
Abstract
Sodium-ion batteries (SIBs) are emerging as a promising alternative to conventional lithium-ion technology, due to the abundance of sodium resources. The major drawbacks for the commercial application of SIBs lie in the slow kinetic processes and poor energy density of the devices. Molybdenum sulfide (MoS2), a graphene-like material, is becoming a promising anode material for SIBs, because of its high theoretical capacity (670 mAh g–1) and layered structure that suitable for Na+ intercalation/extraction. However, the intrinsic properties of MoS2, such as low conductivity, slow Na+ diffusion kinetics and large volume change during charging/discharging, restrict its rate capability and cycle stability. Here, molybdenum disulfide and graphene oxide (3D MoS2/GO) with excellent conductivity were fabricated through layer-by-layer method using amino-functionalized SiO2 nanospheres as templates. The 3D MoS2/GO composite demonstrates excellent cycling stability and capacity of 525 mA h g–1 at 500 mA g–1 after 100 cycles, which mainly due to the integrated MoS2/GO components and unique 3D macroporous structure, facilitating the material conductivity and Na+ diffusion rate, while tolerating the volume expansion of MoS2 during the charge/discharge processes.
Similar content being viewed by others

Introduction
The development of sustainable low-carbon energy society requires the efficient utilization of renewable power sources1,2,3. Sodium-ion batteries (SIBs) have similar reaction mechanism as lithium-ion batteries (LIBs)4,5,6. Therefore, SIBs are growing remarkably as promising candidates to replace LIBs in large-scale energy storage systems, because of the abundant sodium reserves on earth5,7,8,9,10,11,12,13,14. However, the commercial graphite employed in LIBs has been shown to be unsuitable for SIBs, because the radius of Na+ (1.06 Å) is much larger than that of Li+ (0.76 Å), which slows down the dynamism of the reaction and causes big volume expansion, leading to irreversible structural decomposition and capacity decay of SIBs12,15,16,17,18,19. Therefore, it is urgent to find suitable anode materials with high conductivity, fast Na+ diffusion channels and bouncy structure to tolerant volume change during charging and discharging processes.
Nowadays, the promising anode materials for SIBs include carbon-based materials18,20,21,22,23,24, titanium-based materials25,26,27,28,29, metal sulfides30,31,32,33,34,35, metal oxides36,37,38,39,40, alloys and organic compounds41,42. Among them, transition metal chalcogenide, a 2D-layered material, has attracted much attention due to its excellent physical and chemical properties. For example, MoS2 has high theoretical capacity (670 mAh g−1) and large interlayer spacing (6.2 Å), which allows sodium-ions (1.06 Å) for rapid and reversible insertion/extraction. Nevertheless, due to its inherent low conductivity and inevitable instability, molybdenum disulfide cannot be used as SIB anode material alone. At present, the most common strategy to conquer the above limitations is to add carbonaceous materials, such as carbon, graphene and graphene oxides39,40. Graphene oxides have excellent optical, electrical and mechanical properties, and GO composites have been proved to exhibit enhanced conductivity and bouncy structure that diminish volume change41,43. However, MoS2/GO composites that with integrated MoS2/GO components and well organized 3D macroporous structure that not only facilitate Na+ diffusion but also tolerant volume change are rarely reported.
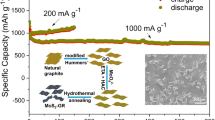
Herein, we prepared 3D MoS2/Graphene oxide (3D MoS2/GO) composite with MoS2 supported on 3D GO for sodium-ion batteries anode material, which showed excellent reversible capacity and cyclic stability. Porous foam structure provides abundant specific surface facilitating Na+ diffusion and attachment, meanwhile, rich channels promote the contact between electrolyte and electrode, accelerating the electrolyte infiltration. The 3D GO framework ensures the high electrode conductivity, speeds up the electron transmission efficiency, effectively alleviates the volume changes, and increases the structural stability and reversible circulation capacity. In this work, the 3D MoS2/GO electrode delivers a high discharge capacity of 525 mAh g–1 at 500 mA g–1 after 100 cycles and a reversible capacity of 277 mAh g–1 at 5000 mA g–1 after 3000 cycles with Coulombic efficiency of 99.99%.
Experimental
Material synthesis
Sample preparation of 3D MoS2/GO
Synthesis of SiO2 nanospheres
The SiO2 nanospheres with a diameter of 100 nm were synthesized according to a previously reported method44. Briefly, 9.6 g TEOS was mixed with 200 mL ethanol, and then the solution was added into 8 mL deionized water and 8 mL ammonia solution (25 wt%). The mixed solution was stirred for 5 h at room temperature, then removed the solvent and dried the solution at 80 °C to obtain SiO2 nanospheres templates.
Synthesis of GO wrapped SiO2 nanospheres
The GO can be wrapped on the on the surface of amino-functionalized silica on the basis of the electrostatic interaction between the negatively charged GO and positively charged amino-functionalized SiO2 nanospheres. GO was synthesized by a modified Hummer’s method45. The acquisition of GO wrapped SiO2 nanospheres refers to Meng46 but has made some changes as follows. The SiO2 nanospheres (0.4 g) were ultrasonically dispersed into Ethanol solution (200 mL), 3-aminopropyltrimethoxysilane (2 mL) was added drop by drop, the solution was refluxed for 5 h under an argon flow (5 L min–1), and then cooled down to RT, the particles were collected and washed three times with ethanol to get amino-functionalized silica (denoted as NH2-SiO2). 40 mg GO was put into 10 mL deionized water for ultrasonic treatment (30 min), then the suspension was added into NH2-SiO2 dispersion in water (200 mL), after fully stirred for 1 h, obvious delamination was found. Finally, the product was collected by centrifugation and washed with deionized water for few times.
Synthesis of 3D MoS2/GO
(NH4)6Mo7O24·4H2O (50 mg) was mixed with the suspension of GO wrapped NH2-SiO2 (in 20 mL water solution) that obtained from step 2.1.1.2. Stir them to dry under room temperature, and collect the solid powder. The solid powder and CS2 (10 mL) were put into 40 mL stainless steel autoclave and maintained at 400 °C for 4 h. Finally, the solid powder was collected and soaked with hydrofluoric acid solution (20 wt%) for 1 day to remove the SiO2 nanospheres. Wash the powder with deionized water for 4–5 times (~ 1000 mL), and dry it (100 ℃ 24 h) to achieve the final product.
Sample preparation of 3D MoS2
For comparison, the 3D MoS2 was made by similar experimental procedures but without the addition of graphene oxide. (NH4)6Mo7O24·4H2O 50 mg was mixed with SiO2 nanospheres suspension (400 mg in 20 mL water solution) under room temperature. After stirring the solution to dry, the obtained powder and CS2 were mixed and transferred into the autoclave, and kept at 400 °C for 4 h, the 3D MoS2 powder was obtained after HF solution (20 wt%) etching for 24 h, deionized water washing for 4–5 times (~ 1000 mL) and drying processes (100 ℃ 24 h).
Sample preparation of 2D MoS2/GO
The 2D MoS2/GO was prepared by same experimental procedures as above for preparing 3D MoS2/GO but without the addition of SiO2. (NH4)6Mo7O24·4H2O 50 mg and 40 mg graphene oxide were dispersed in deionized water (10 mL), the solvent was removed by constant stirring. Then the obtained powder and CS2 were transferred to the autoclave and kept at 400 °C for 4 h, and then cleaned and dried (100 ℃ 24 h).
Materials characterization
High resolution scanning electron microscopy (HRSEM) was conducted on Hitachi S5500. High resolution transmission electron microscopy (HRTEM) was conducted on TECNAI G2 F30. X-ray diffraction (XRD) measurements were conducted on Rigaku D/Max 2500/PC. Raman spectroscopy was performed on a LabRAM HR 800 Raman spectrometer. XPS was carried out on KRATOS Axis UltraDLD. The vacuum degree in the analysis room was 9.8 × 10–10 Torr.
Electrochemical measurements
The battery was assembled with CR2032 button cell shell and its electrochemical performance was tested. The batteries were assembled in a glove box. The electrode was made by mixing the active material, acetylene black, and polyvinylidene fluoride binder at a weight ratio of 7:2:1, and then pasted onto a Cu foil current collector and dried in a vacuum oven at 60 °C. Then cut the Cu foil into pieces with diameter of 12 cm. The mass loading was approximately 1.0 mg cm−2. The capacities described were achieved based on the total weight of 3D MoS2/GO composite. Metallic Na pieces were used as counter electrodes and reference electrodes. A glass fiber membrane (Whatman/F) was used as separator. The 1 M NaClO4 (Aldrich) in Ethylene carbonate (EC): Propylene carbonate (PC) (1:1 vol%) + 5 wt% Fluoroethylene carbonate (FEC) as the electrolyte. The galvanostatic discharge/charge measurements were performed in the voltage range of 0.01–3 V (vs. Na/Na+) on a NEWARE battery testing system. Cyclic voltammetry (CV), and electrochemical impedance spectroscopy (EIS) tests were performed on an electrochemical workstation (Gamry). CV was measured in a voltage range of 0.01–3 V at room temperature. EIS of the electrodes were measured, applying a 5 mV amplitude signal in the frequency of 0.01 Hz–100 kHz.
Results and discussions
3D MoS2/GO were prepared by layer-by-layer method from using the SiO2 nanospheres as 3D templates, GO and MoS2 as coating layers. Figure 1 shows the typical preparation process of the 3D MoS2/GO. Firstly, SiO2 nanospheres are functionalized by amino groups, and GO can be electrostatically attached with it, thus completing the coating of SiO2 by GO. Then, heating at 400 ℃ for 4 h by solvothermal method to grow molybdenum disulfide on the surface of graphene oxide. Finally, the final product 3D MoS2/G was obtained by etching the SiO2 with HF solution.

Schematic illustration of the preparation process of the 3D MoS2/GO composite.
The scanning electron microscope (SEM) images show that SiO2 nanospheres with an average diameter of 100 nm possess very smooth un-textured surfaces, Fig. S1a,b. After grafted with amino- groups on SiO2, the surface of SiO2 is rough instead of smooth, Fig S1c,d. When electrostatic adsorption self-assemble between GO and NH2-SiO2 is completed, the surface of NH2-SiO2 is covered by wrinkled GO sheets, as can be seen in Fig S1e,f. High-resolution scanning electron microscopy (HRSEM) image Fig. 2a shows uniform macroporous structure of 3D MoS2/GO. Three-dimensional foam structure of 3D MoS2/GO provides high specific surface area which facilitates Na+ diffusion and attachment. Also rich channels promotes the contact between electrolyte and electrode, accelerating the electrolyte infiltration. The high-resolution transmission electron microscopy (HRTEM) images shown in Fig. 2b and c reveal the hollow structure with layered lattice spacing of 0.65 nm, which match the (002) plane of 2H-MoS2, indicating the layered structure of MoS2 is well preserved in 3D MoS2/GO. The atomic resolution HAADF-STEM image in Fig. 2d and EDS mapping results in Fig. 2e–g confirm the uniform dispersion of S, Mo and C elements.

Morphology and nanostructure characterizations of 3D MoS2/GO, (a) HRSEM of 3D MoS2/GO, (b,c) HRTEM of 3D MoS2/GO, (d) HAADF-STEM image of 3D MoS2/GO, and (e–g) EDS mapping results of S, Mo and C.
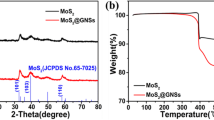
The structure of as-prepared samples were investigated by X-ray diffraction (XRD). The main peaks of the 3D MoS2/GO match the peaks of 2H-MoS2 (JCPDS card No.37-1492) and some weak peaks correspond to peaks of graphitic carbon, confirming the coexistence of MoS2 and graphene oxide, Fig. 3a. The 3D MoS2/GO composite basically retain the layered crystallinity and the diffraction peaks of MoS2. Notably, from the XRD results of 3D MoS2/GO, 3D MoS2, 2D MoS2/GO (Supporting information, Fig. S2), it can be seen that with the addition of graphene oxide in the composite, the intensity of all diffraction peaks of MoS2 decreases, especially the (002) plane peak. Figure 3b is the Raman spectrum of 3D MoS2/GO, displaying the fingerprint peaks locating at 385 cm–1 and 409 cm–1, corresponding to the E12g in-plane mode and the A1g out-of-plane mode respectively47. In addition, there are also two characteristic peaks at 1355 cm–1 and 1595 cm–1, which attribute to D (disordered) band and G (graphite) band of carbon materials respectively48, confirming the presence of GO in 3D MoS2/GO. The chemical configuration and surface electron valence states of MoS2 in 3D MoS2/GO were further determined by X-ray photoelectron spectroscopy (XPS). The XPS survey spectrum in Fig. 3c shows that the elements in the sample are S, Mo, C and O. The high-resolution spectrum of Mo in Fig. 3d shows three peaks at 226.8 eV, 229.2 eV and 232.4 eV, corresponding to S 2 s, Mo 3d5/2 and Mo 3d3/2 respectively. In addition, the peak located at 235.5 eV is the 3d3/2 of Mo6+, which is mainly caused by the surface oxidation of Mo4+ in air during sample characterization49,50. Figure 3e is the S 2p high-resolution spectrum, and two peaks are observed at 162.1 eV and 163.3 eV, assigning to S 2p3/2 and S 2p1/2 respectively49,50,51. Meanwhile, the wide peak ranging from 165.9 to 171.5 eV may be caused by the partial oxidation of S on the surface of the sample in air51,52. Figure 3f shows three peaks at 284.6 eV, 285.4 eV and 288.5 eV, attributing to C–C, C–O and C=O respectively51. Both 3D MoS2 and 2D MoS2/GO XPS spectra show similar peaks, confirming the same MoS2 configuration with 3D MoS2/GO, as shown in Figs. S3, 4.

Structure and composition characterizations of 3D MoS2/GO. (a) XRD pattern of 3D MoS2/GO. (b) Raman spectrum of 3D MoS2/GO. (c) XPS survey spectrum of 3D MoS2/GO and high-resolution XPS spectra of Mo 3d (d), S 2p (e) and C 1 s (f).
To determine the loading amount of MoS2, thermogravimetric analysis (TGA) was performed. As can be seen in Fig. S5, there are three steps of mass loss in 3D MoS2/GO. The first small mass loss (less than 200 ℃) dues to the evaporation of adsorbed water. The second mass loss between 200 ℃ and 400 ℃ belongs to the oxidation of MoS2 (MoS2 + O2 \(\to\) MoO3 + SO2). The third mass loss from 400 ℃ to 490 ℃ should be attributed to the combustion of the GO46. Based on TGA data, assuming that the product at 600 ℃ is pure MoO3, the calculated contents of MoS2 in 3D MoS2/GO, 3D MoS2 and 2D MoS2/GO are about 51.47%, 69.13% and 62.58%, respectively47. Both 3D MoS2/GO (67.0 m2/g) and 3D MoS2 (43.5 m2/g) exhibit high surface area compared with 2D MoS2/GO (8.7 m2/g), verifying the rich porous structure that derived from the SiO2 nanosphere templates, Supporting Information, Fig. S6. 3D MoS2/GO shows the highest surface area, owing to the contribution of GO and its support effect for MoS2 loading. Therefore, through a layer-by-layer method using SiO2 nanospheres as templates, we have successfully fabricated 3D MoS2/GO with high surface area and MoS2 loading.
The electrochemical properties of the 3D MoS2/GO as the anode material for SIBs were investigated, as shown in Fig. 4. Figure 4a shows the cyclic voltammograms (CV) of the 3D MoS2/GO for the first five cycles at a scan rate of 0.5 mV s−1 at voltage window of 0.01–3 V. The peak at 0.75 V is related to the insertion of Na+ between MoS2 and the formation of solid electrolyte interface (SEI) layer formed by electrolyte decomposition. The reduction peak at 0.2 V is related to the further formation of Mo and Na2S by the conversion reaction (NaxMoS2 + Na+ + e– → 2Na2S + Mo) and the insertion of Na+ into graphene52,53. In the first charge process, the prominent oxidation peak at 1.88 V corresponds to the oxidation conversion reaction of Mo with partial Na2S to reform MoS254. After the first cycle, the second to fifth discharging/charging curves almost overlapped, suggesting high reversibility and cycling stability of Na+ storage in the 3D MoS2/GO composite. Figure 4b shows the galvanostatic charge/discharge curves at a current density of 500 mA g–1. In the initial discharge (1500 mAh g–1), two voltage plats appears at 1.0 V and 0.2 V respectively, while in the first charge, the voltage platform at 1.9 V appears, which agrees well with the CV result. Besides, from 40th to 100th cycle, the charge and discharge profiles are almost overlapped, which further proves that the material has good cycling stability during the sodiation/desodiation processes54. Cycling performance at a current density of 500 mA g–1 is shown in Fig. 4c. After 100 cycles, the reversible discharge capacity of 3D MoS2/GO, 2D MoS2/GO and 3D MoS2 are 525 mAh g–1, 324 mAh g–1, and 90 mAh g–1, respectively. The capacity retention rate of 3D MoS2/GO, 2D MoS2/GO and 3D MoS2 are 95.6%, 18.8% and 93.6%, respectively. The rate capabilities are evaluated at current densities ranging from 0.05 to 2 A g−1 as shown in Fig. 4d. When cycled at various current densities of 50, 100, 300, 500, 1000 and 2000 mA g–1, the 3D MoS2/GO electrode delivers the discharge capacities of 599, 599, 577, 559, 531, 492 mAh g–1, respectively, which are superior to those of 3D MoS2 (389, 158, 48, 15, 4 and 2 mAh g–1) and 2D MoS2/GO (281, 267, 225, 202, 165 and 138 mAh g–1) electrodes. When the current density of 3D MoS2/GO returns to 300 mA g–1, the capacity retention rate is 96.8%, further proves its excellent rate property. In addition, as shown in Fig. 4e, the long-term cycling stability of 3D MoS2/GO under high current (5000 mA g–1) was tested. After 3000 cycles, 3D MoS2/GO showed a high capacity of 277 mAh g–1 and a high coulombic efficiency of 99.99%, with capacity retention rate of 37.0%. Compared with other MoS2/carbon-based composites (Table S1), 3D MoS2/GO showed super performance in both large current density and long cycling stability55,56,57,58,59,60,61,62. By comparing 3D MoS2/GO with control samples, we assume the reasons for the excellent performance of 3D MoS2/GO are as follows: (1) The intense contact between MoS2 and GO substrate can enhance the electrical conductivity of the whole material; (2) The addition of GO effectively alleviated the volume expansion of MoS2 and prevented the capacity attenuation; (3) The macroporous structure generated by SiO2 nanospheres templates can provide higher specific surface area and expose more active sites, which are conducive to the infiltration of electrolyte and Na+ transmission, leading to accelerated reaction rate and promoted storage ability of Na+.

Electrochemical performance of different samples. (a) CV curves of the 3D MoS2/GO for the first five cycles at a scan rate of 0.5 mV s–1, (b) Galvanostatic discharge–charge curve of 3D MoS2/GO at 500 mA g–1, (c) Cycling performance of the 3D MoS2/GO, 3D MoS2 and 2D MoS2/GO at a current density of 500 mA g–1 (The first two cycles are activated at a current density of 50 mA g–1), (d) Rate performances of the 3D MoS2/GO, 3D MoS2 and 2D MoS2/GO, (e) Long-term cycling property and Coulombic efficiency of the 3D MoS2/GO at a current density of 5000 mA g–1 (The first two cycles are activated at a current density of 50 mA g–1).
To reveal more information about the high performance of 3D MoS2/GO, the 3D MoS2/GO electrode was recycled after 100 cycles at 500 mA g–1. From the SEM image (Fig. S8a), rich macropores with sizes of 100 nm–2 μm are clearly observed on the surface of the recycled 3D MoS2/GO sample. The non-uniform pore size distribution of the macropores are probably caused by the material treatment processes during the preparation of electrode and the long cycling test of battery. Furthermore, the nanostructure of the recycled 3D MoS2/GO was characterized by HRTEM. From the HRTEM image, the layered structure with d-spacing corresponding to GO (0.40 nm) and MoS2 (0.67 nm) are clearly observed, showing the well-preserved structure of GO and MoS2 after long cycling test. Further confirming the excellent stability of 3D MoS2/GO, and this will benefit for its long cycling stability.
The electrochemical impedance spectroscopy (EIS) was measured before charging/discharging as shown in Fig. 5a. The high-frequency (the semicircular) part is the charge-transfer impedance (Rct), of which 3D MoS2/GO is significantly lower than that of 3D MoS2 and 2D MoS2/GO. The low-frequency part is the Warburg impedance (Zw)63, which relates to Na+ diffusion coefficient D. The equations are as follows54,64:

(a) EIS Nyquist plots of 3D MoS2/GO, 3D MoS2 and 2D MoS2/GO, (b) The relationship between Z’ and ω−1/2 for 3D MoS2/GO, 3D MoS2 and 2D MoS2/GO, (c) CV curves of 3D MoS2/GO at different scan rates, (d) Log (i)–log (v) plots at different cathodic/anodic peaks.
D is the diffusion coefficient of Na+, R is the gas constant, T is the absolute temperature, A is the surface area of the electrode, n is the number of the electrons per molecule involved in the electronic transfer reaction, F is the Faraday constant, C is the concentration of Na+, \({\upsigma }_{\text{w}}\) is the slope of the line Z´ ∼ ω–1/2. As shown in Fig. 5b, the slopes \({\upsigma }_{\text{w}}\) of 3D MoS2/GO, 3D MoS2 and 2D MoS2/GO are 99, 319 and 1030, respectively. According to the equation, the Na+ diffusion coefficient of 3D MoS2/GO electrode is higher than 3D MoS2 and 2D MoS2/GO. Besides, the CV curves at different scan rates are used to explore the charge storage behavior of 3D MoS2/GO, as shown in Fig. 5c and d. The relationship between peak current and scanning rate is as follows65:
i is peak current, a is adjustable parameters, v is scanning rate and b is the slope of the plot of log (i) vs log (v). According to the equation, b value based on cathodic/anodic peaks for are 0.929 and 0.80, respectively. Both are closer to 1, indicating that the capacitive contribution is dominant in the 3D MoS2/GO electrodes. Compared with the intercalation contribution, the capacitive contribution causes less damage to the electrode and is more conducive to the cycling stability. This further explains the excellent cycling stability of 3D MoS2/GO electrode.
Conclusion
In conclusion, 3D MoS2/GO composite has been prepared using SiO2 nanospheres as template through layer-by-layer method. 3D MoS2/GO possesses 3D macroporous structure with rich channels and high specific surface area, and shows super performance as electrode for sodium-ion batteries. It has showed superior specific capacity (277 mAh g–1) and cycling stability (3000 cycles) at high current (5000 mA g–1), with a high Coulombic efficiency of 99.99% in sodium-ion batteries. The excellent rate performance and cycling stability of 3D MoS2/GO may be originated from its unique 3D structure, including rich surface area, abundant 3D transportation channels, less volume change during charging/discharging and high conductivity that inherited from the intense contact between MoS2 and GO. This strategy is also applicable to other transition metal sulfides. Therefore, we believe this work can inspire the development of other alternative electrode materials for high-performance sodium ion batteries.
Data availability
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
References
Bruce Dunn, H. K. & Tarascon, J.-M. Electrical energy storage for the grid: A battery of choices. Science 334, 928–935 (2011).
Poullikkas, A. A comparative overview of large-scale battery systems for electricity storage. Renew. Sustain. Energy Rev. 27, 778–788 (2013).
Zhang, C., Wei, Y.-L., Cao, P.-F. & Lin, M.-C. Energy storage system: Current studies on batteries and power condition system. Renew. Sustain. Energy Rev. 82, 3091–3106 (2018).
Tu, Y. et al. Recent advances on liquid intercalation and exfoliation of transition metal dichalcogenides: From fundamentals to applications. Nano Res. 17, 2088–2110 (2023).
Mathiyalagan, K., Shin, D. & Lee, Y.-C. Difficulties, strategies, and recent research and development of layered sodium transition metal oxide cathode materials for high-energy sodium-ion batteries. J. Energy Chem. 03, 40–57 (2024).
Kong, L.-Y. et al. Layered oxide cathodes for sodium-ion batteries: Microstructure design, local chemistry and structural unit. Sci. China Chem. 01, 191–213 (2024).
Delmas, C. Sodium and sodium-ion batteries: 50 years of research. Adv. Energy Mater. 8, 1703137 (2018).
Deng, J., Luo, W.-B., Chou, S.-L., Liu, H.-K. & Dou, S.-X. Sodium-ion batteries: From academic research to practical commercialization. Adv. Energy Mater. 8, 1701428 (2018).
Eftekhari, A. & Kim, D.-W. Sodium-ion batteries: New opportunities beyond energy storage by lithium. J. Power Sources 395, 336–348 (2018).
Li, F. et al. Sodium-based batteries: From critical materials to battery systems. J. Mater. Chem. A 7, 9406–9431 (2019).
Pan, H., Hu, Y.-S. & Chen, L. Room-temperature stationary sodium-ion batteries for large-scale electric energy storage. Energy Environ. Sci. 6, 2338–2360 (2013).
Sawicki, M. & Shaw, L. L. Advances and challenges of sodium ion batteries as post lithium ion batteries. RSC Adv. 5, 53129–53154 (2015).
Slater, M. D., Kim, D., Lee, E. & Johnson, C. S. Sodium-ion batteries. Adv. Funct. Mater. 23, 947–958 (2013).
Bai, H. et al. Advances in sodium-ion batteries at low-temperature: Challenges and strategies. J. Energy Chem. 03, 518–539 (2024).
Hong, S. Y. et al. Charge carriers in rechargeable batteries: Na ions vs. Li ions. Energy Environ. Sci. 6, 2067–2081 (2013).
Hwang, J. Y., Myung, S. T. & Sun, Y. K. Sodium-ion batteries: Present and future. Chem. Soc. Rev. 46, 3529–3614 (2017).
Raccichini, R., Varzi, A., Passerini, S. & Scrosati, B. The role of graphene for electrochemical energy storage. Nat. Mater. 14, 271–279 (2015).
Wahid, M., Puthusseri, D., Gawli, Y., Sharma, N. & Ogale, S. Hard carbons for sodium-ion battery anodes: Synthetic strategies, material properties, and storage mechanisms. ChemSusChem 11, 506–526 (2018).
Yabuuchi, N., Kubota, K., Dahbi, M. & Komaba, S. Research development on sodium-ion batteries. Chem. Rev. 114, 11636–11682 (2014).
Hou, H., Qiu, X., Wei, W., Zhang, Y. & Ji, X. Carbon anode materials for advanced sodium-ion batteries. Adv. Energy Mater. 7, 201602898 (2017).
Kim, D. Y. et al. Nano hard carbon anodes for sodium-ion batteries. Nanomaterials 9, 793–801 (2019).
Kumar, N. A. et al. Sodium ion storage in reduced graphene oxide. Electrochim. Acta 214, 319–325 (2016).
Zhang, J. et al. 3D free-standing nitrogen-doped reduced graphene oxide aerogel as anode material for sodium ion batteries with enhanced sodium storage. Sci. Rep. 7, 4886 (2017).
Senthil, C., Park, J. W., Shaji, N., Sim, G. S. & Lee, C. W. Biomass seaweed-derived nitrogen self-doped porous carbon anodes for sodium-ion batteries: Insights into the structure and electrochemical activity. J. Energy Chem. 01, 286–295 (2022).
Doeff, M. M., Cabana, J. & Shirpour, M. Titanate anodes for sodium ion batteries. J. Inorg. Organomet. Polym. Mater. 24, 5–14 (2013).
Guo, S., Yi, J., Sun, Y. & Zhou, H. Recent advances in titanium-based electrode materials for stationary sodium-ion batteries. Energy Environ. Sci. 9, 2978–3006 (2016).
Mei, Y., Huang, Y. & Hu, X. Nanostructured Ti-based anode materials for Na-ion batteries. J. Mater. Chem. A 4, 12001–12013 (2016).
Wu, L., Buchholz, D., Bresser, D., Gomes Chagas, L. & Passerini, S. Anatase TiO2 nanoparticles for high power sodium-ion anodes. J. Power Sources 251, 379–385 (2014).
Zhai, H., Xia, B. Y. & Park, H. S. Ti-based electrode materials for electrochemical sodium ion storage and removal. J. Mater. Chem. A 7, 22163–22188 (2019).
Deng, J. et al. Graphene layer reinforcing mesoporous molybdenum disulfide foam as high-performance anode for sodium-ion battery. Mater. Today Energy 8, 151–156 (2018).
Hu, Z., Liu, Q., Chou, S. L. & Dou, S. X. Advances and challenges in metal sulfides/selenides for next-generation rechargeable sodium-ion batteries. Adv. Mater. 29, 201700606 (2017).
Hu, Z. et al. MoS2 nanoflowers with expanded interlayers as high-performance anodes for sodium-ion batteries. Angew. Chem. Int. Ed. 126, 13008–13012 (2014).
Liu, Y. et al. WS2 nanowires as a high-performance anode for sodium-ion batteries. Chemistry 21, 11878–11884 (2015).
Wang, T., Chen, S., Pang, H., Xue, H. & Yu, Y. MoS2-based nanocomposites for electrochemical energy storage. Adv. Sci. 4, 1600289 (2017).
Xiao, Y., Lee, S. H. & Sun, Y.-K. The application of metal sulfides in sodium ion batteries. Adv. Energy Mater. 7, 201601329 (2017).
Hasa, I., Verrelli, R. & Hassoun, J. Transition metal oxide-carbon composites as conversion anodes for sodium-ion battery. Electrochim. Acta 173, 613–618 (2015).
Jiang, Y. et al. Transition metal oxides for high performance sodium ion battery anodes. Nano Energy 5, 60–66 (2014).
Alcantara, M. J. R. & Lavela, P. Tirado, NiCo2O4 Spinel_First report on a transition metal oxide for the negative electrode of sodium-ion batteries. Phys. Inorg. Chem. 14, 2847–2848 (2002).
Wang, Y. et al. Erratum: A zero-strain layered metal oxide as the negative electrode for long-life sodium-ion batteries. Nat. Commun. 4, 2365 (2013).
Xiong, H., Slater, M. D., Balasubramanian, M., Johnson, C. S. & Rajh, T. Amorphous TiO2 nanotube anode for rechargeable sodium ion batteries. J. Phys. Chem. Lett. 2, 2560–2565 (2011).
Banda, H., Damien, D., Nagarajan, K., Hariharan, M. & Shaijumon, M. M. A polyimide based all-organic sodium ion battery. J. Mater. Chem. A 3, 10453–10458 (2015).
Zhang, Y. et al. A calcium organic salt/rGO composite with low solubility and high conductivity as a sustainable anode for sodium-ion batteries. ChemSusChem 12, 4160–4164 (2019).
Wasalathilake, K. C., Li, H., Xu, L. & Yan, C. Recent advances in graphene based materials as anode materials in sodium-ion batteries. J. Energy Chem. 03, 91–107 (2020).
Lin, X., Zhou, L., Huang, T. & Yu, A. Hierarchically porous honeycomb-like carbon as a lithium–oxygen electrode. J. Mater. Chem. A 1, 1239–1245 (2013).
Daniela, D. V. K. et al. Improved synthesis of graphene oxide. ACS Nano 4, 4806–4814 (2010).
Meng, X. et al. Three-dimensionally hierarchical MoS2/graphene architecture for high-performance hydrogen evolution reaction. Nano Energy 61, 611–616 (2019).
Gołasa, K. et al. Resonant Raman scattering in MoS2—From bulk to monolayer. Solid State Commun. 197, 53–56 (2014).
Xie, X. et al. MoS2 nanosheets vertically aligned on carbon paper: A freestanding electrode for highly reversible sodium-ion batteries. Adv. Energy Mater. 6, 1502161 (2016).
Chen, B. et al. Efficient reversible conversion between MoS2 and Mo/Na2 S enabled by graphene-supported single atom catalysts. Adv. Mater. 33, e2007090 (2021).
Wu, X., Xie, X., Zhang, H. & Huang, K. J. Engineering stable and fast sodium diffusion route by constructing hierarchical MoS2 hollow spheres. J. Colloid Interface Sci. 595, 43–50 (2021).
Yu, H., Wang, Z., Ni, J. & Li, L. Freestanding nanosheets of 1T–2H hybrid MoS2 as electrodes for efficient sodium storage. J. Mater. Sci. Technol. 67, 237–242 (2021).
Li, Y. et al. Compositing reduced graphene oxide with interlayer spacing enlarged MoS2 for performance enhanced sodium-ion batteries. J. Phys. Chem. Solids 136, 109163 (2020).
Lamuel David, R. B. & Singh, G. MoS2/graphene composite paper for sodium-ion battery electrodes. ACS Nano 8, 1759–1770 (2014).
Wang, X., Hao, H., Liu, J., Huang, T. & Yu, A. A novel method for preparation of macroposous lithium nickel manganese oxygen as cathode material for lithium ion batteries. Electrochim. Acta 56, 4065–4069 (2011).
Geng, X. et al. Freestanding metallic 1T MoS2 with dual ion diffusion paths as high rate anode for sodium-ion batteries. Adv. Funct. Mater. 27, 1702998 (2017).
Zheng, F. et al. 3D MoS2 foam integrated with carbon paper as binder-free anode for high performance sodium-ion batteries. J. Energy Chem. 2, 26–33 (2022).
Tang, W. J. et al. Hollow metallic 1T MoS2 arrays grown on carbon cloth: A freestanding electrode for sodium ion batteries. J. Mater. Chem. A 6, 18318–18324 (2018).
Ni, Q. et al. Carbon nanofiber elastically confined nanoflowers: A highly efficient design for molybdenum disulfide-based flexible anodes toward fast sodium storage. ACS Appl. Mater. Interfaces 11, 5183–5192 (2019).
Yang, H., Wang, M., Liu, X., Jiang, Y. & Yu, Y. MoS2 embedded in 3D interconnected carbon nanofiber film as a free-standing anode for sodium-ion batteries. Nano Res. 11, 3844–3853 (2018).
Choi, S. H., Ko, Y. N., Lee, J. K. & Kang, Y. C. 3D MoS2–graphene microspheres consisting of multiple anospheres with superior sodium ion storage properties. Adv. Funct. Mater. 25, 01402428 (2015).
Anwer, S. et al. Nature-inspired, graphene-wrapped 3D MoS2 ultrathin microflower architecture as a high-performance anode material for sodium-ion batteries. ACS Appl. Mater. Interfaces 11, 22323–22331 (2019).
Zhu, M. et al. 3D reduced graphene oxide wrapped MoS2@Sb2S3 heterostructures for high performance sodium-ion batteries. Appl. Surf. Sci. 624, 157106 (2023).
Chen, C. et al. Chemical vapor deposited MoS2/electrospun carbon nanofiber composite as anode material for high-performance sodium-ion batteries. Electrochim. Acta 222, 1751–1760 (2016).
Huang, B., Liu, S., Li, H., Zhuang, S. & Fang, D. Comparative study and electrochemical properties of LiFePO4F synthesized by different routes. Bull. Korean Chem. Soc. 33, 2315–2319 (2012).
John Wang, J. P., Lim, J. & Dunn, B. Pseudocapacitive contributions to electrochemical energy storage in TiO2 (anatase) nanoparticles. J. Phys. Chem. C 111, 14925–14931 (2007).
Acknowledgements
This work was supported financially by the National Natural Science Foundation of China (No. 21905035), Liaoning Revitalization Talents Program (XLYC1907093).
Author information
Authors and Affiliations
Contributions
Yan Yang and Fangying Zheng wrote the main manuscript text and Lei Wang prepared the figures. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yang, Y., Zheng, F., Wang, L. et al. 3D MoS2/graphene oxide integrated composite as anode for high-performance sodium-ion batteries. Sci Rep 14, 19231 (2024). https://doi.org/10.1038/s41598-024-69959-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-69959-3
- Springer Nature Limited