
Abstract
Achieving methane-to-methanol is challenging under mild conditions. In this study, methanol is synthesized by one-step direction conversion of CH4 with H2O at room temperature under atmospheric pressure in non-thermal plasma (NTP). This route is characterized by the use of methane and liquid water as the reactants, which enables the transfer of the methanol product to the liquid phase in time to inhibit its further decomposition and conversion. Therefore, the obtained product is free of carbon dioxide. The reaction products include gas and liquid-phase hydrocarbons, CO, CH3OH, and C2H5OH. The combination of plasma and semiconductor materials increases the production rate of methanol. In addition, the addition of Ar or He considerably increases the production rate and selectivity of methanol. The highest production rate of methanol and selectivity in liquid phase can reach 56.7 mmol gcat−1 h−1 and 93%, respectively. Compared with the absence of a catalyst and added gas, a more than 5-fold increase in the methanol production rate is achieved.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Introduction
Methanol is an important raw material for chemical production, and the direct conversion of methane into methanol has commercial value. Currently, the industrial process to synthesize methanol involves the use of syngas1, which is usually prepared by methane reforming2,3, but the cost of this process is high4. The direct one-step oxidation of methane to methanol is more advantageous than the syngas route. Noble metals (e.g., Au, Pd, and Rh) have been reported to exhibit excellent catalytic properties for the conversion of methane5,6,7,8,9,10. Agarwal et al.5 oxidized methane to methanol using Pd–Au colloidal nanoparticles in the presence of H2O2 and O2. Xiao et al.7 designed a “molecular fence” catalyst synthesized by fixation of AuPd alloy nanoparticles within aluminosilicate zeolite crystals followed by modification of the external zeolite surface with organosilanes. Methane was converted into methanol with high efficiency, with a production rate of 91.6 mmol gAuPd−1 h−1. In addition, some non-noble metals have also been reported to show potential for this reaction. Inspired by the biocatalysis of methane monooxygenase (MMOS)11,12, Grundner et al.13 prepared a Cu-MOR catalyst by ion exchange, with active sites similar to those present in MMOS. The conversion of methane to methanol was realized by multiple reaction steps (activating active sites under an O2 atmosphere, methane reaction, and methanol hydrolysis). Except for intermittent reactions, Narsimhan et al.14 reported the simultaneous passage of methane, oxygen, and water into the reactor for the reaction. Although the reported conversion was not high, the oxidation of methane to methanol under continuous conditions was realized for the first time.
Besides the complexity of the operation process, a high reaction temperature is employed due to the intrinsic characteristics of methane, which is difficult to activate15,16,17, and the oxidation of the product methanol is easier than that of methane, typically leading to the continuous oxidation of methanol to CO2 under the reaction conditions. Hence, unsatisfactory results are obtained18,19. NTP is a form of plasma with the characteristics of low temperature and high-energy electrons20. Therefore, the introduction of NTP into the reaction system can convert some difficult-to-activate molecules into active groups21 and considerably accelerate the reaction speed. Nevertheless, the inhibition of the over-oxidation of methane is still an urgent issue.
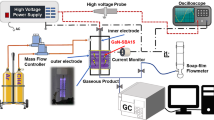
In this study, a new route to synthesize methanol from methane using NTP is designed, which reasonably and completely exploits the catalytic characteristics of plasma. At room temperature under atmospheric pressure, the formation of methanol is realized by directly mixing CH4 and a weak oxidant H2O without cumbersome operational steps. The reaction between CH4 and H2O to afford methanol is performed in a dielectric barrier discharge (DBD) reactor (Fig. 1). The reaction environment involves three parts: the plasma phase, the liquid phase, and the plasma-liquid interface. Each of these parts corresponds to the formation of methyl radicals (CH3·), formation of hydroxyl radicals (·OH), and production of methanol. Methane in the plasma phase is activated by inelastic collision with high-energy electrons, becomes an excited state (CH4*), and is decomposed to methyl radicals (CH3·). These activated molecules and radicals contribute predominantly to methanol production. Liquid-phase H2O provides the required ·OH for the reaction. By constructing a TiO2 surface heterojunction in the liquid phase and using the photons and electrons22 produced in the plasma, the formation of hydroxyl radicals is significantly promoted. In addition, the liquid-phase environment is conducive to the instantaneous transfer of methanol produced at the liquid-plasma interface, inhibiting the decomposition and transformation of methanol.

The reaction between CH4 and H2O to afford methanol is performed in a dielectric barrier discharge (DBD) reactor.
Results and discussion
Catalytic performance
Methanol can be produced in all experiments by this route, and it is crucial that CO2 is not produced. The presence of a small amount of methanol is detected under the plasma-only mode (Fig. 2). On the one hand, the CH3· produced by CH4 activation can directly react with H2O to form methanol; on the other hand, the plasma decomposes H2O molecules when they reach the phase interface, and the gas phase comprises a small amount of water vapor, which is also converted into ·OH by high-energy electrons. Clearly, the addition of the TiO2 catalyst in the DBD plasma enhances the production rate of methanol to 29.7 mmol gcat−1 h−1 (Supplementary Fig. 1). Compared with the absence of a catalyst, a 3-fold increase in the methanol production rate is achieved. The selectivity of gas phase and liquid phase products are shown in Table S1, it can be seen that 4.5–12.7% of methane is converted to methanol.

Effects of different catalysts on the reaction (CH4 flow rate of 5 mL min−1, discharge power of 30 W, ca. 5 mg catalyst, error bars obtained from repeated three sets of experiments on the same catalyst).
X-ray diffraction (XRD) was employed to characterize the chemical structures of a series of TiO2-t samples. Peaks at 25.3°, 37.8°, 48.0°, and 62.7° correspond to the characteristic diffraction peaks of anatase, while those at 27.4°, 36.1°, 41.2°, and 54.3° correspond to those of rutile. The XRD spectra reveal that the catalyst contains both anatase and rutile and that the rutile is the main crystal form (Supplementary Fig. 2). High-resolution transmission electron microscopy (HRTEM) reveals that the TiO2-700 shows crystal plane spacings at ~2.44 and 3.52 Å, corresponding to the (101) crystal plane of the rutile phase and the (101) crystal plane of the anatase phase (Fig. 3f), respectively. This is consistent with the XRD analysis. This catalyst is advantageous as the hybrid catalyst consisting of anatase and rutile can form a heterojunction. Hence, electrons and holes can be effectively separated and lead to better catalyst performance. The surface phase composition of TiO2 is the key factor that affects the catalytic performance21. Visible Raman and UV Raman spectra (Fig. 3a, b) reveal the presence of anatase and rutile phase junctions on the catalyst surface, and the characteristic peak value of anatase in the TiO2-700 sample decreases, indicating that the surface phase changes. The samples show similar band gaps in the UV–Vis spectra (Fig. 3c), but the light absorption intensity of TiO2 gradually increases with increasing treatment temperature, corresponding to the maximum at 700 °C. The X-ray photoelectron spectroscopy valence band (XPS-VB) spectra (Fig. 3d) reveal that the valence band energy of each sample after treatment gradually increases and that the peak value is attained at 700 °C. Figure 3e shows a schematic of the band-gap structure. The VB energy value of 2.43 eV for unprocessed raw powder TiO2 changes to 2.69 eV for TiO2 treated at 700 °C, indicative of the improved oxidation performance of the holes formed by the treated catalyst. The catalytic performance from experiments is consistent with the characterization results. With the increase in the treatment temperature, the production rate of methanol increases to the peak value and then decreases, and TiO2-700 exhibits excellent performance.

a Visible Raman spectra, b UV Raman spectra, c UV–Vis spectra, d XPS spectra showing the valence band (VB) levels, e schematic of the band-gap structure, and f HRTEM image of the TiO2-700 sample.
Possible reaction pathways
Figure 4 shows the possible reaction pathways for the formation of methanol and ethanol. Isotope tracer experiments were performed with D2O and analyzed by gas chromatography-mass spectrometry (GC-MS) (Fig. 5). The GC-MS spectra reveal that most of the produced methanol is CH3OD and that few molecules are CH3OH. The results reveal that the methanol produced by the reaction mainly originates from the combination of methyl radicals (CH3·) and hydroxyl radicals (·OH) (Eq. 10). Moreover, few CH3· radicals combine with ·O to form CH3O· and then combine with H· to form CH3OH (Eqs. 7-9). Experimental results show that in addition to CH3OH, C2H5OH is a main by-product. Therefore, increasing the CH3OH production rate and making efficient use of CH3· and ·OH are the key issues for improving methanol production rate and selectivity.

The methanol produced by the reaction mainly originates from the combination of methyl radicals (CH3·) and hydroxyl radicals (·OH).

The GC-MS spectra reveal that most of the produced methanol is CH3OD and that few molecules are CH3OH.
Effect of adding inert gas on methanol production rate
The addition of an inert gas (e.g., Ar, He) to the reaction feed to improve the reaction performance was explored from two aspects. On the one hand, in DBD plasma, CH3· radicals constitute the main free radicals produced from CH423, but other free radicals are also present (e.g., CH2· and CH·) (Eqs. 1–3). At this time, due to the dilution of the original radical composition by Ar or He, the self-coupling of methyl radicals is reduced (Eq. 4). Hence, the produced methyl radicals are utilized for methanol synthesis to a greater extent, and the selectivity of methanol is subsequently considerably improved. On the other hand, the added Ar or He gas can produce metastable atoms with a longer average life span, thereby leading to Penning ionization. Ar or He activated by electrons transfers its energy to CH4 for the activation of an increased number of CH4 molecules24 (Eqs. 15, 16). The formation of CH3· from CH4 in a plasma is caused by the energetic electron-initiated dissociation of CH4 (bond dissociation energy: 4.52 eV). Furthermore, ·OH can be formed by the electron impact dissociation of H2O (bond dissociation energy: 5.19 eV). Therefore, the average electron energy in a DBD plasma is a key factor affecting the reaction. The electrons in the high-energy tail in the Maxwellian distribution are responsible for the dissociation of CH4 and H2O. Introducing He or Ar can increase the average electron energy in the reactor, which leads to higher CH4 and H2O conversions. The addition of Ar or He considerably increased the production rate of methanol. With the addition of Ar, the production rate reached 58.1 mmol gcat−1 h−1 (Fig. 6a). Compared to previous studies, this system exhibits an excellent methanol production rate, and most of the previous studies used O2 or H2O2 as the oxidant (Table 1). With the addition of He, the production rate reaches 56.7 mmol gcat−1 h−1. Although the production rate increase with He is slightly lower than that with Ar, the selectivity of methanol clearly increases from 65 to 93% in the liquid products under CH4/He plasma. After the addition of Ar or He, the plasma discharge current waveform was monitored (Supplementary Fig. 3). The addition of Ar increases the electron density25, which in turn increases the collision probability between electrons and CH4 in the reaction. As a result, an increased number of CH4 molecules are activated and transformed (Supplementary Fig. 4). The addition of He leads to an increase in the average electron energy in the plasma region but reduces the electron density of the system26. Hence, the CH4 conversion increase is less than that with the addition of Ar.

a Effect of the addition of He or Ar on the reaction at total flow rate of 15 mL min−1. b Effect of the He/CH4 feed ratio on the reaction at CH4 flow rate 5 mL min−1 (discharge power of 30 W, ca. 5 mg TiO2-700 catalyst, error bars obtained from repeated three sets of experiments on the same catalyst).
Effect of the flow ratio of He/CH4 on the methanol production rate
The addition of He improves methanol production rate as well as methanol selectivity. Next, the effects of the addition of different amounts of He on the methanol production rate and selectivity were investigated (Fig. 6b). The results reveal that the selectivity of methanol increases with the increase in He addition. At a He/CH4 ratio of 6:1, the selectivity of methanol reaches 96%. Simultaneously, with the increase in the He/CH4 feed ratio, the methanol production rate initially increases and reaches a peak at a He/CH4 feed ratio of 2:1. A further increase in the He/CH4 ratio leads to a decrease in methanol production rate due to a possible decrease in the gas–liquid contact time by the excess airspeed.
In conclusion, a new route for the direct conversion of methane to methanol in one step at room temperature under atmospheric pressure was designed and demonstrated with simple operation. The entire reaction was further tailored by considering two aspects: CH3· production and ·OH production. A methanol production rate of 56.7 mmol gcat−1 h−1 and a selectivity of 93% in the liquid products were obtained, and CO2 formation was not observed, successfully inhibiting the over-oxidation of methane. This designed reaction system may also be applied for the selective oxidation of other stable chemicals such as benzene to phenol.
Methods
Catalyst preparation
Purchased TiO2 powders were ground in an agate mortar, then placed in a crucible and transferred to a muffle furnace. The TiO2 samples were heat treated in an air atmosphere at 500 °C, 600 °C, 700 °C, or 800 °C for 4 h to obtain a series of catalysts, which were recorded as TiO2-t, where t is the calcination temperature.
Catalyst characterization
Powder X-ray diffraction (XRD) patterns were measured using a Rigaku Smart Lab diffractometer, with a nickel-filtered Cu Kα X-ray source, at a scanning rate of 0.02° over a 2θ range of 5° to 80°. Transmission electron microscopy (TEM) tests were performed using a Tecnai G2 20 S-twin instrument (FEI Co.) with an accelerating voltage of 200 kV. TEM samples were ultrasonicated in ethanol, dropped onto carbon-coated copper mesh, and dried under ambient conditions. High-resolution transmission electron microscopy (HRTEM) measurements were obtained to determine the lattice spacing of the TiO2 for a comprehensive understanding of the crystal structure. X-ray photoelectron spectroscopy (XPS, Thermo Scientific K-Alpha), with an Al Kα excitation source (hν = 1486.6 eV), was used to determine core level binding energies of surface species. Sample charging was corrected by referencing all measurements to the C (1s) peak at 284.8 eV. Raman spectra were recorded with a Horiba Scientific LabRAM HR Evolution spectrograph using 532 and 325 nm laser lines. UV–Vis absorption spectra were obtained in the range of 200–800 nm using a UV-2600 spectrophotometer. The methanol product in the isotope-labeled reaction was analyzed by a gas chromatograph-mass spectrometer (GCMS) using an Agilent GCMS 7890B-5977B equipped with an HP-5ms chromatographic column.
Experimental
Supplementary Fig. 5 shows a process flow diagram of the reaction system. 5 mg catalyst was ultrasonically dispersed in 3 mL H2O and placed in a dielectric barrier discharge (DBD) reactor. The details of the plasma reaction system are shown in Supplementary Fig. 6. The methane feed into the reactor was controlled by a mass flow meter at a constant rate of 5 mL min−1. The plasma was generated using a high-voltage power supply, and the reaction was allowed to proceed for 20 min. The plasma power (30 W) is the input power and is calculated by multiplying the input voltage (50 V) and the input current (0.6 A). In addition, a Lissajous figure was measured with 30 W input power using an oscilloscope. Supplementary Fig. 7 shows the Lissajous figure at an input power of 30 W. According to Supplementary Fig. 7, the actual output power corresponding to the input power of 30 W can be calculated to be 22.4 W. The applied voltage peak to peak (Vpk-pk) was approximately 27 kV (Supplementary Fig. 8). The reaction solution was filtered and then analyzed by gas chromatography to determine the product composition. Methanol and ethanol were analyzed by a gas chromatograph (GC) equipped with an FID detector and a Porapak Q column. The standard calibration curves of the peak areas and methanol and ethanol concentrations are shown in Supplementary Figs. 9 and 10. Methane was analyzed using a GC equipped with an FID detector and a 30 m PLOT-Q capillary column. CO2 was analyzed using a TCD detector and a TDX-01 molecular sieve column.
Calculation method of discharge power
We calculated the average power by finding the area under the curve for the V–Q Lissajous plot and multiplying it by the frequency.
where P is the average power in W, v(t) is the voltage measured by the oscilloscope, q(t) is the charge measured by the oscilloscope in C, f is the frequency in kHz, and S is the area under the curve for one cycle.
Data availability
The authors declare that the/all other data supporting the findings of this study are available within the paper and its supplementary information files.
References
Wu, C.-T. et al. A non-syn-gas catalytic route to methanol production. Nat. Commun. 3, 1–8 (2012).
Guo, D. et al. Effects of extrinsic defects originating from the interfacial reaction of CeO2−x-nickel silicate on catalytic performance in methane dry reforming. Appl. Catal. B-Environ. 277, 119278 (2020).
Kurlov, A. et al. Exploiting two-dimensional morphology of molybdenum oxycarbide to enable efficient catalytic dry reforming of methane. Nat. Commun. 11, 4920 (2020).
Behrens, M. et al. The active site of methanol synthesis over Cu/ZnO/Al2O3 industrial catalysts. Science 336, 893–897 (2012).
Agarwal, N. et al. Aqueous Au–Pd colloids catalyze selective CH4 oxidation to CH3OH with O2 under mild conditions. Science 358, 223–227 (2017).
Williams, C. et al. Selective oxidation of methane to methanol using supported AuPd catalysts prepared by stabilizer-free sol-immobilization. ACS Catal. 8, 2567–2576 (2018).
Jin, Z. et al. Hydrophobic zeolite modification for in situ peroxide formation in methane oxidation to methanol. Science 367, 193–197 (2020).
Rahim, M. H. A. et al. Oxidation of methane to methanol with hydrogen peroxide using supported gold–palladium alloy nanoparticles. Angew. Chem. Int. Ed. 52, 1280–1284 (2013).
Tang, Y. et al. Single rhodium atoms anchored in micropores for efficient transformation of methane under mild conditions. Nat. Commun. 9, 1–11 (2018).
Luo, L. H. et al. Water enables mild oxidation of methane to methanol on gold single-atom catalysts. Nat. Commun. 12, 1218 (2021).
Blanchette, C. D. et al. Printable enzyme-embedded materials for methane to methanol conversion. Nat. Commun. 7, 1–9 (2016).
Ro, S. Y. et al. Native top-down mass spectrometry provides insights into the copper centers of membranebound methane monooxygenase. Nat. Commun. 7, 1–9 (2016).
Grundner, S. et al. Single-site trinuclear copper oxygen clusters in mordenite for selective conversion of methane to methanol. Nat. Commun. 6, 7546 (2015).
Narsimhan, K., Iyoki, K., Dinh, K. & Román-Leshkov, Y. Catalytic oxidation of methane into methanol over copper-exchanged zeolites with oxygen at low temperature. ACS Cent. Sci. 2, 424–429 (2016).
Schwarz, H. Chemistry with methane: Concepts rather than recipes. Angew. Chem. Int. Ed. 50, 10096–10115 (2011).
Chan, S. I. et al. Efficient oxidation of methane to methanol by dioxygen mediated by tricopper clusters. Angew. Chem. Int. Ed. 52, 3731–3735 (2013).
Guo, X. et al. Direct, nonoxidative conversion of methane to ethylene, aromatics, and hydrogen. Science 344, 616–619 (2014).
Olivos-Suarez, A. I. et al. Strategies for the direct catalytic valorization of methane using heterogeneous catalysis: Challenges and opportunities. ACS Catal. 6, 2965–2981 (2016).
Ravi, M., Ranocchiari, M. & Bokhoven, J. A. V. The direct catalytic oxidation of methane to methanol—a critical assessment. Angew. Chem. Int. Ed. 56, 16464–16483 (2017).
Zhu, B. et al. TiO2-supported Au–Ag plasmonic nanocatalysts achieved by plasma restructuring and activation. ACS Catal. 7, 6514–6524 (2017).
Zhang, J., Xu, Q., Feng, Z. C., Li, M. J. & Li, C. Importance of the relationship between surface phases and photocatalytic activity of TiO2. Angew. Chem. Int. Ed. 47, 1766–1769 (2008).
Hawtof, R. et al. Catalyst-free, highly selective synthesis of ammonia from nitrogen and water by a plasma electrolytic system. Sci. Adv. 5, eaat5778 (2019).
Wang, L., Yi, Y., Wu, C., Guo, H. & Tu, X. One-step reforming of CO2 and CH4 into high-value liquid chemicals and fuels at room temperature by plasma-driven catalysis. Angew. Chem. Int. Ed. 129, 13867–13871 (2017).
Goujard, V., Tatibouët, J.–M. & Batiot-Dupeyrat, C. Carbon dioxide reforming of methane using a dielectric barrier discharge reactor: Effect of helium dilution and kinetic model. Plasma Chem. Plasma Process. 31, 315–325 (2011).
Moss, M. S., Yanallah, K., Allen, R. W. K. & Pontiga, F. An investigation of CO2 splitting using nanosecond pulsed corona discharge: Effect of argon addition on CO2 conversion and energy efficiency. Plasma Sources Sci. Technol. 26, 035009 (2017).
Ramakers, M., Michielsen, I., Aerts, R., Meynen, V. & Bogaerts, A. Effect of argon or helium on the CO2 conversion in a dielectric barrier discharge. Plasma Process Polym. 12, 755–763 (2015).
Tomkins, P. et al. Isothermal cyclic conversion of methane into methanol over copper-exchanged zeolite at low temperature. Angew. Chem. Int. Ed. 55, 5467–5471 (2016).
Shan, J., Li, M., Allard, L. F., Lee, S. & Flytzani-Stephanopoulos, M. Mild oxidation of methane to methanol or acetic acid on supported isolated rhodium catalysts. Letter 551, 605–608 (2017).
Hammond, C. et al. Direct catalytic conversion of methane to methanol in an aqueous medium by using copper-promoted Fe-ZSM-5. Angew. Chem. Int. Ed. 51, 5129–5133 (2012).
Shen, Q. K. et al. Single chromium atoms supported on titanium dioxide nanoparticles for synergic catalytic methane conversion under mild conditions. Angew. Chem. Int. Ed. 59, 1216–1219 (2020).
Kwon, Y., Kim, T. Y., Kwon, G., Yi, J. & Lee, H. Selective activation of methane on single-atom catalyst of rhodium dispersed on zirconia for direct conversion. J. Am. Chem. Soc. 139, 17694–17699 (2017).
Xie, J. et al. Highly selective oxidation of methane to methanol at ambient conditions by titanium dioxide supported iron species. Nat. Catal. 1, 889–896 (2018).
Author information
Authors and Affiliations
Contributions
W.B. synthesized, characterized, and tested the catalysts and drafted the manuscript. Y.T. helped supplement experiments and revise the manuscript. X.L. helped perform the analysis with discussion. C.D. and X.M. provided study resources and financial support, guided the experimental ideas and content, and revised the manuscript. C.S. and X.G. revised the manuscript for important intellectual content and provided constructive views and recommendations.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Chemistry thanks Kui Zhang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bi, W., Tang, Y., Li, X. et al. One-step direct conversion of methane to methanol with water in non-thermal plasma. Commun Chem 5, 124 (2022). https://doi.org/10.1038/s42004-022-00735-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-022-00735-y
- Springer Nature Limited