
Abstract
We aimed to assess the cost-effectiveness of pharmacogenetic-guided dosing of warfarin in patients with atrial fibrillation (AF) in the United Kingdom and Sweden. Data from EU-PACT, a randomized controlled trial in newly diagnosed AF patients, were used to model the incremental costs per quality-adjusted life-year (QALY) gained by pharmacogenetic-guided warfarin dosing versus standard treatment over a lifetime horizon. Incremental lifetime costs were £26 and 382 Swedish kronor (SEK) and incremental QALYs were 0.0039 and 0.0015 in the United Kingdom and Sweden, respectively. The corresponding incremental cost-effectiveness ratios (ICERs) were £6 702 and 253 848 SEK per QALY gained. The ICER was below the willingness-to-pay threshold of £20 000 per QALY gained in 93% of the simulations in the United Kingdom and below 500 000 SEK in 67% of the simulations in Sweden. Our data suggest that pharmacogenetic-guided dosing of warfarin is a cost-effective strategy to improve outcomes of patients with AF treated with warfarin in the United Kingdom and in Sweden.
Similar content being viewed by others
Introduction
Warfarin is widely used to decrease the risk of stroke in patients with atrial fibrillation (AF).1 Warfarin dosing requires frequent monitoring of the International Normalised Ratio (INR), because of the narrow therapeutic window and inter-patient and intra-patient variability in dose requirement. INR values below the therapeutic range (usually 2.0-3.0 in patients with AF) lead to loss of efficacy and an increased risk of thromboembolic events, while INR values above the therapeutic range are associated with an increased risk of bleeding. Major bleeding events associated with warfarin, such as intra-cranial haemorrhage (ICH), can cause high morbidity and mortality, and are costly to manage.2
Genetic polymorphisms have been shown to be associated with warfarin dose requirement and also with the risk of adverse treatment outcomes.3 Polymorphisms in the VKORC1 gene, coding the main target enzyme for warfarin, and the CYP2C9 gene, coding the main enzyme responsible for warfarin metabolism, together account for approximately 40% of the inter-individual variability in warfarin dose requirement.4 Several dosing algorithms have been constructed; these have included genetic information, together with patient characteristics such as age, gender, height and weight.4, 5, 6
By the end of 2013, three large randomized controlled trials of pharmacogenetic-guided dosing of coumarin anticoagulants had been published.7, 8, 9 None of these trials was powered to show a significant difference in clinical endpoints, such as bleeding and stroke, and therefore the main outcome measure in all trials was the percentage time spent in therapeutic INR range (PTIR). However, PTIR is a relevant surrogate measure since it has been shown that a 6-10% improvement in PTIR can have clinically significant impact of the risk of bleeding and stroke.10, 11
One of the trials (EU-PACT) included patients starting warfarin in the United Kingdom and Sweden8 and compared a pharmacogenetic-based algorithm12 with standard dosing. This trial demonstrated that pharmacogenetic-guided dosing increased the PTIR in the first 12 weeks of therapy by 7.0 percentage points (95% confidence interval 3.3 to 10.6). However, since pharmacogenetic-guided dosing requires genotyping which incurs additional costs, it is important to investigate whether this would be cost-effective if it is implemented in routine clinical practice. The aim of this study was therefore to assess the cost-effectiveness of pharmacogenetic-guided dosing versus standard dosing of warfarin in newly diagnosed patients with AF in the United Kingdom and Sweden. We performed two country-specific cost-effectiveness analyses because of between-country differences in the health-care system (that is, structure, cost) and quality of standard anticoagulant care.13
Materials and methods
Model structure
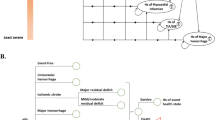
A Markov model was used to analyse the cost-effectiveness of pharmacogenetic-guided dosing versus standard dosing in the United Kingdom and Sweden. The model was similar to the model used in previous studies on this topic14, 15, 16 and developed using Microsoft Excel. The model was used to compare the incidence of adverse events, quality-adjusted life-years (QALYs), and direct medical costs of the two treatment options over a lifetime time horizon. In the EU-PACT trial, the effect of pharmacogenetic-guided dosing was assessed for warfarin, acenocoumarol and phenprocoumon.7 However, data from only the warfarin arm of this trial were used to populate this model, as the warfarin trial was conducted in the United Kingdom and Sweden, and differed in design from the acenocoumarol and phenprocoumon trials. The base-case cohort in the analysis was a hypothetical cohort of patients with AF initiating warfarin treatment; their mean ages were 70.9 in the United Kingdom and 72.5 in Sweden, reflecting the mean ages at start of AF treatment in the EU-PACT trial. Figure 1a shows the different health states according to the Markov model. All patients entered the model in the ‘well’ state and could move to other states at monthly intervals. When an event occurred, the patient would stay in that state for 1 month and then move to ‘well’, ‘disability’ or ‘death’. Patients with a permanent disability after stroke or ICH were assigned to the ‘disability’ state. Patients who recovered from an event were assigned back to the ‘no event state’ but with the possibility of having a ‘recurrent event’ later.

Schematic representation of the Markov model. Patients initiating warfarin entered the model in the ‘well’ state and faced different chances of developing adverse events depending on dosing algorithm.
Thromboembolic events consisted mainly of ischaemic strokes, but 28% were assumed to be transient ischaemic attacks.17, 18 Patients with a stroke had a 10% chance of dying and a 47% chance of disability14, 19 while patients with a transient ischaemic attack were assumed to fully recover. The majority of haemorrhagic events (80%) were assumed to be extra-cranial haemorrhage, and 20% were ICH.19, 20 The risk that an ICH would result in permanent disability was 50% and the chance that it would be fatal was 45%;19, 20; these values were zero for an extra-cranial haemorrhage. Patients were assumed to switch to aspirin after an ICH.21, 22 Input parameters of the model for both the United Kingdom and Sweden are shown in Table 1. Age-specific mortality rates were included in the model using country-specific mortality data, excluding cerebrovascular deaths.23, 24 Country-specific input parameters are presented in Table 2.
Clinical inputs
The percentage time within therapeutic INR range (PTIR) is commonly used as the primary outcome in clinical trials investigating the effect of genotype-guided warfarin dosing.9, 15, 16 We analysed the EU-PACT trial data to determine the percentage time within different INR ranges (<2.0, 2.0–3.0, 3.0–5.0 and >5.0) in the first 3 months of the treatment using the linear interpolation method as described by Rosendaal et al.25 In general, the PTIR was higher in Sweden than in the United Kingdom, mainly due to patients spending less time above the therapeutic range. However, the difference in PTIR between pharmacogenetic-guided dosing and standard care was larger in the United Kingdom than in Sweden (see Table 3). For example, in the first month the PTIR was 52.8% in the pharmacogenetic-guided arm versus 42.6% in the control arm in the United Kingdom, compared with 60.6 versus 56.5% in Sweden. By month 3 the difference was small (approximately 1%) in both countries. We used estimates from literature for the percentage time in the different INR ranges after the first 3 months26 and assumed that this percentage is stable from month 3 onwards and the same in the two arms (conservative assumption of no difference between the two arms after month 3). The risks of adverse events associated with each of the four INR ranges were derived from a meta-analysis of 19 randomized trials and observational studies of coumarin anticoagulants27 (Table 1). The percentages of time spent in the different INR ranges were multiplied by the monthly risk of events associated with these ranges to calculate the incidence of thromboembolic and haemorrhagic events in every cycle.
In the first 3 months of treatment, the INR is measured frequently. In the EU-PACT trial, approximately seven INR measurements were made on average in the first month, followed by three measurements in the second and third months. We assumed one measurement per month thereafter.28, 29
Health state utilities
The baseline utility value in our model was 0.81 for patients with AF.30 To reflect the disutility of blood sampling for INR measurement, a decrement of 0.013 was applied for warfarin use and a decrement of 0.002 for aspirin use31 to reflect the disutility of gastrointestinal effects. Decrements were also ascribed when patients experienced an adverse event (Table 1). In the case of a non-disabling event, these decrements were assumed to last 1 month. For patients in the disability state, a permanent decrement was applied. QALYs were estimated by summing all utility values multiplied by the time spent in each health state.
Costs
The cost of a point-of-care genotyping test, which was used in the EU-PACT trial, was estimated to be approximately US$50 (approximately £35 or 440 SEK),32 although this particular form of testing has not been used in clinical practice yet. The occurrence of a clinical event gave rise to one-time, event-related costs. For less disabling events (transient ischaemic attacks, extra-cranial haemorrhage and non-disabling stroke or ICH), no subsequent costs were applied. Patients with disabling stroke or ICH were assumed to incur monthly costs relating to the management of disability for the remainder of their lifetime. Costs were determined from the perspectives of the National Health Service in the United Kingdom and the health-care sector in Sweden for the year 2014 in the local currency (UK£ and Swedish SEK). Costs and effects were discounted at an annual rate of 3.5% for the United Kingdom and 3.0% for Sweden, in accordance with national guidelines.33, 34
Sensitivity analyses
One-way sensitivity analyses were conducted to evaluate the influence of input parameters on the economic results. The parameters were varied over their 95% confidence intervals or a plausible range (for example, ±20%) if a confidence interval was not available. The costs of genotyping were varied by ±50% because this test is not yet used in clinical practice and there is more uncertainty around this estimate. Annual discount rates applied to costs and effects were varied from 0 to 6% in both countries.
A probabilistic sensitivity analysis was performed using 1000 Monte Carlo simulations to assess the combined uncertainty of multiple model parameters on the estimated cost-effectiveness of genotyping. Values were drawn from Dirichlet distributions for the probabilities of different outcomes of stroke and ICH, beta distributions for all other probabilities and QALYs, and gamma distributions for costs. A normal distribution was used to vary the PTIR, frequency of INR measurements and age.
In the United Kingdom, NICE applies a cost-effectiveness threshold range of £20 000–£30 000 per QALY gained.33 In Sweden, a threshold of 500 000 SEK (approximately £40 000) has been mentioned.35 The probabilistic sensitivity analysis yielded probabilities of genotyping being cost-effective at different threshold values of willingness-to-pay and the results are presented using a cost-effectiveness acceptability curve.36 Given the uncertainty about genotyping costs, we also performed a threshold analysis to identify the highest cost at which genotyping would still be cost-effective, given cost-effectiveness thresholds of £20 000 and 500 000 SEK per QALY gained in the United Kingdom and Sweden, respectively.
Results
Base case
Supplementary figure S1 shows the cumulative risk of haemorrhage (ICH and extra-cranial haemorrhage) and thromboembolism (stroke and transient ischaemic attacks) during the first year of warfarin treatment for standard care and pharmacogenetic-guided dosing algorithms in the United Kingdom and Sweden. The modelled difference between standard care and pharmacogenetic-guided dosing in haemorrhagic event rate was higher in the United Kingdom, where the difference appeared within the first two months and was stable thereafter.
Table 4 presents the first-year incidence of clinical events per 100 patient-years. Genotyping decreased the risk of haemorrhagic events by 0.18% in the United Kingdom and by 0.2% in Sweden. The risk of thromboembolic events decreased by 0.04% in both countries. In the United Kingdom, genotyping increased lifetime costs by £26 and QALYs by 0.0039 (equivalent to1.4 days of full health), resulting in an incremental cost-effectiveness ratio (ICER) of £6 702 per QALY gained (Table 4). In Sweden, the incremental costs and QALYs were 382 SEK and 0.0015 (0.5 days in full health), respectively, with an ICER of 253 848 SEK per QALY gained. Life expectancy (without quality adjustment) in the pharmacogenetic-guided group was 0.0047 years (1.7 days) longer in the United Kingdom and 0.0018 years (0.7 days) longer in Sweden than in the standard care group.
One-way sensitivity analysis
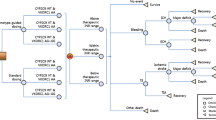
In the UK analysis, none of the parameters increased the ICER above the £20 000 per QALY threshold when they were varied within the specified range. The parameter with the largest influence on the ICER was the risk of stroke when INR was within the therapeutic range. The ICER was £4890 when risk of stroke was set to its lower limit (0.07%) but increased to £14 284 per QALY gained when the risk of stroke was set to its upper limit (0.30%). The costs of genotyping also had a large influence; when these were varied from £17.51 to £52.54 the ICER ranged from £2273 to £11 231 per QALY gained. In Sweden, several parameters changed the results appreciably and led to ICERs higher than the 500 000 SEK per QALY threshold. When INR was in the therapeutic range for 83.9% of the time by month 2 with standard care, the ICER increased to 972 000 SEK per QALY gained. Other parameters with a large influence were: risk of stroke when INR was <2.0, PTIR in month 1 with both pharmacogenetic-guided dosing and standard care, PTIR in month 2 with pharmacogenetic-guided dosing and PTIR in month 3 with standard care. The effect of uncertainty around the 15 most influential parameters on the ICER in the United Kingdom and Sweden is presented in the tornado plot in Figure 2. The one-way sensitivity analyses are especially useful to test the model, that is, to see if the changes in the variables influence the results as expected, which they did for our model.

Tornado plots showing the effect of uncertainty around the most influential parameters on the incremental cost-effectiveness ratio.
Probabilistic sensitivity analysis
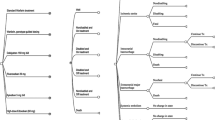
In the probabilistic sensitivity analysis, the incremental costs per QALY gained were below £20 000 in 93% of the simulations in the United Kingdom and below 500 000 SEK in 67% of the simulations in Sweden. The probability that genotyping would be cost-effective at different thresholds of willingness-to-pay is shown in Figure 3.

Cost-effectiveness acceptability curve.
Threshold analysis
Pharmacogenetic-guided dosing is cost-effective if genotyping costs would be no higher than £86 in the United Kingdom (given a cost-effectiveness threshold of £20 000 per QALY gained) or 809 SEK (approximately £64) in Sweden (given a threshold of 500 000 SEK).
Discussion
Our study shows that pharmacogenetic-guided dosing of warfarin can be cost-effective in the management of patients with AF in both the UK and Swedish health-care settings. As the EU-PACT trial demonstrated a larger relative effect of the pharmacogenetic-guided dosing algorithm in comparison to standard care on PTIR in the United Kingdom than in Sweden, the ICER was more favourable in the United Kingdom. The results of our analysis therefore suggest that pharmacogenetic-guided warfarin dosing has a greater likelihood of being cost-effective in the United Kingdom. In the base case analysis, genotyping was also cost-effective in Sweden, but uncertainty around some parameters such as the time spent with an INR<2.0 or time spent within therapeutic INR range led to more uncertainty around the estimated cost-effectiveness. However, the probability that pharmacogenetic-guided dosing would be cost-effective in Sweden, given a threshold of 500 000 SEK per QALY gained, was still 67%.
A limitation to our study was the fact that we were very reliant on modelled extrapolation. First, we extrapolated the results of the EU-PACT study (12 weeks) over a lifetime time horizon (assuming no difference between the two arms after 12 weeks). Second, we extrapolated the intermediate outcome (PTIR) to incidence of clinical events and obtained data on costs, utilities and probabilities from multiple sources which may not necessarily be appropriate for the study population. Although previous studies on this topic14, 37 have relied on the same assumptions, we used more robust evidence to assess the impact of genetic testing on treatment outcomes. It is also however important to note that a recent analysis of the warfarin arm of the ENGAGE AF-TIMI 48 trial showed that patients who carry variants in CYP2C9 and/or VKORC1 were more likely to have unstable INRs and were at increased risk of bleeding events,38 which provides support for our assumptions. Furthermore, it has been shown from the RELY trial that a 10% improvement in PTIR can lead to a 20% improvement clinical outcomes.11 Another limitation is that new direct oral anticoagulants, such as dabigatran or apixaban, can be used instead of warfarin in some but not all AF patients. An assessment of cost-effectiveness of pharmacogenetic-guided warfarin dosing versus these drugs was outside the scope of this study.
To estimate the cost of the genetic test we used a commercial rate for point-of-care testing, although local rates for this test may vary and costs may change (decrease) over time. We therefore performed a threshold analysis and found that pharmacogenetic-guided dosing is cost-effective if genotyping costs would be no higher than £86 in the United Kingdom or 809 SEK in Sweden.
Several studies on the cost-effectiveness of genotyping patients before warfarin initiation have been published, but the results of these studies vary widely.14, 37, 39 In one UK study, the ICER of pharmacogenetic-guided dosing versus clinical dosing was £13 226.39 In a US study by Meckley et al., the costs per QALY gained were $60 750 and the chance that the ICER was below the $50 000 threshold was estimated to be 46%.14 Two other studies reported considerably higher costs per QALY gained than the previous studies ($171 000 to $34 700).37 This variation is mainly due to uncertainty around the effectiveness of genotyping because of the small number of randomized controlled trials, and the heterogeneity of patient populations, trial design, outcome definitions and reporting of results among these randomized controlled trials.37 Our economic evaluation for warfarin is the first one based on an appropriately powered randomised controlled trial, which resulted in considerably less uncertainty around the estimated effectiveness.
The quality of standard treatment in Sweden is high. In the current study, the PTIR in the standard treatment arm varied from 57% in month 1 to 85% in month 3 in Sweden compared with 43% in month 1 to 69% in month 3 in the United Kingdom. This might also explain the lower benefit of pharmacogenetic-guided dosing in Sweden compared with the United Kingdom. This is supported by our one-way sensitivity analysis, where we found that the results were especially sensitive to the PTIR in Sweden.
The total QALY gain in the present study was small (1.4 or 0.5 days in full health in the United Kingdom and Sweden respectively), due to the small difference in incidence of adverse events (patients in both study arms were monitored for 3 months only). We recently conducted a systematic review of economic evaluations of pharmacogenetic tests,40 which identified 10 studies that considered the cost effectiveness of testing prior to prescription of warfarin, eight of which were conducted in the United States. Although the results were mixed (please see Plumpton et al40 for details), the QALY gains ranged from 0.2 to 1.1 days in full health, consistent with our finding.
In summary, our cost-effectiveness analysis based on a real-world clinical trial suggests that genotype-guided dosing of warfarin is cost-effective in both United Kingdom and Sweden, where the trial was conducted. Although pharmacogenetic-guided dosing yields a small health gain when compared with standard care, it would still help to improve the quality of warfarin treatment in the United Kingdom and Sweden. Our study also highlights the fact that small average improvements for the whole population (because the costs are spread among the many patients receiving therapy) hide the fact there will be large health gains in a few individuals – this is an important point in relation to economic evaluation of personalised medicine that needs to be appreciated by all stakeholders, in particular regulators and payers.
References
Lip GY, Lane DA . Stroke prevention in atrial fibrillation: a systematic review. JAMA 2015; 313: 1950–1962.
Wysowski DK, Nourjah P, Swartz L . Bleeding complications with warfarin use: a prevalent adverse effect resulting in regulatory action. Arc int med 2007; 167: 1414–1419.
Schalekamp T, de Boer A . Pharmacogenetics of oral anticoagulant therapy. Curr pharm des 2010; 16: 187–203.
Wadelius M, Chen LY, Lindh JD, Eriksson N, Ghori MJ, Bumpstead S et al. The largest prospective warfarin-treated cohort supports genetic forecasting. Blood 2009; 113: 784–792.
International Warfarin Pharmacogenetics C International Warfarin Pharmacogenetics C Klein TE International Warfarin Pharmacogenetics C Altman RB International Warfarin Pharmacogenetics C Eriksson N International Warfarin Pharmacogenetics C Gage BF International Warfarin Pharmacogenetics C Kimmel SE et al. Estimation of the warfarin dose with clinical and pharmacogenetic data. N Engl J Med 2009; 360: 753–764.
Lenzini P, Wadelius M, Kimmel S, Anderson JL, Jorgensen AL, Pirmohamed M et al. Integration of genetic, clinical, and INR data to refine warfarin dosing. Clin Pharmacol Ther 2010; 87: 572–578.
Verhoef TI, Ragia G, de Boer A, Barallon R, Kolovou G, Kolovou V et al. A randomized trial of genotype-guided dosing of acenocoumarol and phenprocoumon. N Engl j med 2013; 369: 2304–2312.
Pirmohamed M, Burnside G, Eriksson N, Jorgensen AL, Toh CH, Nicholson T et al. A randomized trial of genotype-guided dosing of warfarin. N Engl J Med 2013; 369: 2294–2303.
Kimmel SE, French B, Kasner SE, Johnson JA, Anderson JL, Gage BF et al. A pharmacogenetic versus a clinical algorithm for warfarin dosing. N Engl j med 2013; 369: 2283–2293.
Jones M, McEwan P, Morgan CL, Peters JR, Goodfellow J, Currie CJ . Evaluation of the pattern of treatment, level of anticoagulation control, and outcome of treatment with warfarin in patients with non-valvar atrial fibrillation: a record linkage study in a large British population. Heart (British Cardiac Society) 2005; 91: 472–477.
Van Spall HG, Wallentin L, Yusuf S, Eikelboom JW, Nieuwlaat R, Yang S et al. Variation in warfarin dose adjustment practice is responsible for differences in the quality of anticoagulation control between centers and countries: an analysis of patients receiving warfarin in the randomized evaluation of long-term anticoagulation therapy (RE-LY) trial. Circulation 2012; 126: 2309–2316.
Avery PJ, Jorgensen A, Hamberg AK, Wadelius M, Pirmohamed M, Kamali F et al. A proposal for an individualized pharmacogenetics-based warfarin initiation dose regimen for patients commencing anticoagulation therapy. Clin Pharmacol Ther 2011; 90: 701–706.
Verhoef TI, Redekop WK, van Schie RM, Bayat S, Daly AK, Geitona M et al. Cost-effectiveness of pharmacogenetics in anticoagulation: international differences in healthcare systems and costs. Pharmacogenomics 2012; 13: 1405–1417.
Meckley LM, Gudgeon JM, Anderson JL, Williams MS, Veenstra DL . A policy model to evaluate the benefits, risks and costs of warfarin pharmacogenomic testing. PharmacoEconomics 2010; 28: 61–74.
Verhoef TI, Redekop WK, Veenstra DL, Thariani R, Beltman PA, van Schie RM et al. Cost-effectiveness of pharmacogenetic-guided dosing of phenprocoumon in atrial fibrillation. Pharmacogenomics 2013; 14: 869–883.
Verhoef TI, Redekop WK, de Boer A, Maitland-van der Zee AH . Economic evaluation of a pharmacogenetic dosing algorithm for coumarin anticoagulants in the Netherlands. Pharmacogenomics 2015; 16: 101–114.
Shah SV, Gage BF . Cost-effectiveness of dabigatran for stroke prophylaxis in atrial fibrillation. Circulation 2011; 123: 2562–2570.
O'Brien CL, Gage BF . Costs and effectiveness of ximelagatran for stroke prophylaxis in chronic atrial fibrillation. JAMA: j Am Med Assoc 2005; 293: 699–706.
Federation of Dutch Anticoagulant clinics. Samenvatting medische jaarverslagen 2012. http://www.fnt.nl/media/docs/jaarverslagen/FNT_Medisch_jaarverslag_2012_WEB.pdf.
Fang MC, Go AS, Chang Y, Hylek EM, Henault LE, Jensvold NG et al. Death and disability from warfarin-associated intracranial and extracranial hemorrhages. Am j med 2007; 120: 700–705.
Connolly SJ, Eikelboom J, Joyner C, Diener HC, Hart R, Golitsyn S et al. Apixaban in patients with atrial fibrillation. N Engl j med 2011; 364: 806–817.
Hart RG, Pearce LA, Aguilar MI . Meta-analysis: antithrombotic therapy to prevent stroke in patients who have nonvalvular atrial fibrillation. Ann intern med 2007; 146: 857–867.
Office of National Statistics, life tables. http://www.statistics.gov.uk/.
World Health Organization, Global Health Observatory Data Repository. http://apps.who.int/gho/data/?vid=710#.
Rosendaal FR, Cannegieter SC, van der Meer FJ, Briet E . A method to determine the optimal intensity of oral anticoagulant therapy. Thromb Haemost 1993; 69: 236–239.
Wallentin L, Yusuf S, Ezekowitz MD, Alings M, Flather M, Franzosi MG et al. Efficacy and safety of dabigatran compared with warfarin at different levels of international normalised ratio control for stroke prevention in atrial fibrillation: an analysis of the RE-LY trial. Lancet 2010; 376: 975–983.
Oake N, Jennings A, Forster AJ, Fergusson D, Doucette S, van Walraven C . Anticoagulation intensity and outcomes among patients prescribed oral anticoagulant therapy: a systematic review and meta-analysis. CMAJ 2008; 179: 235–244.
Jowett S, Bryan S, Mahe I, Brieger D, Carlsson J, Kartman B et al. A multinational investigation of time and traveling costs in attending anticoagulation clinics. Value health 2008; 11: 207–212.
Wieloch M, Sjalander A, Frykman V, Rosenqvist M, Eriksson N, Svensson PJ . Anticoagulation control in Sweden: reports of time in therapeutic range, major bleeding, and thrombo-embolic complications from the national quality registry AuriculA. Eur heart j 2011; 32: 2282–2289.
Sullivan PW, Lawrence WF, Ghushchyan V . A national catalog of preference-based scores for chronic conditions in the United States. Med care 2005; 43: 736–749.
Gage BF, Cardinalli AB, Owens DK . The effect of stroke and stroke prophylaxis with aspirin or warfarin on quality of life. Arch intern med 1996; 156: 1829–1836.
Howard R, Leathart JB, French DJ, Krishan E, Kohnke H, Wadelius M et al. Genotyping for CYP2C9 and VKORC1 alleles by a novel point of care assay with HyBeacon(R) probes. Clin Chim Acta 2011; 412: 2063–2069.
NICE National Institute of Health and Care Excellence. Guide to the methods of technology appraisal 2013. http://www.nice.org.uk/media/D45/1E/GuideToMethodsTechnologyAppraisal2013.pdf.
International society for pharmacoeconomics and outcomes research http://www.ispor.org/PEguidelines/index.asp.
Socialstyrelsen, Nationella riktlinjer för prostatacancersjukvård. Medicinskt och hälsoekonomiskt faktadokument. 2007: Stockholm.
Fenwick E, Claxton K, Sculpher M . Representing uncertainty: the role of cost-effectiveness acceptability curves. Health econ 2001; 10: 779–787.
Verhoef TI, Redekop WK, Darba J, Geitona M, Hughes DA, Siebert U et al. A systematic review of cost-effectiveness analyses of pharmacogenetic-guided dosing in treatment with coumarin derivatives. Pharmacogenomics 2010; 11: 989–1002.
Mega JL, Walker JR, Ruff CT, Vandell AG, Nordio F, Deenadayalu N et al. Genetics and the clinical response to warfarin and edoxaban: findings from the randomised, double-blind ENGAGE AF-TIMI 48 trial. Lancet 2015; 385: 2280–2287.
Pink J, Pirmohamed M, Lane S, Hughes DA . Cost-effectiveness of pharmacogenetics-guided warfarin therapy vs. alternative anticoagulation in atrial fibrillation. Clin Pharmacol Ther 2014; 95: 199–207.
Plumpton CO, Roberts D, Pirmohamed M, Hughes DA . A systematic review of economic evaluations of pharmacogenetic testing for prevention of adverse drug reactions. Pharmacoeconomics 2016; doi: 10.1007/s40273-016-0397-9; e-pub ahead of print.
British National Formulary. https://www.medicinescomplete.com/mc/bnf/current/.
Prisdatabas för läkemedel (2015) http://www.apoteket.se.
National Institute of Health and Clinical Excellence. Dabigatran etexilate for the prevention of stroke and systemic embolism in atrial fibrillation. Final appraisal determination. 2011. http://www.nice.org.uk/nicemedia/live/12225/56899/56899.pdf.
National Institute of Health and Clinical Excellence. Apixaban for preventing stroke and systemic embolism in people with nonvalvular atrial fibrillation. NICE technology appraisal guidance 275. 2013. http://www.nice.org.uk/nicemedia/live/14086/62874/62874.pdf.
SBU (2007). Självtestning och egenvård vid användning av blodproppsförebyggande läkemedel. SBU ALERT rapport, NR 2007-05.
Department of Health. National schedule of reference costs 2013/14. https://www.gov.uk/government/publications/nhs-reference-costs-2013-to-2014.
Alberta Health and Wellness. Health costing in Alberta. Annual Report. Edmonton: 2006.
Luengo-Fernandez R, Yiin GS, Gray AM, Rothwell PM . Population-based study of acute- and long-term care costs after stroke in patients with AF. Int j stroke 2013; 8: 308–314.
Ghatnekar O, Carlsson KS Kostnader för insjuknande i stroke år 2009, en incidensbaserad studie. IHE RAPPORT 2012: 2.
Schwander B, Gradl B, Zollner Y, Lindgren P, Diener HC, Luders S et al. Cost-utility analysis of eprosartan compared to enalapril in primary prevention and nitrendipine in secondary prevention in Europe—the HEALTH model. Value health 2009; 12: 857–871.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
This work was supported by the European Community's Seventh Framework Programme under grant agreement HEALTH-F2-2009-223062. Dr Verhoef, Dr Maitland-van der Zee, Dr Wadelius and Dr Pirmohamed report grants from EU Commission FP7 programme, during the conduct of the study; Dr Wadelius reports grants from the Swedish Research Council (Medicine), grants from the Swedish Heart and Lung Foundation, grants from the Clinical Research Support at Uppsala University, during the conduct of the study; Dr Pirmohamed is an NIHR Senior Investigator; and as part of the EU-PACT trial, we worked with LGC (a UK-based company) who were a partner on the project, and developed the point-of-care platform which was used for genotyping in the trial, and on which the results of this cost-effectiveness paper are based. However, we did not receive any funding directly from LGC.
Additional information
Supplementary Information accompanies the paper on the The Pharmacogenomics Journal website
Supplementary information
Rights and permissions
About this article

Cite this article
Verhoef, T., Redekop, W., Langenskiold, S. et al. Cost-effectiveness of pharmacogenetic-guided dosing of warfarin in the United Kingdom and Sweden. Pharmacogenomics J 16, 478–484 (2016). https://doi.org/10.1038/tpj.2016.41
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tpj.2016.41
- Springer Nature Limited
This article is cited by
-
Cost-Effectiveness Analysis of Pharmacogenomics (PGx)-Based Warfarin, Apixaban, and Rivaroxaban Versus Standard Warfarin for the Management of Atrial Fibrillation in Ontario, Canada
PharmacoEconomics (2024)
-
Cost Effectiveness of Strategies to Manage Atrial Fibrillation in Middle- and High-Income Countries: A Systematic Review
PharmacoEconomics (2023)
-
Pharmacogenetic testing for adverse drug reaction prevention: systematic review of economic evaluations and the appraisal of quality matters for clinical practice and implementation
BMC Health Services Research (2021)
-
Implementation of genotype-guided dosing of warfarin with point-of-care genetic testing in three UK clinics: a matched cohort study
BMC Medicine (2019)
-
Cost-effectiveness of precision medicine: a scoping review
International Journal of Public Health (2019)