem cerebellum samples of European descent.31, 32 cDNAs from cultured primary human brain astrocytes and endothelial cells were prepared using the Direct-zol RNA MiniPrep kit (Zymo Research, Irvine, CA, USA) and EasyScript cDNA synthesis kit (Lamda Biotech, Sovereign CT, USA). Human neuron cDNA was purchased from Sciencell Research Laboratories (Carlsbad, CA, USA). Gene expression and splicing were assessed by real-time PCR using an ABI-7900HT Fast real-time PCR system (Supplementary Methods).
Recombinant FMO3 expression
Sequence-verified pVL1393 vectors containing full length FMO3 cDNA with variants were purchased from Life Technologies (Carlsbad, CA, USA). Protein was expressed as previously33 at a multiplicity of infection of 8, with 10 μg ml−1 FAD (Sigma, St. Louis, MO, USA) added 12 h after infection. Supersomes expressing reference FMO3 and FMO1 were purchased from Corning (Corning, NY, USA). FAD content was quantified fluorimetrically34 and used as a measure of FMO holoenzyme content to calculate kcat values. All protein concentrations were determined by Bradford assay.
Cell culture
Cryopreserved primary astrocytes and endothelial cells isolated from human brain (cerebral cortex) were acquired from Sciencell Research Laboratories. Astrocytes (1 × 106 cells, passage 2) were initially cultured on poly-l-lysine coated (2 μg cm−2) T-75 flasks in complete astrocyte medium (Sciencell Research Laboratories) and grown until confluency. Endothelial cells (5 × 105 cells, passage 1) were cultured on a bovine fibronectin coated (2 μg cm−2) T-25 flask in complete endothelial cell medium (Sciencell Research Laboratories) and cultured until confluency.
Liver, brain and cultured astrocyte in vitro nicotine metabolism
Cerebral cortex from a 58 year-old Caucasian male flash-frozen 5.5 h postmortem was obtained from the National Disease Research Interchange (NDRI), Philadelphia PA, USA. Microsomes were prepared (Supplementary Methods) and initial experiments determined an appropriate incubation time and microsomal protein concentrations that yielded metabolic activity within a linear range. Metabolites were analyzed by liquid chromatography tandem mass spectrometry (Supplementary Methods) and quantified using the ratio of metabolite to internal standard, subtracting background from control incubations (without NADPH), and using calibration curves. Further control incubations included with boiled protein or without protein (Supplementary Figure 1).
Nicotine-N-oxide agonism and modulation of neuronal nicotinic acetylcholine receptors
Human α4β2 neuronal nicotinic acetylcholine receptors were expressed in Xenopus oocytes following a protocol approved by the Washington University in St. Louis Animal Care and Use Committee (Supplementary Methods). The cRNAs were produced using mMessage mMachine (Ambion, Austin, TX, USA). Oocytes were injected with a total of 18–20 ng of cRNA in a final volume of 40–60 nl, and incubated in ND96 (96 mm NaCl, 2 mm KCl, 1.8 mm CaCl2, 1 mm MgCl2, 2.5 mm Na pyruvate, 5 mm HEPES; pH 7.4) at 16 °C. Oocytes were used within 2–3 days after injection. To bias receptor stoichiometries, a 9-fold excess of the α4 or β2 subunit cRNA was injected, with stoichiometry verified by sensitivity to acetylcholine (ACh).35 Electrophysiological experiments were conducted using the two-electrode voltage clamp technique (Supplementary Methods).
Statistical analysis
Statistical analyses of pharmacogenetic data were performed using ‘R’ (Vienna, Austria). Metabolism phenotypes analyzed are fractions of deuterated (d2)-nicotine or non-deuterated (d0)-nicotine equivalents converted to d2 nicotine-N-oxide or d0 nicotine-N-oxide respectively, that is, nicotine-N-oxide/(nicotine+nicotine-N-oxide+cotinine+trans-3′-hydroxycotinine+nicotine-glucuronide). Valid measurements below the limits of detection were assumed to be zero. For regression analyses, genetic variables were treated as previously described.24, 28 Current smoking was defined by a mean d0-cot >2 ng ml−1. Measures of statistical significance are not altered to correct for multiple testing. Genetic associations with smoking phenotypes were tested using a linear model with genotype coded additively and sex, age, and study as covariates. In tests of the Wisconsin Index of Smoking Dependence Motives (WISDM) PDM scale the WISDM Secondary Dependence Motives subscale was used as a covariate. Michaelis–Menten parameters for recombinant enzyme experiments were determined by nonlinear regression using SigmaPlot (Systat Software, San Jose, CA, USA).
Results
FMO3 haplotypes associated with nicotine N-oxidation
FMO3 haplotypes defined by the five FMO3 coding SNPs common in Europeans (minor allele frequency ⩾2%) were determined in 170 subjects with measurements for deuterated nicotine metabolites (Supplementary Table 2). We did not identify any subjects with the reportedly common FMO3 variant N61K, but this variant has never been found in studies of trimethylaminuria, nor by an exome sequencing project including >4000 European American genomes ((ESP) NGESP, NHLBI Exome Sequencing Project (ESP) Exome Variant Server). Among our subjects, 59 were current smokers with non-deuterated nicotine-N-oxide measurements. The mean plasma concentration of non-deuterated nicotine-N-oxide in these smokers was 29.4±22.3 pmol ml−1 (5.2±4.0 ng ml−1). The percent of oral deuterated nicotine converted to nicotine-N-oxide after 4 h was highly correlated with the percent of ad libitum smoked nicotine converted to N-oxide in these subjects (R2=0.82). The dominant indirect effect of C-oxidation upon the percent of nicotine remaining available for conversion to N-oxide is demonstrated by the strong association between CYP2A6 genotype and measures of N-oxidation. Overall CYP2A6 genotype variables account for 34% (R2=0.340, n=170) of the variance in the percent of oral nicotine converted to N-oxide and 37% (R2=0.367, n=59) of the variance in the percent of ad libitum smoked nicotine converted to N-oxide.
Using the most common FMO3 haplotype (1) as the reference, in a multivariate model including CYP2A6 genotype variables, the third most common haplotype (haplotype 3, K158; G308) defined by the minor allele of rs2266780, is significantly associated with reduced N-oxidation of both oral nicotine and ad libitum smoked nicotine (P=3.3 × 10−5 and P=5.6 × 10−6 respectively, Table 1). rs2266780 is also significantly associated with reduced nicotine N-oxidation when tested individually (P=0.01 and P=0.002, respectively). In the same multivariate models, FMO3 haplotype 5 is also significantly associated with reduced N-oxidation (Table 1), although these associations do not reach significance in single-variable analyses. After accounting for CYP2A6 genotype, rs2266780 alone explained 8.5% (R2=0.085) and 21.6% (R2=0.216) of the remaining variance in the percent of nicotine-N-oxide for oral and smoked nicotine, respectively. Common FMO3 haplotypes 2 (K158) and 4 (M257) were not significantly associated with altered nicotine-N-oxidation relative to the reference haplotype (Table 1). Neither current smoking nor sex was significantly associated with altered nicotine N-oxidation, consistent with previous reports.36 UGT2B10 haplotypes28 were also not associated.
Although FMO3 genotype demonstrates a significant influence on in vivo nicotine N-oxidation, this pathway represents a small fraction of overall hepatic nicotine metabolism (4–10% of excreted nicotine equivalents5, 19). Although we find CYP2A6 genotype explains the majority (58%) of the variance in total oral nicotine metabolism after four hours (metabolism of nicotine to all measured metabolites, called nicotine equivalents), FMO3 haplotypes explain <1% of that variance. However, relative to the small amount of nicotine typically converted to N-oxide, the impact of rs2266780 genotype (defining haplotype 3) is great. On average 1.6±1.3% of deuterated and 1.3±0.9% of non-deuterated nicotine equivalents were present as nicotine-N-oxide in rs2266780 major allele homozygotes compared with only 0.7±0.4% and 0.4±0.1%, respectively in minor allele homozygotes. Thus, less than half as much nicotine N-oxidation occurs in homozygotes for the reduced function FMO3 haplotype than in individuals homozygous for the reference haplotype (P=5.4 × 10−3 and 3.9 × 10−6 respectively, Figure 1).

rs2266780 genotype associated with (a) deuterated (d2)-nicotine N-oxidation (d2-nicotine-N-oxide/(d2-nicotine+d2-nicotine-N-oxide+d2-cotinine+d2-trans-3'-hydroxycotinine+d2-nicotine-glucuronide) 4 h after oral administration, and (b) ad libitum smoked nicotine N-oxidation (nicotine-N-oxide/(nicotine+nicotine-N-oxide+cotinine+trans-3'-hydroxycotinine+nicotine-glucuronide). The boxplots provide summaries of the data distributions for each group of (n) subjects. A box represents the interquartile range, which includes 50% of values. The line across the box indicates the median. The whisker lines extend to the highest and lowest values that are within 1.5 × the interquartile range. Further outliers are marked with circles.
Nicotine N-oxidation by variant recombinant FMO3 enzymes
One potential explanation for FMO3 variant effects on clinical nicotine N-oxidation is altered FMO3 catalytic activity. Four versions of the FMO3 enzyme corresponding to the reference allele, K158, G308 and K158;G308 haplotypes were expressed to assess their relative nicotine N-oxidation activity in vitro. However, we did not observe a reduction in the nicotine-N-oxidation activity of the variant FMO3s relative to the reference allele in vitro (Supplementary Table 3). The product of FMO3-mediated nicotine oxidation is overwhelmingly the trans rather than the cis stereoisomer. This was true among our four FMO3 haplotypes and commercially available microsomes containing the reference allele (≈79:1), similar to previous reports.37 Incubations with human liver microsomes also primarily produced the trans isomer (ratio≈53:1). Interestingly, we also found that recombinant FMO1-containing microsomes overwhelmingly produce the cis isomer (≈30:1). This suggests nicotine as a convenient probe to distinguish between FMO3 and FMO1 activities.
Haplotypes associated with aberrant FMO3 mRNA splicing in liver and brain
Another potential mechanism of genetically determined variation in FMO3 activity is altered FMO3 mRNA splicing. Aberrant splicing of FMO3 mRNA was previously reported in multiple adult and fetal human tissues; these events result in the omissions of the third and seventh exons, leading to frame-shifts and premature stop codons in the aberrantly spliced mRNAs.38 Custom splicing assays were used to determine the relative amounts of exon skipping of FMO3 exons 3 or 7 in cDNAs from genotyped European American liver biopsy samples. Only rs2266780 (E308G) was independently associated with relative levels of FMO3 cDNAs lacking exons 3 or 7 (P<10−16; Supplementary Figure 2). These differences equate to greater than 14-fold and 6-fold increases in transcripts lacking exons 3 and 7, respectively. We also repeated the experiments in cDNAs from an unrelated set of human cerebellum samples and found a similarly unequivocal difference (P=6.5 × 10−9). PCR amplification using the primers from the original study38 confirmed that the products previously reported are associated with the rs2266780 minor allele (Figure 2). rs2266780 is in high linkage disequilibrium (R2⩾0.95 in Europeans) with at least eighteen reported intronic SNPs in the FMO3 gene, but none of these are strongly predicted to affect splicing in silico.

Aberrant FMO3 splicing in human liver and cerebellum tissues by rs2266780 genotype. (a) Aberrant exon 2–4 splicing (skipping exon 3) versus correct exon 3–4 splicing in liver cDNAs, and (b) aberrant exon 6–8 splicing (skipping exon 7) versus correct exon 7–8 splicing in liver or (c) cerebellum cDNAs. The difference in PCR cycle times (ΔCt) for cDNAs for (n) samples divided by rs2266780 genotype. Relative expression, ΔCt, determined by subtracting the Ct value of the reaction using exon skipping primers from the Ct value of the reaction using correct splicing primers (see methods). The boxplot provides a summary of the data distribution. The box represents the interquartile range, which includes 50% of values. The line across the box indicates the median. The whisker lines extend to the highest and lowest values that are within 1.5 × the interquartile range. Further outliers are marked with circles. (d) The products of PCR primers38 flanking regions of FMO3 including exons 3 or 7 amplified from liver cDNAs heterozygous for s2266780 (AG), homozygous for the s2266780 major allele (AA) or from pooled cDNAs from eight liver samples of various genotypes.
We did not detect significant differences in overall FMO3 transcript expression either in liver or cerebellum samples using TaqMan gene expression assays (data not shown), possibly due to the difficulty of detecting such stable differences in liver biopsy and brain autopsy samples and the demonstrated large effects of diet on FMO3 mRNA expression.39 However FMO3 was detected in all of the liver and cerebellum samples tested with expression levels approximately six hundred-fold higher in liver than in cerebellum, similar to prior reports.20
FMO3 genotype associated with nicotine dependence
Despite the modest role of N-oxidation to hepatic nicotine metabolism, prior studies have indicated a potential association between FMO genotype and smoking behaviors.40, 41, 42 Therefore, we tested for an association between rs2266780 (E308G) and cigarette consumption and nicotine dependence in a well-ascertained sample of European Americans seeking smoking cessation treatment. Interestingly, we found a significant association (P=0.0036, n=1558) between rs2266780 and nicotine dependence as measured by the Fagerström Test of Nicotine Dependence (FTND). rs2266780 was not significantly associated with cigarette consumption, whether measured as cigarettes per day or using a biomarker of cigarette smoke exposure, exhaled carbon monoxide (P>0.2). Other FMO3 variants were not associated with nicotine dependence or cigarette consumption.
The FTND contains six questionnaire items that measure physical dependence and tolerance on an overall 0–10 scale. Analyses of these items showed that the association between rs2266780 and FTND is driven by item 1; rs2266780 was not significantly associated with other items. Item 1, the ‘time to first cigarette’, elicits information on the time interval a smoker reports between waking in the morning and lighting the first cigarette; the minor allele of rs2266780 is associated with a robust increase in this measure (−0.13 per allele on a 0–3 scale, P=9.0 × 10−4). Among FTND items, time to first cigarette has been demonstrated to be the most informative metric of heritable factors that influence nicotine dependence,43 and distinct from overall cigarette consumption.44, 45 31% of rs2266780 major allele homozygotes reported typically smoking within the first 5 min after awakening, while only 13% of rs2266780 minor allele (haplotype 3) homozygotes reported the same urgency to smoke (Figure 3).

Time to first, the interval in minutes that smokers report typically waiting before lighting their first cigarette after awakening in the morning, for (n) samples divided by rs2266780 genotype.
We subsequently tested and found a significant association (P=0.006) between rs2266780 and The Wisconsin Index of Smoking Dependence Motives (WISDM) Primary Dependence Motives (PDM) scale. The PDM measures core dimensions of dependence characterized by strong craving, perceived loss of control over smoking, and an inability to tolerate the passage of time without smoking; it is especially predictive of smoking cessation.46, 47
To determine whether FMO3 genotype accounts for variance in FTND and PDM independent of other predictive genetic variables,29 we ran multivariable competitive regression models with rs2266780, rs16969968, the key CHRNA5 variant, and a previously described nicotine metabolism metric based on CYP2A6 genotype.13, 29 These models reveal that the FMO3 variant independently predicts variance in the FTND total score (P=0.009), time to first cigarette (P=0.002), and WISDM PDM scale (P=0.027). CHRNA5 and CYP2A6 did not significantly independently predict variance in either the FTND total score or time to first cigarette, but all three genes made independent contributions to the prediction of the WISDM PDM scale (P-values=0.027–0.045).
Nicotine N-oxidation in human brain and astrocytes
The association between nicotine dependence and the reduced function allele led us to investigate novel potential mechanisms for FMO3’s influence. Although FMO3 and FMO1 mRNAs are expressed in the human brain,20 evidence for FMO activity in the human brain is limited.21, 48 To determine whether nicotine N-oxidation occurs in the brain we prepared microsomes from human autopsy cerebral cortex tissue, as well as from primary cultured human astrocytes, and from whole mouse brains. Cultured astrocytes were chosen because of the reported preferential expression of FMO1 in mouse astrocytes relative to other brain cell types (FMO3 is not reported expressed at detectable levels in any mouse brain cell type).49 We attempted to detect FMO3 mRNA in human primary cultured astrocytes, as well as in human cultured primary neurons and brain endothelial cells, but found the level of expression was below the detection limits of our assay.
Nicotine N-oxidation activity was detected in human brain tissue (Supplementary Figure 1). Microsomes were incubated with nicotine or benzydamine, a common FMO substrate and N-oxide metabolites were detected for both substrates (benzydamine data not shown). Primary human astrocyte and cerebral cortex microsomes both produce trans and cis nicotine-N-oxide at ratios of approximately 4:1 and 1:1 respectively (at 5 mm nicotine, Table 2), indicating the activities of both FMO3 and FMO1. This is in contrast to mouse brain microsomes, which overwhelmingly produce cis nicotine-N-oxide. Production of cis nicotine-N-oxide by human brain microsomes followed classic Michaelis–Menten kinetics, but the production of the trans isomer was non-saturable (Supplementary Figure 2) as previously reported for recombinant FMO3.40 Microsomes prepared from different passages of cultured primary astrocytes (passages 3–7) did not show a reduction in FMO activity.
Activation and modulation of α4β2 nicotinic acetylcholine receptors by trans-nicotine-N-oxide
An alternative hypothesis to explain the association between FMO3 genotype and time to first cigarette would be that nicotine-N-oxide, whether generated in the liver or brain, has pharmacological activity relevant to tobacco use, such as a direct effect on neuronal nicotinic acetylcholine receptors. Therefore, we measured direct activation and modulation of currents elicited by ACh in human α4β2 nicotinic receptors expressed in Xenopus oocytes. In receptors expressed in the 3:2 (α4:β2) stoichiometry, exposure to 200 μm trans-nicotine-N-oxide resulted in small (peak amplitude: 87±10 nA, mean±s.e.m., n=8 cells) inward currents. Each cell was also tested with a saturating concentration (1 mm) of ACh. The relative peak response to trans-nicotine-N-oxide was 1.7±0.3% of the response to ACh. Sample current traces are shown in Figure 4a.

Direct activation and modulation of human α4β2 receptors by trans-nicotine-N-oxide. (a) Sample traces from Xenopus oocytes expressing human α4β2 nicotinic receptors in the stoichiometry of 3:2. Direct activation by 200 μm nicotine-N-oxide was minimal compared to the response to 1 mm acetylcholine (ACh) from the same cell. Co-application of 200 μm nicotine-N-oxide reduced the peak response to 1 μm ACh. (b) Sample traces from Xenopus oocytes expressing human α4β2 nicotinic receptors in the stoichiometry of 2:3. Direct activating and modulating effects of nicotine-N-oxide are qualitatively similar to those observed in a. (c) Concentration–response relationships for nicotine-N-oxide-mediated modulation of currents elicited by 1 μm ACh in oocytes expressing α4β2 receptors in 3:2 or 2:3 stoichiometry. The data show the response ratio (peak amplitude in the presence vs absence of nicotine-N-oxide) in %. All data points were collected from the same set of cells (5 for 3:2 and 4 for 2:3). Statistical analysis compares the response ratio to 100% (that is, no effect) using a paired t-test. NS, not significant, **P<0.01, ***P<0.001.
Co-application of trans-nicotine-N-oxide modulated current responses to ACh. The (α4)3(β2)2 receptors activated by 1 μm ACh (approximately EC3) in the presence of 200 μm trans-nicotine-N-oxide showed a mean peak response of 44±5% (n=5) of control (Figure 4a). The concentration–response data for inhibition of currents elicited by 1 μm ACh are shown in Figure 4c. Currents from receptors activated by 1 mm ACh (a saturating concentration) were not modulated by 200 μm trans-nicotine-N-oxide (98±2% of control, four cells, P>0.4, paired t-test). We infer that the inhibitory effect is competitive by nature.
The (α4)2(β2)3 receptors exhibit higher sensitivity to ACh, and changes in modulation by Zn2+ and Ca2+ permeability compared with (α4)3(β2)2 receptors.35 To increase assembly of (α4)2(β2)3 receptors, we injected oocytes with an excess of β2 subunit cRNA (see Methods). The resulting receptors showed larger direct activation (5.7±1.1% of the response to saturating ACh; n=5) compared with (α4)3(β2)2 receptors (P<0.01; two-sample t-test) in the presence of 200 μm trans-nicotine-N-oxide. Co-application of trans-nicotine-N-oxide resulted in depressed peak response. Sample current traces are shown in Figure 4b. The inhibitory effect of the drug was similar for (α4)3(β2)2 and (α4)2(β2)3 receptors (Figure 4c).
Discussion
In this first comprehensive analysis of FMO3 genotype and in vivo nicotine N-oxidation, we provide strong evidence that a common haplotype (K158; G308) is associated with significantly reduced activity. It was our hypothesis that by precisely determining the FMO3 polymorphisms that affect nicotine N-oxidation we might refine a genetic model of nicotine metabolism13, 24, 29 and see a modest improvement in its prediction of cigarette consumption over a model based on CYP2A6 genotype alone. However, to our surprise, we discovered a robust association between the reduced-function FMO3 haplotype and reduced nicotine dependence as defined by the FTND. In particular, we find a striking relationship with longer time to first cigarette, demonstrated as the most informative measure of heritable factors from the FTND.43
Evidence that differences in cytochrome P450 enzyme activity in the brain influences substance abuse phenotypes is scant, but the activity of FMOs in the brain is well established21 and we have now demonstrated nicotine N-oxidation in human brain microsomes. A prominent role for FMO3-mediated nicotine metabolism in the brain could explain the association with nicotine dependence and time to first cigarette. In the absence of CYP2A6 activity in the brain,22 differences in N-oxidation may determine a large portion of the variance in local nicotine clearance, as the reduced activity FMO3 allele causes a twofold decrease in nicotine N-oxidation in liver.
A relatively straightforward explanation for FMO3’s influence on nicotine dependence is via heritable differences in nicotine clearance, perhaps locally in the brain; but it is also possible that differences in levels of the metabolite, nicotine-N-oxide, are responsible. While the modest response elicited by trans-nicotine-N-oxide in vitro probably rules out direct activation of α4β2 receptors by nicotine-N-oxide as an important mechanism, the significant inhibition of receptor activation by ACh in the presence of the metabolite warrants further study. In particular, we are interested to understand how nicotine-N-oxide may modulate the activity of receptors containing the α5 subunit, demonstrated to influence nicotine-related phenotypes.
Selective inhibition of CYP2A6 has been investigated as a potential strategy to improve the bioavailability of nicotine, but selective inhibition of FMO activity might lengthen nicotine perdurance specifically in the brain, the site of nicotine’s pharmacological activity associated with dependence, while avoiding increased peripheral effects and risk for cardiovascular events. Complete FMO3 deficiency in humans is not associated with symptoms other than trimethylaminuria and therefore FMO inhibition may allow a wide therapeutic window. Furthermore, the results reported here indicate that differences in FMO3 activity influence aspects of nicotine dependence beyond levels of consumption, suggesting that targeting FMOs may have other advantages over more broadly targeting hepatic nicotine metabolism as a smoking cessation pharmacotherapy.
References
Mokdad AH, Marks JS, Stroup DF, Gerberding JL . Actual causes of death in the United States, 2000. JAMA 2004; 291: 1238–1245.
WHO. World Health Statistics Report—2008 2008.
Prevention CfDCa. Quitting Smoking Among Adults—United States, 2001–2010 2010.
Cahill K, Stevens S, Perera R, Lancaster T . Pharmacological interventions for smoking cessation: an overview and network meta-analysis. Cochrane Database Syst Rev 2013; 5: CD009329.
Hukkanen J, Jacob P 3rd, Benowitz NL . Metabolism and disposition kinetics of nicotine. Pharmacol Rev 2005; 57: 79–115.
von Weymarn LB, Brown KM, Murphy SE . Inactivation of CYP2A6 and CYP2A13 during nicotine metabolism. J Pharmacol Exp Ther 2006; 316: 295–303.
Ho MK, Tyndale RF . Overview of the pharmacogenomics of cigarette smoking. Pharmacogenomics J 2007; 7: 81–98.
Lerman C, Jepson C, Wileyto EP, Patterson F, Schnoll R, Mroziewicz M et al. Genetic variation in nicotine metabolism predicts the efficacy of extended-duration transdermal nicotine therapy. Clin Pharmacol Ther 2010; 87: 553–557.
Schnoll RA, Patterson F, Wileyto EP, Tyndale RF, Benowitz N, Lerman C . Nicotine metabolic rate predicts successful smoking cessation with transdermal nicotine: a validation study. Pharmacol Biochem Behav 2009; 92: 6–11.
Thorgeirsson TE, Gudbjartsson DF, Surakka I, Vink JM, Amin N, Geller F et al. Sequence variants at CHRNB3-CHRNA6 and CYP2A6 affect smoking behavior. Nat Genet 2010; 42: 448–453.
TAG-Consortium. Genome-wide meta-analyses identify multiple loci associated with smoking behavior. Nat Genet 2010; 42: 441–447.
Cho MH, Castaldi PJ, Wan ES, Siedlinski M, Hersh CP, Demeo DL et al. A genome-wide association study of COPD identifies a susceptibility locus on chromosome 19q13. Hum Mol Genet 2012; 21: 947–957.
Bloom AJ, Harari O, Martinez M, Madden PA, Martin NG, Montgomery GW et al. Use of a predictive model derived from in vivo endophenotype measurements to demonstrate associations with a complex locus, CYP2A6. Hum Mol Genet 2012; 21: 3050–3062.
Saccone NL, Wang JC, Breslau N, Johnson EO, Hatsukami D, Saccone SF et al. The CHRNA5-CHRNA3-CHRNB4 nicotinic receptor subunit gene cluster affects risk for nicotine dependence in African-Americans and in European-Americans. Cancer Res 2009; 69: 6848–6856.
Thorgeirsson TE, Geller F, Sulem P, Rafnar T, Wiste A, Magnusson KP et al. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature 2008; 452: 638–642.
Bierut LJ, Madden PA, Breslau N, Johnson EO, Hatsukami D, Pomerleau OF et al. Novel genes identified in a high-density genome wide association study for nicotine dependence. Hum Mol Genet 2007; 16: 24–35.
Berg JZ, Mason J, Boettcher AJ, Hatsukami DK, Murphy SE . Nicotine metabolism in African Americans and European Americans: variation in glucuronidation by ethnicity and UGT2B10 haplotype. J Pharmacol Exp Ther 2010; 332: 202–209.
Benowitz NL, Perez-Stable EJ, Fong I, Modin G, Herrera B, Jacob P 3rd . Ethnic differences in N-glucuronidation of nicotine and cotinine. J Pharmacol Exp Ther 1999; 291: 1196–1203.
Murphy SE, Park SS, Thompson EF, Wilkens LR, Patel Y, Stram DO et al. Nicotine N-glucuronidation relative to N-oxidation and C-oxidation and UGT2B10 genotype in five ethnic/racial groups. Carcinogenesis 2014; 35: 2526–2533.
Zhang J, Cashman JR . Quantitative analysis of FMO gene mRNA levels in human tissues. Drug Metab Dispos 2006; 34: 19–26.
Bhagwat SV, Bhamre S, Boyd MR, Ravindranath V . Cerebral metabolism of imipramine and a purified flavin-containing monooxygenase from human brain. Neuropsychopharmacology 1996; 15: 133–142.
Ferguson CS, Tyndale RF . Cytochrome P450 enzymes in the brain: emerging evidence of biological significance. Trends Pharmacol Sci 2011; 32: 708–714.
Shimizu M, Yano H, Nagashima S, Murayama N, Zhang J, Cashman JR et al. Effect of genetic variants of the human flavin-containing monooxygenase 3 on N- and S-oxygenation activities. Drug Metab Dispos 2007; 35: 328–330.
Bloom J, Hinrichs AL, Wang JC, von Weymarn LB, Kharasch ED, Bierut LJ et al. The contribution of common CYP2A6 alleles to variation in nicotine metabolism among European-Americans. Pharmacogenet Genomics 2011; 21: 403–416.
McCarthy DE, Piasecki TM, Lawrence DL, Jorenby DE, Shiffman S, Fiore MC et al. A randomized controlled clinical trial of bupropion SR and individual smoking cessation counseling. Nicotine Tob Res 2008; 10: 717–729.
Piper ME, Federman EB, McCarthy DE, Bolt DM, Smith SS, Fiore MC et al. Efficacy of bupropion alone and in combination with nicotine gum. Nicotine Tob Res 2007; 9: 947–954.
Piper ME, Smith SS, Schlam TR, Fiore MC, Jorenby DE, Fraser D et al. A randomized placebo-controlled clinical trial of 5 smoking cessation pharmacotherapies. Arch Gen Psychiatry 2009; 66: 1253–1262.
Bloom AJ, von Weymarn LB, Martinez M, Bierut LJ, Goate A, Murphy SE . The contribution of common UGT2B10 and CYP2A6 alleles to variation in nicotine glucuronidation among European Americans. Pharmacogenet Genomics 2013; 23: 706–716.
Bloom AJ, Hartz SM, Baker TB, Chen LS, Piper ME, Fox L et al. Beyond cigarettes per day. A genome-wide association study of the biomarker carbon monoxide. Ann Am Thorac Soc 2014; 11: 1003–1010.
Stephens M, Donnelly P . A comparison of bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet 2003; 73: 1162–1169.
Bloom AJ, Harari O, Martinez M, Zhang X, McDonald SA, Murphy SE et al. A compensatory effect upon splicing results in normal function of the CYP2A6*14 allele. Pharmacogenet Genomics 2013; 23: 107–116.
Wang JC, Cruchaga C, Saccone NL, Bertelsen S, Liu P, Budde JP et al. Risk for nicotine dependence and lung cancer is conferred by mRNA expression levels and amino acid change in CHRNA5. Hum Mol Genet 2009; 18: 3125–3135.
Gadel S, Friedel C, Kharasch ED . Differences in methadone metabolism by CYP2B6 variants. Drug Metab Dispos 2015; 43: 994–1001.
Aliverti A, Curti B, Vanoni MA . Identifying and quantitating FAD and FMN in simple and in iron-sulfur-containing flavoproteins. Methods Mol Biol 1999; 131: 9–23.
Li P, Ann J, Akk G . Activation and modulation of human alpha4beta2 nicotinic acetylcholine receptors by the neonicotinoids clothianidin and imidacloprid. J Neurosci Res 2011; 89: 1295–1301.
Kang J, Chung W, Lee K, Park C, Kang J, Shin I et al. Phenotypes of flavin-containing monooxygenase activity determined by ranitidine N-oxidation are positively correlated with genotypes of linked FM03 gene mutations in a Korean population. Pharmacogenetics 2000; 10: 67–78.
Park SB, Jacob P 3rd, Benowitz NL, Cashman JR . Stereoselective metabolism of (S)-(-)-nicotine in humans: formation of trans-(S)-(-)-nicotine N-1'-oxide. Chem Res Toxicol 1993; 6: 880–888.
Lattard V, Zhang J, Cashman JR . Alternative processing events in human FMO genes. Mol Pharmacol 2004; 65: 1517–1525.
Katchamart S, Stresser DM, Dehal SS, Kupfer D, Williams DE . Concurrent flavin-containing monooxygenase down-regulation and cytochrome P-450 induction by dietary indoles in rat: implications for drug-drug interaction. Drug Metab Dispos 2000; 28: 930–936.
Hinrichs AL, Murphy SE, Wang JC, Saccone S, Saccone N, Steinbach JH et al. Common polymorphisms in FMO1 are associated with nicotine dependence. Pharmacogenet Genomics 2011; 21: 397–402.
Bloom AJ, Murphy SE, Martinez M, von Weymarn LB, Bierut LJ, Goate A . Effects upon in vivo nicotine metabolism reveal functional variation in FMO3 associated with cigarette consumption. Pharmacogenet Genomics 2013; 23: 62–68.
Chenoweth MJ, Zhu AZ, Sanderson Cox L, Ahluwalia JS, Benowitz NL, Tyndale RF . Variation in P450 oxidoreductase (POR) A503V and flavin-containing monooxygenase (FMO)-3 E158K is associated with minor alterations in nicotine metabolism, but does not alter cigarette consumption. Pharmacogenet Genomics 2014; 24: 172–176.
Haberstick BC, Timberlake D, Ehringer MA, Lessem JM, Hopfer CJ, Smolen A et al. Genes, time to first cigarette and nicotine dependence in a general population sample of young adults. Addiction 2007; 102: 655–665.
Borland R, Yong HH, O'Connor RJ, Hyland A, Thompson ME . The reliability and predictive validity of the Heaviness of Smoking Index and its two components: findings from the International Tobacco Control Four Country study. Nicotine Tob Res 2010; 12: S45–S50.
Branstetter SA, Mercincavage M, Muscat JE . Predictors of the nicotine dependence behavior time to the first cigarette in a multiracial cohort. Nicotine Tob Res 2014.
Piasecki TM, Piper ME, Baker TB . Tobacco dependence: insights from investigations of self-reported smoking motives. Curr Dir Psychol Sci 2010; 19: 395–401.
Piasecki TM, Piper ME, Baker TB . Refining the tobacco dependence phenotype using the Wisconsin Inventory of Smoking Dependence Motives: II. Evidence from a laboratory self-administration assay. J Abnorm Psychol 2010; 119: 513–523.
Bhamre S, Bhagwat SV, Shankar SK, Boyd MR, Ravindranath V . Flavin-containing monooxygenase mediated metabolism of psychoactive drugs by human brain microsomes. Brain Res 1995; 672: 276–280.
Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 2014; 34: 11929–11947.
Acknowledgements
We thank Dr Jeffery Milbrandt and his lab for their advice and assistance. We acknowledge the use of tissues procured by the National Disease Research Interchange (NDRI) with support from NIH grant 2 U42 OD011158. The Collaborative Genetic Study of Nicotine Dependence was supported by National Cancer Institute grant P01 CA089392. The University of Wisconsin Transdisciplinary Tobacco Use Research Center was supported by National Institute on Drug Abuse grant P50 DA019706 and National Cancer Institute grant P50 CA084724 (TBB). This work was also supported by the National Institute on Drug Abuse grants T32 DA007261 (AMT & AJB), R01 DA036583 (LJB), R01 DA14211 (EDK), R01 DA25931 (EDK) and K01 DA034035 (AJB), National Cancer Institute grant K05 CA139871 (TBB), National Institute of Heart, Lung, and Blood Disease grant R01 HL109031 (TBB), and the Taylor Family Institute for Innovative Psychiatric Research (GA). Genotyping services for the UW-TTURC sample were provided by the Center for Inherited Disease Research (CIDR) and funding support for CIDR was provided by NIH grant U01 HG004438 and NIH contract HHSN268200782096C to The Johns Hopkins University. Assistance with genotype cleaning was provided by the Gene Environment Association Studies (GENEVA) Coordinating Center (U01 HG004446). LC/MS/MS analysis carried out in the Analytical Biochemistry Core of the University of Minnesota Cancer Center, was supported in part by National Cancer Institute grant CA-77598.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
LJB and AG are listed as an inventors on Issued US Patent 8,080,371, ‘Markers for Addiction’ covering the use of certain SNPs in determining the diagnosis, prognosis and treatment of addiction.
Additional information
Supplementary Information accompanies the paper on the The Pharmacogenomics Journal website
Rights and permissions
About this article

{kind=link}
{kind=link}
Cite this article
Teitelbaum, A., Murphy, S., Akk, G. et al. Nicotine dependence is associated with functional variation in FMO3, an enzyme that metabolizes nicotine in the brain. Pharmacogenomics J 18, 136–143 (2018). https://doi.org/10.1038/tpj.2016.92
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tpj.2016.92
- Springer Nature Limited