
Abstract
Porcine epidemic diarrhea virus (PEDV) is a serious disease that poses a significant threat to the pig industry. This study focused on analyzing the Spike protein of PEDV, which harbors crucial antigenic determinants, in identifying dominant epitopes. Immunoinformatics tools were used to screen for B-cell, CD4+ and CD8+ predominance epitopes. These epitopes were then connected to the N-terminal of ferritin to form a self-assembled nanoparticle vaccine. Various physical and chemical properties of the candidate vaccine were analyzed, including secondary structure prediction, tertiary structure modeling, molecular docking, immune response simulation and computer cloning. The results demonstrated that the candidate vaccine was antigenic, soluble, stable, non-allergic, and formed a stable complex with the target receptor TLR-3. Immune simulation analysis showed that the candidate vaccine effectively stimulated both cellular and humoral reactions, leading to increased related cytokines production. Furthermore, efficient and stable expression of the candidate vaccine was achieved through reverse translation in the Escherichia coli K12 expression system following codon optimization and in silico cloning. The developed nanoparticle candidate vaccine in this study holds promise as an effective PEDV vaccine candidate, offering a new approach for the research, development and improvement of vaccines targeting porcine enteric diarrhea coronavirus.
Similar content being viewed by others

Introduction
Porcine epidemic diarrhea (PED) is an acute intestinal infection caused by the porcine epidemic diarrhea virus (PEDV)1. It affects pigs of all ages, manifesting primarily through symptoms such as watery diarrhea, vomiting, anorexia, dehydration, and weight loss, particularly in piglets. The mortality rate can reach up to 100%, resulting in significant economic losses within the global pig-intensive farming industry2. Extensive research has been conducted on PEDV, focusing on its pathogenic mechanisms, diagnostic approaches, and detection techniques. However, the virus continues to evolve and mutate in nature, giving rise to new strains and frequently triggering fresh outbreaks2,3. Vaccination stands as a crucial measure in preventing and managing infectious diseases, providing protection for the body and enhancing specific immunity4. Presently, various vaccines are tailored to combat PEDV, including whole virus-inactivated vaccines and improved live vaccines5,6. While the inactivated vaccines boast safety and a straightforward production process, they solely induce humoral immunity. Conversely, live attenuated vaccines provoke robust cellular and humoral immune responses but pose the risk of virulence reversal7. Hence, it is imperative to vigorously advocate for the development and enhancement of novel vaccines to combat PEDV effectively.
PEDV is classified within the genus Alpha Coronavirus in the Coronaviridae family and falls under the order Nidovirales. It is characterized as a single-stranded RNA virus with a plus-stranded genome, measuring approximately 28 kb in size5. The virus contains seven open reading frameworks (ORFs) encoding four structural proteins: spiking (S), capsular (M), envelope (E) and nucleocapsid (N). Additionally, it encodes three non-structural proteins: open reading frame 1a/b and ORF36. The S protein, present as a trimeric glycoprotein on the viral surface, assumes a pivotal role in viral functionality4. It consists of two domains, S1 and S2, which facilitate virus entry, attachment to host cell receptors, and the induction of neutralizing antibody production. The S2 domain is responsible for membrane fusion and viral replication8. The S protein also determines the tissue tropism and host range of the virus, making it an attractive target for attenuated coronavirus and subunit vaccines9. However, employing a full-length S protein vaccine may elicit harmful immune responses and toxic side effects10. To mitigate this concern, the screening and design of dominant epitopes recognized by T and B cells can be effectively achieved through immunoinformatics11. Moreover, enhancing the antigenicity of these epitopes can be achieved through efficient delivery platforms. Ferritin, a self-assembled, symmetrical octahedral protein, is ideal for multivalently displaying antigens in highly ordered and repetitive arrays12. This study aims to control the sustained spread of PEDV by constructing a self-assembled nanoparticle vaccine targeting the dominant epitope of the S protein.
Materials and methods
Based on theoretical guidance, the study mainly described the prediction of PEDV S protein dominant epitopes (B cells, CD4+ and CD8+), vaccine design, 3D structural modeling, molecular docking, protein expression and immune simulation summary, and the results were shown in Fig. 1.

The flow chart was used to describe the screening of the PEDV S protein dominant epitope and the construction of a nanoparticle candidate vaccine (Biorender drawn).
T cell and B cell epitope prediction
The PEDV-S protein (GenBankID: AAK38656) was retrieved in FASTA file format from the NCBI database (https://www.ncbi.nlm.nih.gov/protein) to serve as a template for cellular epitope prediction. For CD4+ epitope prediction, the NetMHCIIpan-4.0 server (https://services.healthtech.dtu.dk/service.php?NetMHCIIpan-4.0) was utilized, with adjustments made to the epitope length to ensure a strong binding epitope of 1513. Subsequently, the induction of IFN-γ and IL-4 was evaluated using the IFNepitope tool (https://webs.iiitd.edu.in/raghava/ifnepitope/index.php) and IL-4pred (http://crdd.osdd.net/raghava/il4pred/)14,15. CD8+ epitopes were predicted using the NetMHCpanEL4.1 server (http://tools.iedb.org/mhci/) and the NetMHCpan4.1 server (https://services.healthtech.dtu.dk/service.php?NetMHCpan-4.1)16,17. Epitope length was adjusted to 14, with the allele threshold set to approach 0, and binding sequences ranked in the top 5% based on their scores. B cell epitopes were predicted using the ABCpred server (https://webs.iiitd.edu.in/raghava/abcpred/ABC_submission.html) and the PEPTIDES server (http://imed.med.ucm.es/Tools/antigenic.html)18,19. For ABCpred, the length was adjusted to 16, and the threshold was set to 0.8. PEPTIDES performed predictive analysis on the entire amino acid sequence. All predicted epitopes were further evaluated for immunogenicity and toxicity using the IEDBMHC-I server (http://tools.iedb.org/immunogenicity/) and the ToxinPred server (https://webs.iiitd.edu.in/raghava/toxinpred/design.php)20,21. The final result consisted of non-toxic epitopes with immunogenicity scores greater than 0.25.
Screening and conservativeness analysis of dominant epitopes
The B cell epitopes, CD4+ epitopes, and CD8+ epitopes generated by the server underwent comparative screening analysis. Overlapping segments among these epitopes were identified and selected as dominant epitopes. The conservation of these dominant epitopes was evaluated using the IEDB server (http://tools.iedb.org/conservancy/) and 169 PDCoV-S protein amino acid sequences retrieved from the NCBI database22 (refer to Supplementary Table S1 for detailed information of GenBankID). Analysis parameters were set to ‘linear’ for the analysis type and ‘100%’ for the sequence identification threshold, with identical template sequences removed. The PEDV-S protein crystal structure (PDBID:7W6M) was selected from the PDB database (https://www.rcsb.org), and spatial mapping of the dominant epitopes was carried out using Pymol.
Vaccine design and evaluation
CD8+, CD4+, and B-cell dominant epitopes were tandemly linked using AAY, GPGPG and KK flexlinker, respectively, as previously described23. The C-terminus of the construct was ligated to ferritin (GenBank: WP_000949190) via (GGGGS)3 linker to present a multivalent display of the dominant epitope. The candidate vaccine was analyzed for antigenicity using the ANTIGENpro server (http://scratch.proteomics.ics.uci.edu/)24, while solubility during overexpression was assessed using the SOLpro server (http://scratch.proteomics.ics.uci.edu/index.html)24. Allergenicity was determined using the AllerTop Server (http://www.ddg-pharmfac.net/AllerTOP/)25, hydrophobicity was examined using the ProtScale server (https://web.expasy.org/cgi-bin/protscale/protscale.pl#opennewwindow)26, and transmembrane structural domains was performed using the TMHMM-2.0 server (https://services.healthtech.dtu.dk/services/TMHMM-2.0/)27. Additionally, physicochemical properties were assessed using the ExPASy server (https://web.expasy.org/protparam/)26, while the secondary structure was analyzed using the PSIPRED 4.0 server (http://bioinf.cs.ucl.ac.uk/psipred)28.
Vaccine tertiary structure prediction, refinement and validation
The candidate vaccine is modeled using the trRosetta server (https://yanglab.nankai.edu.cn/trRosetta/)29. Subsequently, the initial model was refined using the GalaxyRefine server (http://galaxy.seoklab.org/cgi-bin/submit.cgi?)30. Additionally, the quality of the model was assessed and compared using the PROCHECK server (https://saves.mbi.ucla.edu/) and authenticated using the ProSA-web server (https://prosa.services.came.sbg.ac.at/prosa.php)31,32.
Molecular docking, immunosimulation and computerized cloning
In this study, TLR-3 (PDBID:2A0Z) and the candidate vaccine were subjected to molecular docking using the pyDockWEB server (https://life.bsc.es/pid/pydockweb)33. The resulting complex was visualized using LigPlot+ to analyze the two-dimensional interworking interface. Immunization simulations were conducted using the C-ImmSim server (https://150.146.2.1/C-IMMSIM/index.php)34 with step-time settings of 1, 25 and 50, while other parameters were kept at their default values to assess the candidate vaccine's efficacy in inducing both humoral and cellular immunity. Codon optimization and reverse translation of the candidate vaccine amino acid sequences were accomplished using the JCat server (http://www.jcat.de/)35. The ExpOptimizer server (https://www.novopro.cn/tools/codon-optimization.html) (NOVOPRO, China) validated CAI values against GC content. Visualization of restriction endonuclease sites was facilitated using SnapGene, followed by their insertion into the adapted expression vector system.
Results
Screening of S protein dominant epitopes of PEDV
In order to enhance the reliability of the dominant epitope screening strategy, this study employed multiple immunological tools to analyze the same dominant epitope. The results revealed that all dominant epitopes were non-toxic and displayed high immunogenicity, as summarized in Table 1. A total of 5 B-cell dominant epitopes were obtained through ABCpred and PEPTIDES screening. Additionally, 3 CD8+ cell dominant epitopes were obtained using NetMHCpanEL4.1 and NetMHCpan4.1, while 3 CD4+ cell dominant epitopes were obtained through NetMHCIIpan-4.0 followed by IFNepitope and IL-4pred analysis. The B-cell epitopes, denoted as B1, B2, B3, B4 and B5, exhibited minimum identities of 93. 75%, 75.00%, 75.00%, 81.25% and 80.00%, respectively. Similarly, the CD4+ epitopes (CD4-1, CD4-2, and CD4-3) and CD8+ epitopes (CD8-1, CD8-2, and CD8-3) displayed minimum identities of 53.33%, 93.33%, 78.57%, 64.29% and 92.86%, respectively. These results indicate a relatively high level of identity for the dominant epitopes. Spatial mapping of the dominant epitopes in PyMOL revealed their presence on the surface of the S protein, as depicted in Fig. 2. These short surface peptides contribute to antigenic chimerism and stimulate the immune response. However, the exact spatial location of the CD4-3 and CD8-3 epitopes remains unclear due to the incomplete resolution of the crystal structure of the S protein (detailed data are shown in Supplementary Table S2).

Spatial mapping of dominant epitopes of PDDV-S protein. (A) PEDV structure; (B) S protein tertiary structure; (C) Red labeled B cell epitope, green labeled CD4+ epitope, purple labeled CD8+ epitope.
Construction of PEDV-S protein vaccine
Based on the results of the screening analysis, we used KK, AAY, GPGPG, and (GGGGS)3 flexible linkers to connect B cell, CD8 and CD4 epitopes in a specific order to construct a candidate vaccine (detailed data are shown in Supplementary Table S3). To enhance the immunogenicity of the candidate vaccine, a double lysine KK linker was utilized for the B cell dominant epitope to maintain its independent immunogenic activity. Additionally, AAY and GPGPG linkers were used for the T cell dominant epitope to improve the recognition of candidate vaccine subunits. The (GGGGS)3 linker was utilized to connect epitopes to ferritin, ensuring stability and proper folding rate. Our analysis revealed that using a single GGGGS flexible linker resulted in low protein solubility during the construction of the candidate vaccine. However, when we introduced (GGGGS)3, protein solubility significantly increased (amino acid sequence in Fig. 3C).

Results of candidate vaccine evaluation. (A) Candidate vaccine hydrophobicity prediction; (B) Candidate vaccine transmembrane domain prediction; (C) Candidate vaccine secondary structure analysis.
Evaluation of the candidate vaccine of PEDV-S protein
According to ANTIGENpro, the candidate vaccine exhibited a predicted immunogenicity of 0.606918. Additionally, SOLpro predicted an overexpression solubility of 0.584405 for the candidate. AllerTOPV2.0 classified the vaccine candidate as a non-sensitizing protein. ExPASy analysis revealed that the candidate vaccine has a full length of 374 amino acids, a molecular weight of 40.89 kDa, an isoelectric point of 5.48 and a molecular formula of C1855H2850N476O553S7. The antigenic stabilization index of the candidate was calculated to be 32.98, with a lipid index of 89.39, classifying the candidate vaccine as a stable protein. The results presented in Fig. 3A reveal that most of the amino acids in the hydrophobicity of the candidate vaccine exhibit negative values, indicative of hydrophilic characteristics. Figure 3B illustrates that none of the candidate vaccines possess transmembrane domains according to the TMHMM-2.0 prediction; instead, all are extracellular regions supporting soluble expression. The PSIPRED secondary structure prediction indicates that the candidate vaccine mainly consists of helical structures. The secondary structure prediction results are shown in Fig. 3C.
Modeling, refinement and evaluation of tertiary structures of vaccine candidates
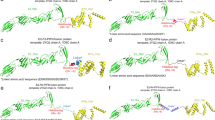
TrRosetta utilizes homology modeling to generate 2D views (contact map, distance map, and orientation map) of the candidate vaccine (Fig. 4A–E) and the tertiary structure model (Fig. 4F). However, the initial model exhibited instability. To address this issue, the crude model underwent refinement using Galaxy-Refine, and a cartoon comparison diagram was generated using Pymol (Fig. 5B). PROCHECK was employed to generate Ramachandran diagrams for evaluating the model's quality before and after refinement. The Ramachandran score results for the initial model (Fig. 5A) revealed the most favored regions (A, B, L) at 89.8% and additional allowed regions (a, b, l, p) at 9.5%. After refinement, the Ramachandran scoring results (Fig. 5C) indicated an increase in most favored regions (A, B, L) to 93.3%, with additional allowed regions (a, b, l, p) at 6.0%. These scores indicate an improvement in the model's quality after refinement (Detailed data are shown in Supplementary Table S4). ProSAweb was utilized to enhance the quality of the refined model further. The model structures were scored and validated. The structural accuracy analysis reveals a z value of − 5.85 (Fig. 5D), indicating a more satisfactory native protein conformation. Additionally, predominantly negative energy scores for the local model quality (Fig. 5E) signify an ideal model suitable for further analysis.

Three-level structure modeling of the candidate vaccine. (A–E) are the Omega, Phi, Distance, Theta and Contact parameters of the two-dimensional structure; ferritin is red; B cell epitope is purple and green; CD4+ epitope is orange; CD8+ epitope is sea blue; TM-score: 0.488.

Modeling and optimization of the three-level structure of candidate vaccine. (A) Ramachandran diagram of the crude model; (B) Comparison of the structure of the crude model and the refined model, the crude model and refined model are green and blue, respectively; (C) Ramachandran diagram of the refined model; (D) ProSA SEB refined model evaluation; (E) Local model quality assessment.
Candidate vaccine docking with TLR3
TLR-3, a member of the TLR family known for its role in inducing the transcription of type I interferons, proinflammatory cytokines, and chemokines crucial for initiating the host's antiviral response, was investigated. Approximately 102 composite docking complexes were generated using pyDockWEB docking. The complex with the highest composite ranking was chosen from the docking results, as depicted in Fig. 6A. The composite scores for this complex were as follows: Electrostatics: − 13.194; Desolvation: − 32.294; Vdw: 40.519; Total: − 41.436. Utilizing Pymol, we zoomed in on the interacting amino acid residues, with corresponding results presented in Fig. 6B,C. LigPlot+ was employed to generate a two-dimensional interaction map based on the docking results of the candidate vaccine and TLR3, and the outcomes are displayed in Fig. 6D. This map allows clear observation of hydrogen-bonded amino acid residues and hydrophobic interacting amino acid residues.

Docking results of candidate vaccine with TLR3 molecules. (A) Docking of a candidate vaccine to TLR3 (candidate vaccine structure in red, TLR3 in green); (B) local zoom in to show amino acid residues; (C) amino acid residues for specific interactions; (D) 2D interactions schematic diagram.
Immunosimulation and computerized cloning
Both specific and non-specific immune responses play a vital role in the process of vaccine immunity. Immune simulations on the candidate vaccine were conducted using C-ImmSim (Figs. 7, 8, 9). These simulations revealed that the candidate vaccine activated NK cells, giant cells and phages. Additionally, dendritic cells stimulated the differentiation and proliferation of B lymphocytes, leading to a significant increase in B cell populations and levels of IgG1 + IgG2, IgM and IgG + IgM antibodies. Similar trends were observed for CD4+ T cells (Th1 and Th2 cells) and CD8+ T cells (CTL), as well as immunoglobulins and immune complexes. The candidate vaccine also induced the production of IFN-γ by immune cells, forming three peaks. These findings indicate that after three rounds of immune stimulation, the simulation demonstrated a robust induction, thereby generating a potent immune response. Overall, the simulated immune response indicates that the vaccine can induce both cellular and humoral immunity, making it an excellent candidate.

Display of changes in the number of B cells and Th. (A) B lymphocytes; (B) Plasma B lymphocytes; (C) B lymphocyte population; (D) CD4 helper T lymphocytes; (E) CD4 helper T lymphocytes; (F) CD4T regulatory lymphocytes.

Display of changes in the number of CTL, NK, MA, DC, and EP cells. (A) CD8 T cytotoxic lymphocytes; (B) Per CD8 T cytotoxic lymphocytes; (C) Natural killer cells; (D) Total macrophage count; (E) dendritic cells; (F) epithelial cells.

Trends in changes in immunoglobulins, immune complexes, and concentrations of cytokines and interleukins. (A) Antigens and immunoglobulins; (B) Antigens and immunoglobulins (excluding ferritin); (C) Concentrations of cytokines and interleukins.
Computer simulation clones of candidate vaccines
JCat reverse translation of the candidate vaccine amino acid sequence resulted in a candidate vaccine nucleotide sequence of 1122 bp. To enhance the expression of the candidate vaccine, codon optimization was performed in E. coli K-12. The optimized sequence showed a CAI improvement value of 1.0, which is considered optimal. With a GC content of 48.395%, falling within the recommended range of 30–70% for protein expression, the sequence proved suitable for expression in E. coli. Validation by the ExpOptimizer server revealed a value of 0.74, while the GC content remained consistent at 48.40% (Fig. 10A,B). SnapGene was used to insert the optimized sequence into the prokaryotic expression vector pET-32a(+) using BamHI and XhoI sites. The results of the constructed prokaryotic expression plasmid are depicted in Fig. 10C. The plasmids were subjected to PCR simulation, identified by double digestion and analyzed using 1% agarose gel electrophoresis. The results are shown in Fig. 10D,E. The candidate vaccine was successfully inserted, and the bands appeared in the correct position after double enzyme digestion (nucleic acid sequences and original images of simulated gel electrophoresis are shown in supplement).

Construction of candidate vaccine expression system. (A,B) ExpOptimizer server analyzed results of vaccine candidates; (C) Candidate vaccine (red) inserted into pET-32a(+); (D) PCR simulation analysis of candidate vaccines; (E) PET-32a-Vaccine plasmid double digested.
Discussion
Coronaviruses are pathogens that originate from bats and have a wide range of hosts and tissue tropism2. The ongoing coronavirus disease 2019 (COVID-19) caused by SARS-CoV-2 poses a significant threat to the global economy and public health36. PEDV, another member of the coronavirus family, is notorious for its recurrent outbreaks in swine herds, causing widespread concern in the hog industry4. Prior to 2010, PEDV only had sporadic incidents and was limited to certain regions8. However, in October 2010, an epidemic with acute diarrhea symptoms broke out in southern provinces of China and quickly spread across the country37. The mutant strains of PEDV caused high mortality rates among piglets, reaching levels of 80–100%2. By 2013, the mutated virus strain had disseminated globally after being initially reported in the United States, exacerbating the detrimental impact of PEDV on the global pig industry and food safety38. The emergence of highly pathogenic PEDV strains underscores the limitations of the original vaccine strategy in providing cross-protection and managing virus escape39. Therefore, there is an urgent need to develop and improve vaccines to control and prevent PEDV. Ferritin possesses the capability to self-assemble and attach target antigens onto its surface via adsorption and covalent binding mechanisms40. This results in the formation of multivalent display nanoparticles, which play a crucial role in activating antigen-presenting cells (APCs)41. Additionally, it enhances antigen uptake by APCs, assists in antigen processing, fosters dendritic cell (DC) maturation, and augments B-cell activation42. Nanoparticle-based candidate vaccines are constructed through the integration of reverse vaccinology for comprehensive gene sequence screening and the incorporation of multiple B-lymphocyte or T-lymphocyte epitopes from viral proteins43. This approach addresses challenges associated with epitope shortness, poor stability, and low in vivo immunogenicity, thereby effectively eliciting both humoral and cellular immune responses.
The study was conducted to achieve immune balance, using multiple immunoinformatics tools to screen and construct the nanoparticle candidate vaccine for S-protein dominant epitopes. We identified and analyzed 5 B cell epitopes, 3 CD4+ and 3 CD8+ dominant epitopes. The B2 dominant epitope overlaps with the currently reported neutralizing epitope 499–638 aa44, suggesting strong potential for the candidate vaccine to neutralize the virus effectively. These dominant epitopes were then fused to ferritin in a specific order. Our analysis revealed that the candidate vaccine exhibited an immunogenicity score of 0.606918 and an overexpression solubility score of 0.584405, both exceeding the server's threshold for determination. The majority of the secondary structure of the vaccine consists of random coils, indicating a solid structural foundation and a loose spatial arrangement that facilitates epitope formation45. In the tertiary structure modeling, the refined model was increase in most favored regions (A, B, L) to 93.3%, with additional allowed regions (a, b, l, p) at 6.0%. Meanwhile, it was similar to the natural protein. The reliable structure of the energy score indicates that the structure of the model is stable, and it is helpful for the following research. Molecular docking investigations clearly demonstrate that the candidate vaccine forms a stable complex with TLR3. These findings imply that the candidate vaccine has the potential to activate intracellular signaling pathways, including NF-κB and cytokine release, thereby eliciting innate immune system activation and fostering durable adaptive immunity46. Although the C-IMMSIM immune simulation failed to simulate the self-assembly of ferritin into nanoparticles, it still illustrated significant antibody titer production against the antigen by the candidate vaccine. Furthermore, secondary and tertiary immune responses led to a gradual increase in active Th cell populations, total and memory TH cell counts, and cytotoxic T cells. Notably, after the three stages of immunity, IFN-γ exhibited a robust response among cytokines and interleukins, playing a crucial role in inhibiting viral replication, promoting T cell and natural killer cell production and aiding in viral clearance. Simulation data suggests that the candidate vaccine has the potential to induce both cellular and humoral immune responses. Additionally, these results are consistent with previous research on COVID-19 ferritin nanoparticle vaccines47, which have shown strong immune responses in animals. Therefore, it is anticipated that the actual immune response to the candidate vaccine will exceed expectations. Finally, the candidate vaccine was reverse-translated and molecularly cloned. The optimized sequence exhibited a CAI improvement value of 1.0 and a GC content of 48.395%, indicating high-level expression of the protein. However, to determine the practical application value of the candidate vaccine, further experiments involving protein expression and purification, as well as in vitro and in vivo studies, will be necessary to assess its ability to stimulate the immune response effectively.
Conclusions
This study successfully designed a recombinant multi-epitope nanoparticle vaccine targeting the PEDV S protein. The design process involved considering T/B cell dominant epitopes, spatial mapping, allele selection, homology modeling, molecular interactions and computer cloning. The innovative and logically structured design adopted in this study offers valuable insights for vaccine developers. The predictive evaluation and immune simulation enable the rapid design of vaccine molecules based on immunoinformatics. The constructed nanoparticle candidate vaccine provides a theoretical basis and offers data support for establishing a new vaccine development platform for PEDV.
Data availability
The datasets used and/or analyzed in the current study are available from the first or corresponding author upon reasonable request.
References
Zhao, Y., Zhou, C., Guo, B., Yang, X. & Wang, H. Pyrococcus furiosus Argonaute-mediated porcine epidemic diarrhea virus detection. Appl. Microbiol. Biotechnol. 108(1), 137. https://doi.org/10.1007/s00253-023-12919-0 (2024).
Li, Z., Ma, Z., Li, Y., Gao, S. & Xiao, S. Porcine epidemic diarrhea virus: Molecular mechanisms of attenuation and vaccines. Microb. Pathog. 149, 104553. https://doi.org/10.1016/j.micpath.2020.104553 (2020).
Sun, Y. et al. Porcine epidemic diarrhea virus in Asia: An alarming threat to the global pig industry. Infect. Genet. Evol. 70, 24–26. https://doi.org/10.1016/j.meegid.2019.02.013 (2019).
Sekhon, S. S. et al. Porcine epidemic diarrhea (PED) infection, diagnosis and vaccination: A mini review. Toxicol. Environ. Health Sci. 8(5), 277–289. https://doi.org/10.1007/s13530-016-0287-8 (2016).
Qin, Z. et al. The oral inactivated porcine epidemic diarrhea virus presenting in the intestine induces mucosal immunity in mice with alginate-chitosan microcapsules. Animals (Basel) 13(5), 889. https://doi.org/10.3390/ani13050889 (2023).
Hou, Y. & Wang, Q. Emerging highly virulent porcine epidemic diarrhea virus: Molecular mechanisms of attenuation and rational design of live attenuated vaccines. Int. J. Mol. Sci. 20(21), 5478. https://doi.org/10.3390/ijms20215478 (2019).
Wei, M. Z. et al. Overview of the recent advances in porcine epidemic diarrhea vaccines. Vet. J. 304, 106097. https://doi.org/10.1016/j.tvjl.2024.106097 (2024).
Escalera, A. et al. The impact of S2 mutations on Omicron SARS-CoV-2 cell surface expression and fusogenicity. Emerg. Microbes Infect. 13(1), 2297553. https://doi.org/10.1080/22221751.2023.2297553 (2024).
Muñoz-Gómez, M. J. et al. Immune response against the SARS-CoV-2 spike protein in cancer patients after COVID-19 vaccination during the Omicron wave: A prospective study. J. Infect. Public Health 17(7), 102473. https://doi.org/10.1016/j.jiph.2024.102473 (2024).
Golob, J. L., Lugogo, N., Lauring, A. S. & Lok, A. S. SARS-CoV-2 vaccines: A triumph of science and collaboration. JCI Insight 6(9), e149187. https://doi.org/10.1172/jci.insight.149187 (2021).
Sahu, L. K. & Singh, K. Cross-variant proof predictive vaccine design based on SARS-CoV-2 spike protein using immunoinformatics approach. Beni Suef Univ. J. Basic Appl. Sci. 12(1), 5. https://doi.org/10.1186/s43088-023-00341-4 (2023).
Pan, J. et al. An intranasal multivalent epitope-based nanoparticle vaccine confers broad protection against divergent influenza viruses. ACS Nano 17(14), 13474–13487. https://doi.org/10.1021/acsnano.3c01829 (2023).
Andreatta, M. et al. An automated benchmarking platform for MHC class II binding prediction methods. Bioinformatics 34(9), 1522–1528. https://doi.org/10.1093/bioinformatics/btx820 (2018).
Dhanda, S. K., Vir, P. & Raghava, G. P. Designing of interferon-gamma inducing MHC class-II binders. Biol. Direct 8, 30. https://doi.org/10.1186/1745-6150-8-30 (2013).
Dhanda, S. K., Gupta, S., Vir, P. & Raghava, G. P. Prediction of IL4 inducing peptides. Clin. Dev. Immunol. 2013, 263952. https://doi.org/10.1155/2013/263952 (2013).
Trolle, T. et al. Automated benchmarking of peptide-MHC class I binding predictions. Bioinformatics 31(13), 2174–2181. https://doi.org/10.1093/bioinformatics/btv123 (2015).
Reynisson, B., Alvarez, B., Paul, S., Peters, B. & Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 48(W1), W449–W454. https://doi.org/10.1093/nar/gkaa379 (2020).
Saha, S. & Raghava, G. P. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins 65(1), 40–48. https://doi.org/10.1002/prot.21078 (2006).
Buchan, D. W., Minneci, F., Nugent, T. C., Bryson, K. & Jones, D. T. Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res. 41(Web Server issue), 349–357. https://doi.org/10.1093/nar/gkt381 (2013).
Calis, J. J. et al. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput. Biol. 9(10), e1003266. https://doi.org/10.1371/journal.pcbi.1003266 (2013).
Gupta, S. et al. In silico approach for predicting toxicity of peptides and proteins. PLoS One 8(9), e73957. https://doi.org/10.1371/journal.pone.0073957 (2013).
Bui, H. H., Sidney, J., Li, W., Fusseder, N. & Sette, A. Development of an epitope conservancy analysis tool to facilitate the design of epitope-based diagnostics and vaccines. BMC Bioinform. 8, 361. https://doi.org/10.1186/1471-2105-8-361 (2007).
Tahir Ul Qamar, M. et al. Reverse vaccinology assisted designing of multiepitope-based subunit vaccine against SARS-CoV-2. Infect. Dis. Poverty 9(1), 132. https://doi.org/10.1186/s40249-020-00752-w (2020).
Cheng, J., Randall, A. Z., Sweredoski, M. J. & Baldi, P. SCRATCH: A protein structure and structural feature prediction server. Nucleic Acids Res. 33(Web Server issue), 72–76. https://doi.org/10.1093/nar/gki396 (2005).
Dimitrov, I., Bangov, I., Flower, D. R. & Doytchinova, I. AllerTOP vol 2—A server for in silico prediction of allergens. J. Mol. Model. 20(6), 2278. https://doi.org/10.1007/s00894-014-2278-5 (2014).
Duvaud, S. et al. Expasy, the Swiss Bioinformatics Resource Portal, as designed by its users. Nucleic Acids Res. 49(W1), W216–W227. https://doi.org/10.1093/nar/gkab225 (2021).
Ikeda, M., Arai, M., Lao, D. M. & Shimizu, T. Transmembrane topology prediction methods: A re-assessment and improvement by a consensus method using a dataset of experimentally-characterized transmembrane topologies. In Silico Biol. 2(1), 19–33 (2002).
Karypis, G. YASSPP: Better kernels and coding schemes lead to improvements in protein secondary structure prediction. Proteins 64(3), 575–586. https://doi.org/10.1002/prot.21036 (2006).
Yang, J. et al. Improved protein structure prediction using predicted interresidue orientations. Proc. Natl. Acad. Sci. USA 117(3), 1496–1503. https://doi.org/10.1073/pnas.1914677117 (2020).
Heo, L., Park, H. & Seok, C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 41(Web Server issue), W384–W388. https://doi.org/10.1093/nar/gkt458 (2013).
Laskowski, R. A., Rullmannn, J. A., MacArthur, M. W., Kaptein, R. & Thornton, J. M. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8(4), 477–486. https://doi.org/10.1007/BF00228148 (1996).
Lovell, S. C. et al. Structure validation by Calpha geometry: Phi, psi and Cbeta deviation. Proteins 50(3), 437–450. https://doi.org/10.1002/prot.10286 (2003).
Jiménez-García, B., Pons, C. & Fernández-Recio, J. pyDockWEB: A web server for rigid-body protein-protein docking using electrostatics and desolvation scoring. Bioinformatics 29(13), 1698–1699. https://doi.org/10.1093/bioinformatics/btt262 (2013).
Rapin, N., Lund, O., Bernaschi, M. & Castiglione, F. Computational immunology meets bioinformatics: The use of prediction tools for molecular binding in the simulation of the immune system. PLoS One 5(4), e9862. https://doi.org/10.1371/journal.pone.0009862 (2010).
Grote, A. et al. JCat: A novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 33(Web Server issue), W526–W531. https://doi.org/10.1093/nar/gki376 (2005).
Song, G., Li, R. & Cheng, M. Q. Safety, immunogenicity, and protective effective of inhaled COVID-19 vaccines: A systematic review and meta-analysis. J. Med. Virol. 96(4), e29625. https://doi.org/10.1002/jmv.29625 (2024).
Wang, D., Fang, L. & Xiao, S. Porcine epidemic diarrhea in China. Virus Res. 226, 7–13. https://doi.org/10.1016/j.virusres.2016.05.026 (2016).
Zhuang, H. et al. Molecular characterization and phylogenetic analysis of porcine epidemic diarrhea virus strains circulating in China from 2020 to 2021. BMC Vet. Res. 18(1), 392. https://doi.org/10.1186/s12917-022-03481-4 (2022).
He, D. et al. Establishment and application of a multiplex RT-PCR to differentiate wild-type and vaccine strains of porcine epidemic diarrhea virus. J. Virol Methods 272, 113684. https://doi.org/10.1016/j.jviromet.2019.113684 (2019).
Yang, D. et al. Construction and immune effect evaluation of the S protein heptad repeat-based nanoparticle vaccine against porcine epidemic diarrhea virus. Virology 596, 110113. https://doi.org/10.1016/j.virol.2024.110113 (2024).
Choi, B., Kim, H., Choi, H. & Kang, S. Protein cage nanoparticles as delivery nanoplatforms. Adv. Exp. Med. Biol. 1064, 27–43. https://doi.org/10.1007/978-981-13-0445-3_2 (2018).
Palombarini, F. et al. Self-assembling ferritin-dendrimer nanoparticles for targeted delivery of nucleic acids to myeloid leukemia cells. J. Nanobiotechnol. 19(1), 172. https://doi.org/10.1186/s12951-021-00921-5 (2021).
Omoniyi, A. A. et al. In silico design and analyses of a multi-epitope vaccine against Crimean-Congo hemorrhagic fever virus through reverse vaccinology and immunoinformatics approaches. Sci. Rep. 12(1), 8736. https://doi.org/10.1038/s41598-022-12651-1 (2022).
Wang, X. et al. Oral delivery of probiotics expressing dendritic cell-targeting peptide fused with porcine epidemic diarrhea virus COE antigen: A promising vaccine strategy against PEDV. Viruses 9(11), 312. https://doi.org/10.3390/v9110312 (2017).
Kumar, K. M. et al. Immunoinformatic exploration of a multi-epitope-based peptide vaccine candidate targeting emerging variants of SARS-CoV-2. Front. Microbiol. 14, 1251716. https://doi.org/10.3389/fmicb.2023.1251716 (2023).
Liu, B. M. et al. Key roles for phosphorylation and the coiled-coil domain in TRIM56-mediated positive regulation of TLR3-TRIF-dependent innate immunity. J. Biol. Chem. 300(5), 107249. https://doi.org/10.1016/j.jbc.2024.107249 (2024).
Ma, X. et al. Nanoparticle vaccines based on the receptor binding domain (RBD) and heptad repeat (HR) of SARS-CoV-2 elicit robust protective immune responses. Immunity 53(6), 1315-1330.e9. https://doi.org/10.1016/j.immuni.2020.11.015 (2020).
Funding
This research was supported by the conservation of germplasm resources of Xinjiang black pig, Genetic improvement and utilization, Plan for tackling key scientific and technological problems in key areas of the XPCC (2022AB012).
Author information
Authors and Affiliations
Contributions
S.L. (Shinian Li) and W. C. (Chaoli Wang) came up with and designed the study; Formal analysis X. B. (Xue Bai); writing—original draft, S. L; writing—review and editing, W C. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, S., Bai, X. & Wang, C. Epitopes screening and vaccine molecular design of PEDV S protein based on immunoinformatics. Sci Rep 14, 19537 (2024). https://doi.org/10.1038/s41598-024-70579-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-70579-0
- Springer Nature Limited