
Abstract
Layered strategies to combat hypoxia provide flexibility in dynamic oxygen environments. Here we show that multiple miRNAs are required for hypoxic survival responses during C. elegans embryogenesis. Certain miRNAs promote while others antagonize the hypoxic survival response. We found that expression of the mir-35 family is regulated by hypoxia in a HIF-1-independent manner and loss of mir-35–41 weakens hypoxic survival mechanisms in embryos. In addition, correct regulation of the RNA binding protein, SUP-26, a mir-35 family target, is needed for survival in chronic hypoxia. The identification of the full mRNA target repertoire of these miRNAs will reveal the miRNA-regulated network of hypoxic survival mechanisms in C. elegans.
Similar content being viewed by others


Introduction
Oxygen (O2) is crucial for cellular survival. Its availability has had an enormous impact on shaping life in the variety of ecosystems and many organisms have evolved to live and reproduce under the ambient O2 concentration (21%). The correct level of O2 in tissues during embryogenesis is particularly important for the successful execution of a variety of developmental events1. However, abnormally low O2 levels (hypoxia) during development can lead to permanent defects and even premature termination of embryogenesis2,3. Therefore, embryos have developed molecular strategies to cope with inherent as well as exogenous hypoxic conditions4,5. The core hypoxic pathway relies on stabilization of the hypoxia-inducible factor HIF-1, which regulates the transcription of many protein-coding genes and microRNAs (miRNAs) responsible for the hypoxic response6,7. In addition, hif-1-independent hypoxic pathways have been identified in different models8,9. This suggests that other hif-1 independent pathways may exist to regulate aspects of the hypoxic response.
miRNAs are small (18–25nt) non-coding RNA molecules that were first discovered in Caenorhabditis elegans10. They function mainly as negative post-transcriptional regulators by binding to the 3’ untranslated region (UTR) of target mRNAs11. miRNAs are grouped in distinct families that share the same seed sequence, which is an important determinant for mRNA target specificity. Therefore, members of a miRNA family can conceptually act in a redundant manner. To date, miRNAs have been implicated in a range of developmental paradigms in C. elegans10,12,13,14, in addition to stress-related phenomena such as the regulation of lifespan5,15,16,17. Moreover, miRNAs18 and other non-coding RNA species19 function in the hypoxic response20. Their active turnover and ability to simultaneously regulate more than one target are properties that make miRNAs potentially useful stress response molecules21,22. Although they appear to regulate the majority of mRNAs in a cell23, information is often lacking on their specific role in different stress responses. We therefore sought to identify miRNAs that are important for the regulation of the hypoxic response in C. elegans.
We screened 40 C. elegans miRNA mutants covering 22 miRNA families for the ability to control embryonic survival in hypoxia. We found that loss of 14 miRNA families caused embryos to be either sensitive or resistant to hypoxic exposure. Focusing on the mir-35 family, we found that expression of all members of this family is induced in embryos exposed to hypoxia in a hif-1-independent manner. The mir-35 family is required for hypoxic survival as deletion mutants that remove mir-35–41 exhibit >90% embryonic lethality under hypoxia. Finally, we identified sup-26, which encodes an RNA binding protein, as a target of the mir-35 family that is, at least in part, required for embryonic survival in chronic hypoxia. Our study is the first to offer insights into miRNA regulation of the embryonic hypoxic response in C. elegans and further work will identify targets of these miRNAs that together provide appropriate survival mechanisms in hypoxic conditions.
Results and Discussion
Multiple miRNAs are important for the embryonic hypoxic response
In an effort to identify miRNAs important for the control of hypoxic responses in C. elegans embryos, we screened a collection of mutants that lack individual or multiple miRNAs for embryonic survival under hypoxia (Figure S1, Table S1 and Table S2). We exposed semi-synchronized populations of embryos to 0.5% O2 for 24 hours at 20 oC and after recovery for 24 hrs at 21% O2 embryos were scored for survival (Figure S1). To exclude phenotypes caused by background mutations, we outcrossed the mutants and used multiple alleles or strains where possible. Using this approach, we were able to identify multiple gene families that regulate the ability of embryos to survive a hypoxia insult (Table S1). Loss of certain miRNAs or miRNA families led to hypoxia sensitivity (mir-2, mir-35, mir-44, mir-49, mir-51, mir-60, mir-63 and mir-67) and others to hypoxia resistance (let-7, mir-58, mir-67, mir-79, mir-237, mir-246, mir-359). Finally, we found that 20 miRNA mutant strains exhibited similar embryonic survival rates to wild type animals (Table S2). These data suggest that multiple miRNAs are required for C. elegans embryos to respond appropriately to hypoxia.
The mir-35–41 family regulates the embryonic response to hypoxia
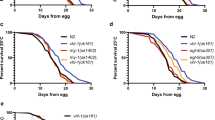
We focused our studies on the mir-35–41(nDf50) mutant strain due to its high sensitivity to hypoxia (Fig. 1 and Table S1). The mutant strain carrying the nDf50 deficiency lacks 7 (mir-35–41) of the 8 members of mir-35 family (Fig. 1A). The embryonic lethality observed in this mutant under ambient O2 concentration (21% O2) at 20 °C is approximately 50%, in agreement with previous reports24, however, lethality is increased to over 90% when freshly-laid embryos are exposed to 0.5% O2 for 24 hrs at 20 °C (Fig. 1B). Hypoxia sensitivity was also phenocopied in the independently isolated mir-35–41(gk262) mutant strain (Fig. 1A–B). A higher percentage of nDf50 mutant embryos die younger in hypoxia than in normoxia, implying that hypoxia affects development at an earlier stage in this mutant (Fig. 1C). Indeed, when we subjected older embryos (6–9 h old) to hypoxia we observed significantly lower embryonic lethality compared to when we subjected early embryos (0–3 h old) (Figure S2A). The similarity between the elevated nDf50 embryonic lethality we found in hypoxia and the known high lethality of this mutant in high temperature24 may reflect common components of hypoxia and heat stress such as challenges in protein folding and stability and induction of heat shock proteins25. However, when we subjected both mir-35–41 deletion mutant strains to 2 g/l sodium sulfite, which mimics hypoxic stress26, we also observed a significant increase in embryonic lethality (Figure S3A) indicating that mir-35 family has a specific role in survival of embryos in hypoxia.

The mir-35 family is required for embryonic hypoxic survival.
(A) mir-35–41 locus, mutant alleles and genomic rescue fragment. nDf50 (blue) and gk262 (red) alleles remove the entire mir-35–41 locus and part of the Y62F5A.9 gene. The genomic rescue fragment used in (D) is marked in green, which includes a 602 bp upstream region and the mir-35 hairpin. (B) At 21% O2, two mir-35–41 mutant alleles, nDf50 and gk262, exhibit 50% and 75% embryonic lethality respectively, whereas, wild type and hif-1(ia4) mutant embryos exhibit minimal lethality. At 0.5% O2, both mir-35–41 mutants approach 100% embryonic lethality (n = 176–242), whereas wild type and hif-1(ia4) mutant embryos exhibit 10% (n = 974) and 40% (n = 1132) lethality respectively. These data partially overlap with Table S1. (C) An increased percentage of nDf50 mutant embryos die younger in hypoxia than in normoxia. ‘Malformed’ refers to embryos with severe defects in their overall structure that do not permit stage identification. (D) Normoxic and hypoxic lethality of nDf50 mutant embryos is rescued by transgenic expression of mir-35. The sequence used to rescue mir-35 is shown as a green line in (A). n = 96–125. # refers to independent transgenic lines. Contingency table values are presented and Fischer exact test applied for statistical evaluation. ***≤0.001, ****≤0.0001. n.s. = not significant.
The mir-35–41 cluster is located within an intron of a worm specific gene (Y62F5A.9) and both nDf50 and gk262 lesions affect exonic sequences of this gene (Fig. 1A). We therefore performed rescue experiments by expressing either mir-35 alone or the entire mir-35–41 cluster under the control of mir-35-locus upstream sequence located within the intron of its host gene (Fig. 1 and Figure S2B–C). We rescued embryonic lethality in both normoxic and hypoxic conditions using these strategies (Fig. 1D and S2C). This indicates that the mir-35 family is required for the hypoxic response of embryos and implies that a single mir-35 family member can rescue phenotypes of mir-35–41 mutant animals, in accordance to previous reports24,27,28. However, we wished to exclude the possibility that the hypoxia-induced embryonic lethality of mir-35–41 mutant embryos is due to general sensitivity of strains that exhibit a high embryonic lethality in ambient O2 conditions. Therefore, we analyzed the hypoxic response of mutants of three unrelated genes (plk-1, spd-2 and mex-1) that exhibit high normoxic embryonic lethality. We exposed embryos of these mutant strains to 0.5% O2 and did not observe any significant hypoxia-induced enhancement of embryonic lethality (Figure S3B, Table S3). These data point to a specific role for the mir-35 family in the embryonic hypoxic response.
Hypoxic induction of the mir-35 family is independent of HIF-1
The mir-35 family is predominantly expressed during embryogenesis24. We confirmed this expression pattern by constructing a transgene using a 602 bp intronic promoter driving expression of yfp fused to a nuclear localization signal (NLS) (Fig. 2). This promoter region was sufficient to rescue the nDf50 hypoxic phenotype when driving the mir-35–41 cluster (Figure S2). We detected YFP expression in virtually all cells starting from ∼20 cell embryos and persisting throughout mid- and late embryogenesis (Fig. 2). We also detected expression in L1 animals but the signal drops in later larval stages. Thus, the rescuing mir-35 intronic promoter drives expression throughout embryogenesis in most, if not all cells.

The mir-35–41 promoter drives ubiquitous expression in the embryo.
A mir-35–41prom::2xNLS::yfp transcriptional reporter drives expression throughout the embryo from the 20 cell stage. The region used to drive yfp expression is shown in yellow on the genomic view (top). Upper panels are Nomarski micrographs and bottom panels are fluorescence images of the same embryos. Anterior is to the left. Scale bar 10 μm.
We have shown that the mir-35 family is required for hypoxic survival of embryos and is expressed throughout embryogenesis. Next, we asked whether the level of each mir-35 family member is regulated by hypoxia. We therefore subjected early embryos to 0.5% O2 for 20 mins or 4 hrs, to test for acute and chronic responsiveness and measured the levels of the mature sequence of each family member by quantitative real-time PCR (qRT-PCR). We found that the expression of the mature sequence of all eight members of the mir-35 family was induced by approximately 2-fold after 4 hrs but not after 20 mins (Fig. 3A–B). Whereas, expression of the Y62F5A.9 host gene is not induced (Figure S4C) and expression of all three reference miRNAs (mir-34, mir-86 and mir-1829c) is stable in these conditions (geNorm M value <0.5, Coefficiency of variation <0.2). Interestingly, the expression of predicted primary and precursor forms of the mir-35–41 locus measured by a previously described qPCR-based method29, using primers flanking the middle member of the cluster (mir-38), is decreased after 4h in hypoxia (Figure S4D). This suggests post-transcriptional regulation of this locus upon hypoxia. As previous studies in C. elegans have shown that HIF-1 stabilization and general downregulation of translation are important requirements for animals to survive hypoxic insults25,30,31, such regulation of the mir-35 family could be dependent on HIF-1 transcription. Therefore we subjected hif-1(ia4) mutant embryos to 4 hrs of 0.5% O2 and quantified induction of the mir-35 family. We still detected induction of mir-35–42 indicating that this is independent of HIF-1 (Figure S4A). Previous work has shown that C. elegans embryos are more sensitive to hypoxia when they are exposed directly to low O2 rather than in utero32. This prompted us to ask whether hypoxia-induced mir-35–42 induction may differ in utero. We therefore, subjected late L4 animals to 0.5% O2 for 15 hrs and developing embryos were subsequently extracted from the gonad. Using qRT-PCR analysis we observed an approximate 4-fold induction in expression of all mir-35 family members except for mir-38 and mir-41 (Figure S4B). These data suggests that post-transcriptional mechanisms within the mother regulate the differential expression of the mir-35 family under hypoxic stress. Taken together, these results show that embryonic expression of the mir-35 family is induced by hypoxia in a hif-1-independent manner and that mir-38 and mir-41 are differentially regulated by hypoxia in utero.

Expression of the mir-35 family is regulated by chronic hypoxia.
(A–B) qRT-PCR showing mir-35 family member expression levels in wild type embryos exposed to 21% O2 (black bars) or 0.5% O2 (orange bars) for 20 mins (A) or 4 hrs (B). The level of normoxic expression was set to 1 for each of the three repetitions. (C) Graphical representation of mir-35 family member abundance in normoxia. mir-38 and mir-41 are less abundant than the other family members, even from those miRNAs transcribed in the same cluster. Values on the graph are logarithmic functions with base 10 of the fold change value for each miRNA. mir-41 showed the lowest relative abundance and was arbitrarily set as the value 1. Primer efficiencies are 116%, 118%, 116%, 113%, 93%, 93%, 109% and 118% for each respective miRNA. (D) Intensity of GFP expression driven by the mir-35–41 promoter is unaffected by 4 hrs of hypoxic exposure. The transgene used is wwIs8[pmir-35–41::GFP + unc-119(+)]. (E–F) gfp transcription, driven by the mir-35–41 promoter in wild type embryos exposed to 21% O2 (black bars) or 0.5% O2 (orange bars) for 20 mins (E) or 4 hrs (F). Data are presented as means of at least 3 independent repetitions and error bars represent ± SD. Students t-test was used to assess for statistical significance. *p ≤ 0.05, **≤0.01, ***p ≤ 0.001, ****≤ 0.0001, n.s. = not significant.
During our qRT-PCR analysis, we observed additional evidence that the mir-35 cluster is potentially post-transcriptionally regulated. We observed that the expression levels of the mature miRNAs differ between members of the mir-35 family. In particular, based on the measured Cq values, mature mir-38 and mir-41 are much less abundant than the other members, 117- and 215-fold less abundant respectively (in accordance with previous reports33) when compared to the average abundance of the other 6 members (Fig. 3C). This observation further supports the possibility that mir-38 and mir-41 are differentially processed after transcription of the locus.
We next asked whether hypoxic induction of the mir-35 family is due to enhanced transcription of the mir-35–41 locus. To answer this question, we used a strain carrying an integrated gfp transgene driven by genomic region upstream of the mir-35 locus34. We subjected embryos of this strain to 0.5% O2 for 4 hrs and found that the intensity of mir-35–41-promoter driven GFP was similar to that of embryos cultivated in normoxia (Fig. 3D). To confirm these data at the RNA level we subjected the same strain to 0.5% O2 for 20 mins or 4 hrs and found that the level of gfp mRNA is not elevated in hypoxia in either condition (Fig. 3E–F). Taken together, these results suggest that the induction of the mir-35 family in hypoxia is controlled by a post-transcriptional mechanism such as increased protection by RNA binding proteins35,36.
sup-26 is a potential mir-35–41 direct target
Although the involvement of mir-35 family in embryonic development has been observed in the past24,37, there is a lack of information as to the downstream regulatory targets of these miRNAs during embryogenesis. Using miRNA target prediction logarithms (mirSOM, TargetScan) we extracted a list of potential direct targets of the mir-35 family. We screened this list for genes that are potentially related to hypoxia based on published literature. We noticed that the human homolog (RBMS1) of one of the candidates, namely SUP-26, is involved in the brain ischemic response38. The 3’UTR of sup-26 contains a sequence complementary to the seed sequence of all the mir-35 family members. In addition, SUP-26 co-purifies with mir-35-42 in embryos as part of miRISC39. SUP-26 is an RNA binding protein that acts in the sex determination pathway in C. elegans40.
As a potential target of the mir-35 family one would expect sup-26 to be expressed during embryogenesis, the period at which the mir-35 family predominates. We therefore generated a transgene using the sup-26 promoter to drive nuclear-localized YFP followed by the native sup-26 untranslated region (UTR). We detected ubiquitous expression of YFP throughout embryogenesis indicating that sup-26 is expressed in a common temporal and spatial window to that of the mir-35 family (Fig. 4).

The sup-26 promoter drives ubiquitous expression in the embryo.
A sup-26prom::2xNLS::yfp transcriptional reporter drives expression throughout the embryo from the 80 cell stage. Region used to drive YFP expression is shown in yellow on the genomic view (top). Upper panels are Nomarski micrographs and bottom panels are fluorescence images of the same embryos. Anterior to the left. Scale bar 10 μm.
To test the possibility that sup-26 is a direct mir-35 target we conducted sensor experiments in a heterologous tissue, the pharynx, after failing to do so in embryos due to specific transgene toxicity (data not shown). We drove expression of mir-35 in the pharynx in combination with a RFP reporter under the control of the unrelated unc-54 3’UTR and a GFP reporter under the control of the sup-26 3’UTR (wild type or mir-35 binding site mutated) (Fig. 5B–D). We found that mir-35 robustly downregulates GFP expression (sup-26 3’UTR) and not the RFP sensor (control 3’UTR) (Fig. 5B–D). Further, regulation via the sup-26 3’UTR is dependent on the predicted mir-35 binding site (Fig. 5B–D) strongly suggesting a direct interaction between mir-35 and the sup-26 3’UTR. Interestingly, a genetic interaction between these two molecules was recently observed independently by the Ambros group41.

sup-26 is a mir-35–41 target and is required for hypoxic survival.
(A) The C. elegans sup-26 3’ UTR contains a single mir-35 family binding site (only mir-35 is shown). The mir-35 seed sequence is shown in blue and the predicted sup-26 3’UTR is shown in red. (B) Sensor experiment constructs. mir-35 was expressed in the pharynx together with a RFP reporter controlled by the unregulated unc-54 3’UTR and a GFP reporter controlled by the sup-26 3’UTR (wild type or mir-35 binding site mutated). The mir-35 binding site in the sup-26 3’UTR was mutated from CCCGGUG to CCatGgG to prevent binding of mir-35 family miRNAs. (C) Representative picture of the sensor experiment results. mir-35 downregulates GFP expression (sup-26 3’UTR) and not the RFP sensor (control 3’UTR). Regulation via the sup-26 3’UTR is dependent on the mir-35 binding site. (D) Quantification of the sensor experiment results. n > 50. Fischer exact test was used for statistical evaluation. # refers to independent transgenic lines. ****≤0.0001.
Finally, we asked whether sup-26 plays a role in the hypoxic response of embryos as one may expect as a potential mir-35 family target. We therefore subjected two independent sup-26 loss-of-function alleles, gk403 and gk426, to embryonic hypoxia (Figure S5). Both mutants exhibited the wild type level of embryonic lethality when exposed to 0.5% O2 for 24 hrs, however, when we exposed embryos to chronic hypoxia (45 hrs), we observed a significant increase in embryonic lethality (Figure S5 and data not shown). This increase was reduced to the wild type levels when the sup-26 product was reintroduced to the sup-26(gk403) mutant (Figure S5B). These data suggest that sup-26 plays a role in the embryonic response to hypoxia. However, since reduced SUP-26 levels resulted in embryonic lethality under low oxygen conditions, we hypothesize that the mir-35 family negatively regulates sup-26 in order to keep its expression to a physiological level, but not to eliminate its expression, for optimal survival of the embryos in hypoxia. Thus, defective regulation of sup-26 levels may be one of the causes of hypoxic lethality exhibited by loss of the mir-35 family. Other mir-35 target genes are also very likely to be involved in this process since the embryonic lethality of the sup-26 mutants is not significantly increased by the milder oxygen conditions (0.5% for 24 hrs).
miRNAs, such as miR-210 were previously shown to play a pivotal role in hypoxic response in mammals and other organisms by regulating key factors and also being regulated by hif-142. Our study offers insights into the role of additional miRNA families in hypoxia in a highly attractive experimental system, the C. elegans embryo and we hope that further work based on these findings will extend our current knowledge on the role of miRNAs in protecting tissues when challenged with decreased oxygen tension.
Methods
Strains used
All strains used in this study are listed in Table S4.
Molecular biology and transgenic lines
The oligonucleotides sequences used in this study are listed in Table S5.
Embryonic lethality in hypoxia
See Supplementary information for details. Briefly, plates containing embryos were placed for 24 hrs at 0.5% O2 (nitrogen balanced). After 24 hrs recovery in ambient O2 conditions, embryonic lethality was scored (Figure S1). For the in utero hypoxia experiment, mid-L4 animals were placed in 0.5% O2 for 15 hrs and eggs were removed from mothers by bleaching followed by immediate RNA extraction.
mRNA isolation and qRT-PCR analysis
miRNA qPCRs were performed as described in42. All qRT-PCR primers were designed using the on line tool described in43. See Supplementary information for further details.
Statistical analysis
See Supplementary information.
Microscopy
Animals were anaesthetized on 5% agarose pads using 20 mM NaN3. Images were taken using a fluorescence microscope and the Zen software (Zeiss, AXIO Imager M2).
Additional Information
How to cite this article: Kagias, K. and Pocock, R. microRNA regulation of the embryonic hypoxic response in Caenorhabditis elegans. Sci. Rep. 5, 11284; doi: 10.1038/srep11284 (2015).
References
Simon, M. C. & Keith, B. The role of oxygen availability in embryonic development and stem cell function. Nat Rev Mol Cell Biol 9, 285–96 (2008).
Semenza, G. L. Oxygen homeostasis. Wiley Interdiscip Rev Syst Biol Med 2, 336–61 (2010).
Pocock, R. Invited review: decoding the microRNA response to hypoxia. Pflugers Archiv : European journal of physiology 461, 307–15 (2011).
Dunwoodie, S. L. The role of hypoxia in development of the Mammalian embryo. Dev Cell 17, 755–73 (2009).
Kagias, K., Nehammer, C. & Pocock, R. Neuronal responses to physiological stress. Front Genet 3, 222 (2012).
Benita, Y. et al. An integrative genomics approach identifies Hypoxia Inducible Factor-1 (HIF-1)-target genes that form the core response to hypoxia. Nucleic Acids Res 37, 4587–602 (2009).
Nallamshetty, S., Chan, S. Y. & Loscalzo, J. Hypoxia: a master regulator of microRNA biogenesis and activity. Free Radic Biol Med 64, 20–30 (2013).
Arsham, A. M., Howell, J. J. & Simon, M. C. A novel hypoxia-inducible factor-independent hypoxic response regulating mammalian target of rapamycin and its targets. J Biol Chem 278, 29655–60 (2003).
Lee, J. & Lee, J. Hypoxia-inducible Factor-1 (HIF-1)-independent hypoxia response of the small heat shock protein hsp-16.1 gene regulated by chromatin-remodeling factors in the nematode Caenorhabditis elegans. J Biol Chem 288, 1582–9 (2013).
Lee, R. C., Feinbaum, R. L. & Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75, 843–54 (1993).
Bartel, D. P. MicroRNAs: target recognition and regulatory functions. Cell 136, 215–33 (2009).
Johnston, R. J. & Hobert, O. A microRNA controlling left/right neuronal asymmetry in Caenorhabditis elegans. Nature 426, 845–9 (2003).
Pedersen, M. E. et al. An epidermal microRNA regulates neuronal migration through control of the cellular glycosylation state. Science 341, 1404–8 (2013).
Zou, Y., Chiu, H., Domenger, D., Chuang, C. F. & Chang, C. The lin-4 microRNA targets the LIN-14 transcription factor to inhibit netrin-mediated axon attraction. Sci Signal 5, ra43 (2012).
de Lencastre, A. et al. MicroRNAs both promote and antagonize longevity in C. elegans. Current biology : CB 20, 2159–68 (2010).
Boulias, K. & Horvitz, H. R. The C. elegans MicroRNA mir-71 Acts in Neurons to Promote Germline-Mediated Longevity through Regulation of DAF-16/FOXO. Cell Metabolism 15, 439–450 (2012).
Shen, Y., Wollam, J., Magner, D., Karalay, O. & Antebi, A. A steroid receptor-microRNA switch regulates life span in response to signals from the gonad. Science 338, 1472–6 (2012).
Kulshreshtha, R. et al. A microRNA signature of hypoxia. Mol Cell Biol 27, 1859–67 (2007).
Ferdin, J. et al. HINCUTs in cancer: hypoxia-induced noncoding ultraconserved transcripts. Cell Death Differ 20, 1675–87 (2013).
Huang, X. & Zuo, J. Emerging roles of miR-210 and other non-coding RNAs in the hypoxic response. Acta Biochim Biophys Sin (Shanghai) 46, 220–32 (2014).
Chatterjee, S. & Grosshans, H. Active turnover modulates mature microRNA activity in Caenorhabditis elegans. Nature 461, 546–9 (2009).
Zhang, Z., Qin, Y. W., Brewer, G. & Jing, Q. MicroRNA degradation and turnover: regulating the regulators. Wiley Interdiscip Rev RNA 3, 593–600 (2012).
Friedman, R. C., Farh, K. K., Burge, C. B. & Bartel, D. P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19, 92–105 (2009).
Alvarez-Saavedra, E. & Horvitz, H. R. Many families of C. elegans microRNAs are not essential for development or viability. Curr Biol 20, 367–73 (2010).
Shen, C., Nettleton, D., Jiang, M., Kim, S. K. & Powell-Coffman, J. A. Roles of the HIF-1 hypoxia-inducible factor during hypoxia response in Caenorhabditis elegans. J Biol Chem 280, 20580–8 (2005).
Jiang, B. et al. Sodium sulfite is a potential hypoxia inducer that mimics hypoxic stress in Caenorhabditis elegans. Journal of Biological Inorganic Chemistry 16, 267–274 (2011).
Massirer, K. B., Perez, S. G., Mondol, V. & Pasquinelli, A. E. The miR-35-41 family of microRNAs regulates RNAi sensitivity in Caenorhabditis elegans. PLoS Genet 8, e1002536 (2012).
Liu, M. et al. mir-35 is involved in intestine cell G1/S transition and germ cell proliferation in C. elegans. Cell Res 21, 1605–18 (2011).
Schmittgen, T. D. et al. Real-time PCR quantification of precursor and mature microRNA. Methods 44, 31–38 (2008).
Jiang, H., Guo, R. & Powell-Coffman, J. A. The Caenorhabditis elegans hif-1 gene encodes a bHLH-PAS protein that is required for adaptation to hypoxia. Proc Natl Acad Sci U S A 98, 7916–21 (2001).
Anderson, L. L., Mao, X., Scott, B. A. & Crowder, C. M. Survival from hypoxia in C. elegans by inactivation of aminoacyl-tRNA synthetases. Science 323, 630–3 (2009).
Miller, D. L. & Roth, M. B. C. elegans are protected from lethal hypoxia by an embryonic diapause. Curr Biol 19, 1233–7 (2009).
Kato, M., de Lencastre, A., Pincus, Z. & Slack, F. J. Dynamic expression of small non-coding RNAs, including novel microRNAs and piRNAs/21U-RNAs, during Caenorhabditis elegans development. Genome Biol 10, R54 (2009).
Martinez, N. J. et al. Genome-scale spatiotemporal analysis of Caenorhabditis elegans microRNA promoter activity. Genome Res 18, 2005–15 (2008).
Pilotte, J., Dupont-Versteegden, E. E. & Vanderklish, P. W. Widespread regulation of miRNA biogenesis at the Dicer step by the cold-inducible RNA-binding protein, RBM3. PLoS One 6, e28446 (2011).
van Kouwenhove, M., Kedde, M. & Agami, R. MicroRNA regulation by RNA-binding proteins and its implications for cancer. Nat Rev Cancer 11, 644–56 (2011).
Brenner, J. L., Kemp, B. J. & Abbott, A. L. The mir-51 family of microRNAs functions in diverse regulatory pathways in Caenorhabditis elegans. PLoS One 7, e37185 (2012).
Doeppner, T. R. et al. The novel proteasome inhibitor BSc2118 protects against cerebral ischaemia through HIF1A accumulation and enhanced angioneurogenesis. Brain 135, 3282–97 (2012).
Wu, E. et al. Pervasive and cooperative deadenylation of 3’UTRs by embryonic microRNA families. Mol Cell 40, 558–70 (2010).
Mapes, J., Chen, J. T., Yu, J. S. & Xue, D. Somatic sex determination in Caenorhabditis elegans is modulated by SUP-26 repression of tra-2 translation. Proc Natl Acad Sci U S A 107, 18022–7 (2010).
McJunkin, K. & Ambros, V. The Embryonic mir-35 Family of MicroRNAs Promotes Multiple Aspects of Fecundity in C. elegans. G3 (Bethesda) (2014).
Kagias, K., Podolska, A. & Pocock, R. Reliable reference miRNAs for quantitative gene expression analysis of stress responses in Caenorhabditis elegans. BMC Genomics 15, 222 (2014).
Gubelmann, C. et al. GETPrime: a gene- or transcript-specific primer database for quantitative real-time PCR. Database (Oxford) 2011, bar040 (2011).
Acknowledgements
We thank members of Pocock lab, Anders Lund and Lisa Salcini for comments on the manuscript, Agnieszka Podolska and Camilla Nehammer for the outcrossing of some miRNA mutant strains, Mikael Pedersen for injection help and the Caenorhabditis Genetics Center (CGC), which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440), for providing worm strains. This work was supported by the European Research Council (ERC) Starting Grant number 260807 to R.P.
Author information
Authors and Affiliations
Contributions
K.K. and R.P. designed the project. K.K. performed the experiments. K.K. and R.P. wrote the paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Kagias, K., Pocock, R. microRNA regulation of the embryonic hypoxic response in Caenorhabditis elegans. Sci Rep 5, 11284 (2015). https://doi.org/10.1038/srep11284
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep11284
- Springer Nature Limited