
Abstract
NR2F2 encodes COUP-TFII, an orphan nuclear receptor required for the development of the steroidogenic lineages of the murine fetal testes and ovaries. Pathogenic variants in human NR2F2 are associated with testis formation in 46,XX individuals, however, the function of COUP-TFII in the human testis is unknown. We report a de novo heterozygous variant in NR2F2 (c.737G > A, p.Arg246His) in a 46,XY under-masculinized boy with primary hypogonadism. The variant, located within the ligand-binding domain, is predicted to be highly damaging. In vitro studies indicated that the mutation does not impact the stability or subcellular localization of the protein. NR5A1, a related nuclear receptor that is a key factor in gonad formation and function, is known to physically interact with COUP-TFII to regulate gene expression. The mutant protein did not affect the physical interaction with NR5A1. However, in-vitro assays demonstrated that the mutant protein significantly loses the inhibitory effect on NR5A1-mediated activation of both the LHB and INSL3 promoters. The data support a role for COUP-TFII in human testis formation. Although mutually antagonistic sets of genes are known to regulate testis and ovarian pathways, we extend the list of genes, that together with NR5A1 and WT1, are associated with both 46,XX and 46,XY DSD.
Similar content being viewed by others

Introduction
Gonadal sex determination is the process by which a sexually reproducing organism initiates differentiation as a male or female. Commitment of a common gonadal progenitor to either male (Sertoli cell) or female (granulosa cell) fate is the outcome of a battle between poorly characterized organ-specific, mutually antagonistic gene regulatory networks that canalize development down one organogenetic pathway, whilst actively repressing the alternate1. In XY individuals, the Sex determining region Y (Sry) gene initiates a genetic cascade that leads to upregulation of SRY-box transcription factor 9 (Sox9) beyond a critical threshold1. This results in the differentiation of bipotential somatic cell precursors into Sertoli cells that orchestrate testicular development. In the absence of SRY, in XX individuals, commitment to the granulosa cell lineage occurs via RSPO1/WNT4/β-CATENIN and RUNX1/FOXL2 gene regulatory networks2,3. Gonadal sex determination is followed by the differentiation of internal and external genitalia regulated by the presence or absence of androgens and anti-Müllerian hormone4.
Errors in the sex determination/development cascades give rise to a heterogeneous group of pathologies termed disorders/differences of sex development (DSD)5. DSD are congenital conditions in which the development of chromosomal, gonadal or anatomical sex is discordant6. DSD can present in isolation, or with multiple somatic anomalies in various syndromes6. Based on chromosomal composition, DSD are classified into three groups—46,XY DSD, 46,XX DSD, and sex chromosome DSD. 46,XY DSD present as genital under-virilization and encompass anomalies of testicular development, androgen synthesis or action, persistent Müllerian duct syndrome and unclassified structural variations7. 46,XX DSD includes testicular or ovotesticular DSD, where XX individuals develop testis or ovotestis, and have virilized genitalia due to excessive androgen8. Despite recent technological advances, a definitive genetic diagnosis is achieved in less than 50% of 46,XY DSD cases and 20% of 46,XX testicular or ovotesticular DSD9,10,11. Etiologically, 46,XY and 46,XX DSD were considered to be distinct pathologies caused by mutations in different groups of genes, however, recent data provide evidence to the contrary. Pathogenic variants in Nuclear receptor subfamily 5 group A member 1 (NR5A1) were initially associated with a wide range of reproductive phenotypes including 46,XY DSD, male infertility and primary ovarian insufficiency in 46,XX individuals12,13,14. Recently, missense variants involving a specific amino acid residue (p.Arg92) were described in association with 46,XX testicular or ovotesticular DSD15,16,17,18. Similarly, pathogenic variants of the Wilms tumor 1 (WT1) gene were first reported in association with syndromic 46,XY DSD19,20,21,22, and subsequently, variants involving the 4th zinc finger of WT1 were described in individuals presenting with 46,XX testicular or ovotesticular DSD23.
Nuclear receptor subfamily 2 group F member 2 (NR2F2) gene encodes the orphan nuclear receptor chicken ovalbumin upstream promoter-transcription factor II (COUP-TFII)24. COUP-TFII is involved in many essential processes such as metabolic homeostasis, angiogenesis, organogenesis, and cell fate determination and differentiation during embryonic development25. In the developing murine and human ovaries, COUP-TFII protein is detected in the interstitial/stromal population, presumed to be the precursor of theca cells26,27. In adult human ovaries, COUP-TFII is co-expressed with the steroidogenic enzyme 17α-hydroxylase in the theca cells28. In fetal human testes, COUP-TFII is detected from 7 weeks of gestation in interstitial cells that later become Leydig cells29, where it physically interacts with NR5A1 to regulate Insulin-like 3 (Insl3) gene expression required for testicular descent in mice30,31,32. COUP-TFII is also expressed in pituitary gonadotropes where it represses NR5A1-mediated activation of rat Luteinizing hormone subunit beta (Lhb) promoter33. Pathogenic variants in NR2F2 have been associated with a range of pathologies, including facial dysmorphism, congenital heart defect, diaphragmatic hernia, asplenia and 46,XX DSD (OMIM#107773)24,26,34,35,36. To date, five patients have been reported with 46,XX testicular or ovotesticular DSD, together with developmental abnormalities of the eyelids, heart and diaphragm. These include two patients carrying de novo NR2F2 frameshift variants, c.103_109delGGCGCCC (p.Gly35Argfs*75), c.97_103delCCGCCCG (p.Pro33Alafs*77), one with the c.97_103delCCGCCCG (p.Pro33Alafs*77) variant (unknown mode of transmission), one patient with a de novo missense c.23G > A variant (p.Trp8Ter), and another with a de novo deletion at 15q26.2 encompassing NR2F2 locus26,34,36.
In a large cohort of DSD patients, we previously observed a de novo heterozygous missense variant in NR2F2 (NM_021005.4:c.737G > A, p.Arg246His) in a 46,XY boy37. In this study, we evaluated the functional consequences of the COUP-TFII p.Arg246His variant. We showed that the mutant protein did not affect the nuclear localization or steady-state level of the protein, nor did it interrupt the protein–protein interaction with a known biologically relevant cofactor, NR5A1. However, we observed that COUP-TFII p.Arg246His significantly lost the inhibitory effect on NR5A1-dependent activation of target promoters including LHB and INSL3. Our data support the pathogenicity of COUP-TFII p.Arg246His variant and expand the list of genes, whose variants are associated with both 46,XX and 46,XY DSD.
Results
Clinical phenotype of the patient
A 2-year-old 46,XY boy, of healthy non-consanguineous parents with North African ancestry, presented with under-virilized genitalia37. DSD was not reported in any other members of the family (Fig. 1A). Clinical examination showed micropenis, middle hypospadias, well-formed scrotum, and inguinal testes. Müllerian structures were not observed on abdo minopelvic ultrasonography and other visceral organs were unremarkable. Hormonal assessment at the presentation and reassessment at 5 years of age showed age-appropriate levels of serum gonadotropins, androgens and anti-Müllerian hormone (Table 1). There was no reported anomaly in other organ systems. The clinical diagnosis was non-syndromic 46,XY DSD of unknown etiology.

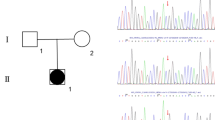
Identification and modeling of the COUP-TFII p.Arg246His variant. (A) The pedigree of the family. Squares denote male and circles denote female family members. Solid square represents the patient. (B) Location of the Arg 246 residue. Chromatogram shows the de novo heterozygous change in NR2F2 (NM_021005: c.G737A > p.Arg246His). Schematic representation of the functional domains of the COUP-TFII protein is shown with an arrow indicating the position of the p.Arg246His mutation. The N-terminus Activation-Function 1 (AF-1), the DNA-Binding domain (DBD), the Ligand-Binding domain (LBD) and the C-terminus Activation-Function 2 (AF-2) domains are indicated. The mutated residue is located in the LBD. (C) Structure of COUP-TFII LBD showing the location and polar interactions of the WT and mutant residues. The LBD structure is drawn as cartoon; wild-type (blue, left), mutated (red, middle) or superimposed (right) amino acids are shown as sticks. The wild-type Arg 246 residue is involved in two polar interactions. One involves the neighboring Val 242 located on the same alpha-helix, the other one, the residue Glu 393 located on an opposite alpha-helix, stabilizing AF-2 domain. The mutant His 246 residue has lost the ability to interact with Glu 393. (D) Evolutionary conservation of Arg 246 residue. Sequence alignment of the partial LBD of human COUP-TFII protein with other species (left) and the LBD of several nuclear receptors (right) shows a high degree of amino acid conservation. The position of the mutation is highlighted by the box.
The mutated residue is located within the ligand binding domain (LBD) and is predicted to affect protein function
Whole exome sequencing identified a novel heterozygous variant in the NR2F2 gene (NM_021005: c.G737A > p.Arg246His) (Fig. 1B). Pathogenic variants in other known 46,XY DSD-causing genes were not observed38. Sanger sequencing of the patient and his parents confirmed that the NR2F2 variant was de novo. It is absent in public databases (https://gnomad.broadinstitute.org/), is classified as likely pathogenic based on the American College of Medical Genetics criteria39, and is predicted to be deleterious by multiple algorithms including PolyPhen (0.988), SIFT (0.03), REVEL (0.922), MutationTaster (0.99) and CADD (32). The mutated residue is localized in the LBD of COUP-TFII (Fig. 1B). In the WT protein, Arg 246 is involved in two polar interactions, one with the neighboring Val 242 (2.8 Å) and a second one with the Glu 393 residue (3.4 Å) located on opposite helix (Fig. 1C). The mutant His 246 cause the loss of hydrogen bound with Glu 393. The residue Arg 246 is highly conserved across species, as well as in the ligand-binding domain of several other nuclear receptors (Fig. 1D). Analysis of the structural effects of the variant on the protein was performed using HOPE (https://www3.cmbi.umcn.nl/hope/)40. Unlike positively charged wild-type (WT) arginine, the mutated histidine residue is neutral, which is predicted to disrupt the ionic interaction with glycine at position 403, affecting the protein conformation.
The mutation does not affect the steady-state level, subcellular localization of COUP-TFII or interaction with NR5A1
We evaluated the effect of the mutation on the production of COUP-TFII protein by Western blot. The whole protein extract was obtained from Human Embryonic Kidney 293 containing the SV40 large T antigen (HEK293-T) cells transiently transfected with plasmids encoding WT or mutant COUP-TFII. Both WT and mutant proteins were detected around 46 kDa at comparable (P = 0.84) levels of expression (Fig. 2A and Supplementary Figure S1). Immunocytochemistry revealed that the mutation did not alter the subcellular localization of the protein with both the WT and mutant COUP-TFII localizing to the nucleus (Fig. 2B). The Arg 246 residue is located within the highly-conserved LBD of the protein, and plays a crucial role in ligand recognition and ligand-dependent activation41. COUP-TFII physically interacts with NR5A1 to regulate gene transcription32. Duolink proximity ligation assay revealed that the COUP-TFII p.Arg246His maintained the binding with NR5A1 similar to that of the WT protein (Fig. 2C).

The mutation does not impact the protein stability, subcellular localization or interaction with NR5A1. (A) Protein stability. Left: Whole cell protein was extracted from the HEK293-T cells transfected with either WT or p.Arg246His COUP-TFII-encoding plasmids. Western blot analysis showed a single band around 46 kDa for both COUP-TFII-WT and COUP-TFII-p.Arg246His. The image is cropped from the original full-length blot shown in Supplementary Figure S1 and is representative of three independent experiments. Right: the band intensity was normalized to that of β-Actin and plotted as mean ± SEM of COUP-TFII/β-Actin ratios obtained from three independent experiments. There was no significant difference between the COUP-TFII-WT and COUP-TFII-p.Arg246His protein production/stability (Student t-test, P = 0.84). (B) Nuclear localization of COUP-TFII. HEK293-T cells were transfected with WT or mutant COUP-TFII expression vectors. Immunocytochemistry showed strong nuclear localization for both WT and mutant proteins (green). Images were taken at 40X magnification (Scale bar: 20 µm). (C) The mutation does not affect the interaction between COUP-TFII and NR5A1. Left: NR5A1 and WT or p.Arg246His COUP-TFII-encoding plasmids were transiently expressed for 48 h in HEK293-T cells. Protein–protein interaction was assessed by the Duolink proximity ligation assay (PLA). Nuclei are stained with DAPI (blue) while each signal/dot (green) represents a COUP-TFII/NR5A1 interaction event. Right: quantification of PLA. For each condition, the number of interactions of at least 50 individual cells was counted and the median (interquartile range) was analyzed. Statistical analysis revealed that the binding of NR5A1 to COUP-TFII-p.Arg246His was comparable to that of COUP-TFII-WT (Mann–Whitney test, P = 0.22). Images were taken at 63X magnification (Scale bar: 20 µm).
Mutant COUP-TFII alters the transactivation activity of NR5A1 on target promoters
We investigated whether the mutation altered the transcriptional activity of COUP-TFII. Lhb, Insl3 and Cyp11a1 promoters are known targets of COUP-TFII and the expression of these genes is activated by NR5A114,32,33,42,43. A previous study by Zheng et al. reported that COUP-TFII repressed NR5A1-dependent activation of rat Lhb promoter33. We assessed the transactivation ability of WT and mutant COUP-TFIIs using rat and human Lhb/LHB promoters in HEK293-T cells. We observed similar inhibition of NR5A1-dependent activation of both the rat (Supplementary Figure S2) and the human LHB promoter (Fig. 3A) by the WT COUP-TFII. The inhibitory effect was also significant with WT COUP-TFII alone, and may reflect repression of endogenous NR5A1 transcriptional activity in HEK293-T cells. However, COUP-TFII p.Arg246His showed a significant decrease (P < 0.001) in inhibition. We then determined if the mutation affected the enhancing activity of COUP-TFII on NR5A1-dependent transcriptional activation of the Leydig cell factor INSL3 that is required for testicular descent30,31,32, and CYP11A1 encoding a steroidogenic enzyme. WT COUP-TFII reduced the NR5A1-mediated activation of the human INSL3 and CYP11A1 promoters. This effect was abolished on INSL3 promoter when using the mutant COUP-TFII protein (P < 0.001) (Fig. 3B), but we did not observe any significant effect on the minimal CYP11A1 promoter (Supplementary Figure S3). We also investigated the effect of WT or/and mutant COUP-TFII, alone or in combination with NR5A1 on endogenous expression of selected target genes (STAR, CYP11A1, CYP17A1 and INSL3) in HEK293-T cells (Supplementary Figure S4). We observed a strong activating effect of NR5A1 on the expression of these genes, whereas there was no significant difference between WT and mutant COUP-TFII alone or in combination with NR5A1.

Mutant COUP-TFII shows loss of inhibitory effect on NR5A1-dependent activation of target promoters. The transcriptional activities of COUP-TFII-WT and COUP-TFII-p.Arg246His were studied using the human (A) LHB and (B) INSL3 promoters as reporters, following transfection in HEK293-T cells. The data shown represent the dot plot of results with mean ± SEM of a minimum of three independent experiments, each with twelve replicates. The reporter constructs were transfected into HEK293-T cells with either the COUP-TFII-WT and COUP-TFII-p.Arg246His expression vector with or without NR5A1-WT expression vector. All data were standardized for Renilla activity. The results are expressed as relative percentage of NR5A1-WT activity (100%). COUP-TFII-WT exerts an inhibitory effect on NR5A1-mediated activation of both (A) LHB and (B) INSL3. However, there is a loss of inhibition by COUP-TFII-p.Arg246His. The statistical significances are marked by the horizontal lines (dash line *, P < 0.05; bold line ***, P < 0.001).
Discussion
We identified a heterozygous missense variant in NR2F2 (NM_021005: c.G737A > p.Arg246His) in a boy presenting with non-syndromic 46,XY DSD. In-silico predictive analysis supported the pathogenicity of the variant. The variant did not affect the steady-state level, subcellular localization, or binding capacity of COUP-TFII with NR5A1. However, as compared to the wild-type, the mutant protein failed to appropriately suppress NR5A1-dependent activation of target promoters.
The biological function of COUP-TFII in testis development has been deduced from murine models, which indicate a key role for the protein in Leydig cell differentiation and androgen production via the regulation of genes encoding steroidogenic enzymes. The inducible depletion of Nr2f2 at E18.5 in XY mice results in Leydig cell hypoplasia, whilst Nr2f2-depletion at pre-pubertal stages results in reduced expression of several genes encoding steroidogenic enzymes, including Star, Cyp11a1, Hsd3b1 and Cyp17a1, as well as decreased testosterone biosynthesis43. In murine immortalized Leydig cell lines (MA-10 and MLTC-1), silencing COUP-TFII reduces the expression of Star, which shuttles cholesterol from the outer to the inner mitochondrial membrane and initiates steroidogenesis, resulting in significantly decreased testosterone production44. This effect may also be caused by the reduced expression of another COUP-TFII target, Glutathione S-Transferase Alpha 3 (Gsta3), that is required for the production of ∆4-androstene-3,17-dione, a precursor of testosterone45. In the human, COUP-TFII expression is detected from gestational week (GW) 7 in the interstitial compartment of fetal testes29. At GW8, Leydig cells can be identified in the interstitial tissue associated with initiation of testosterone production29,46. NR2F2 expression is down-regulated in fetal Leydig cells by GW15 followed by a decrease in the testosterone levels29,47. Taken together these data support a regulatory role for COUP-TFII in testosterone biosynthesis during the first trimester of gestation.
The under-virilization of the external genitalia observed in the boy described in this study could be caused by reduced levels of prenatal testosterone either due to hypogonadotropic hypogonadism or by primary hypogonadism. In humans, testosterone production is regulated by placental human chorionic gonadotropin during the first trimester of gestation, and subsequently by luteinizing hormone of the fetal pituitary from mid-gestation48. Testosterone is required for the formation of male external genitalia within the first trimester, and partly for penile growth and inguinoscrotal descent of testes during the third trimester48. Therefore, anomalies of external genitalia, including micropenis, cryptorchidism and hypospadias are common in patients with 46,XY DSD, in whom testosterone synthesis or action is impaired from an early gestational stage49. In contrast, hypospadias is rarely observed in patients with congenital hypogonadotropic hypogonadism where luteinizing hormone secretion is low and may impact genital development at a later stage48,50,51. In addition, less than 30% of patients with congenital hypogonadotropic hypogonadism present with micropenis and undescended testes51. Therefore, in this case, the under-virilization including micropenis, undescended testes together with hypospadias support the diagnosis of 46,XY DSD due to primary hypogonadism.
Variants in NR2F2 are associated with syndromic testicular or ovotesticular DSD26,34,36. However, the contribution of NR2F2 variants to 46,XY DSD remains unclear. Single nucleotide variants and large genomic rearrangements involving the NR2F2 gene have been reported in association with under-virilization of the external genitalia as well as somatic anomalies in 46,XY individuals35,52,53,54,55,56,57. In mice, the conditional deletion of Nr2f2 causes cryptorchidism in addition to cardiovascular and diaphragmatic defects54. In the human, chromosomal deletions encompassing the 15q26.2 region (the locus for human NR2F2) have been reported in boys with multiple dysmorphic features (Supplementary Table 1)52,53,55,56,57. These rearrangements range from 36 kb to 8.6 Mb in size52,53,55,56,57, and involve approximately 60 genes56. The genital anomalies include undescended testes, hypospadias, micropenis and scrotal hypoplasia. However, due to the size of these deletions and the large number of genes involved, it is not possible to directly correlate the clinical presentation with haploinsufficiency of NR2F2. Two studies described four de novo missense variants, c.269A > G (p.His90Arg), c.287G > A (p.Cys96Tyr), c.1022C > A (p.Ser341Tyr) and c.1097G > C (p.Arg366Pro), and a de novo frameshift c.558dup variant (Arg187Alafs*122) in 46,XY boys with cryptorchidism, glandular hypospadias and multiple congenital anomalies35,36. However, functional studies that support the variant causality on genital phenotypes were unavailable in these cases.
Based on the crystal structure, COUP-TFII is in an auto-repressed conformation in the absence of ligands41. The interaction between the activation-function 2 (AF-2) of the LBD and the co-factor binding site stabilizes the structure and prevents the recruitment of co-activators or co-repressors. The AF-2 conformational states determine the activity of nuclear receptors58. The AF-2 of COUP-TFII is stabilized by a hydrogen bond between the Arg 246 and Glu 393 residues (Fig. 1C)41. Mapping the mutated His 246 residues on the LBD backbone revealed a loss of interaction with Glu 393. In-vitro studies show mutagenesis of residues 249 and 250 which lie adjacent to Arg 246 and line the LBD pocket caused a 50% decrease in transactivation of the target Nerve Growth Factor-Induced protein A (NGFI-A) promoter compared to the WT protein41. Thus, we hypothesized that the COUP-TFII p.Arg246His variant may disrupt the conformation of the AF-2 domain, and consequently alter the transcriptional activity of the protein. This is supported by our in-vitro assays, which show a significant loss of function for the mutant COUP-TFII protein. In pituitary gonadotropes, COUP-TFII acts as a transcriptional repressor of NR5A1 mediated activation of rat Lhb promoter33. Of the two NR5A1-binding elements identified on rat Lhb promoter, COUP-TFII binds to the 3′-domain in competition with NR5A1 and thereby decreases NR5A1-mediated activation33. Unlike the 5′-binding element, the sequence of the 3′-binding element is conserved in human LHB promoter (Supplementary Figure S5). We observed that COUP-TFII repressed the NR5A1-mediated activation of human and rat LHB promoters (Fig. 3A). The COUP-TFII p.Arg246His variant results in the loss of this inhibitory effect on NR5A1-mediated activation of the human LHB promoter. Previous studies have shown that COUP-TFII acts synergistically with NR5A1 to enhance INSL3 promoter activity in mouse Leydig cells and CV-1 fibroblast cells32,59. This synergy is proposed to be due to the recruitment of another transcription factor(s) acting as a co-activator32. In contrast, we observed that in HEK293-T cells COUP-TFII decreases the NR5A1-dependent activation of INSL3 promoter in HEK293-T cells, whereas the COUP-TFII p.Arg246His shows a loss of this repressive activity. COUP-TFII can function as a transcriptional repressor or activator depending on the cellular and genetic context24. Our observation of COUP-TFII acting as a transcription repressor of NR5A1-dependent INSL3 activation could be due to the use of different cell lines that have distinct endogenous transcription factors and cofactors.
Although mutually antagonistic sets of genes are known to regulate testis and ovarian pathways, we extend the list of genes, together with NR5A1 and WT1, that are associated with both 46,XX and 46,XY DSD12,15,19,21,23,26,36. The fact that there is no overlapping variant in these genes causing DSD in both sexes may reflect their distinct roles within testes and ovaries, involving sex/organ-specific mechanisms that remain to be identified.
Materials and methods
Patient samples
The patient met the revised 46,XY criteria of the Pediatric Endocrine Society (LWPES)/European Society for Pediatric Endocrinology (ESPE). The study was approved by the local French ethical committee (2014/ 18NICB-registration number IRB00003835) and all methods were performed in accordance with the relevant guidelines and regulations. Family history including the birthplace, language, and ethnicity of the participant and his parents was obtained by self-reporting.
Whole exome sequencing
Following whole exome sequencing, exon enrichment was performed using Agilent SureSelect Human All Exon V4. Paired-end sequencing with an average sequencing coverage of × 50 was performed on the Illumina HiSeq2000 platform. The sequencing platform of the manufacturer′s proprietary software was applied for generating read files which were then mapped with the Burrows-Wheeler Aligner to the human genome reference (hg38, http://hgdownload.cse.ucsc.edu/goldenPath/hg38/bigZips/analysisSet/hg38.analysisSet.2bit). Duplicate reads were marked by using Picard. Additional BAM files were sorted by SAMtools. The GATK version 1.6 was used for local realignment of the mapped reads around potential insertion/deletion (indel) sites. Single nucleotide polymorphism (SNP) and indel variants of each sample were called by the GATK Unified Genotyper. SNP novelty was determined against dbSNP138. Common variants were filtered using dbSNP (build 138) (www.ncbi.nlm.nih.gov/projects/SNP/), the 1000 Genomes Project (http://www.1000genomes.org/), and the gnomAD database (http://gnomad.broadinstitute.org/). After dataset filtering, novel or rare (minor allele frequency < 0.01) variants were detected and analyzed by using the EnsEMBL SNP Effect Predictor (http://www.ensembl.org/homosapiens/userdata/uploadvariations). The analyses focused on non-synonymous coding, loss-of-function, and splice-site variants. Novel or rare variants were confirmed by visual examination using the IGV browser. The potential pathogenicity of the variants was determined by in silico analysis and potential pathogenic variants were confirmed by Sanger sequencing.
In silico analysis
Predictions on the structural and functional effects of the variant on the COUP-TFII protein (UniProt sequence #P24468) were assessed using freely available HOPE software (http://www3.cmbi.ru.nl/hope/)40. The X-ray structure of the human COUP-TFII ligand binding domain (LBD; PDB no. 3CJW; AA 175-414) was used in PyMOL software (Schrödinger, LLC. 2010. Version 2.6.0) to map the wild-type Arg 246 and mutant His 246 residues and study the resulting polar interactions within the protein structure.
Plasmids and site-directed mutagenesis
Vector containing full-length NR2F2 coding sequence (pCMV6-NR2F2, #SC108069, Origene) was used for evaluating COUP-TFII functions in this study. The pCMX-NR5A1 vector containing NR5A1 WT cDNA sequence has been described elsewhere60. The pCMX (https://www.addgene.org/vector-database/2249/) and pCDNA6 (#V22120, ThermoFisher Scientific) empty vectors were used as controls13. The Luciferase reporter containing INSL3 promoter was a generous gift from Prof. Jacques J. Tremblay (Université Laval, Québec, Canada) and has been described elsewhere45. The Lhb promoter sequences from the rat (-207/+ 5)33, and the equivalent region in the human (− 207/ + 5) were cloned into pGL3-basic Luciferase reporter-vector (E1751, Promega) using the forward primer containing XhoI restriction site: 5′-CGGGCTCGAGTTCCCAATGTCAGTTAAGC-3′ and the reverse primer containing SacI restriction site: 5′-GGTACCGAGCTCTTCCCAATGTCAGTTAAGC-3′ for the rat sequence, and the forward primer containing XhoI restriction site: 5′-CGGGCTCGAGGTCTCTGCCTCACCTCT-3′ and the reverse primer containing SacI: 5′-GGTACCGAGCTCGTCTCTGCCTCACCTCT-3′ for the human sequence.
NR2F2 expression vector containing the NM_021005.4:c.737G > A (p.Arg246His) variant was generated by site-directed mutagenesis (QuikChange II, Stratagene), using the forward primer: 5′-GACCAGGTGGCCCTGCTTCACCTCACCTGGAGCGAGC-3′ and the reverse primer 5′-GCTCGCTCCAGGTGAGGTGAAGCAGGGCCACCTGGTC-3′.
Plasmids were amplified following heat-shock transformation of NEB-5alpha competent cells (#C2992H, BioLabs), and purified with the NucleoBond Xtra Maxi Plus kit (#740416, Macherey–Nagel). The sequence of all plasmids was confirmed by direct sequencing before performing functional studies.
Cell culture
HEK293-T cells were cultured in DMEM medium (#31966-021, Gibco) supplemented with 10% fetal bovine serum (#10270-106, Gibco) and 1% Penicillin/Streptomycin 10,000 U/mL (#P06-07100, PAN Biotech). The cells were passaged every two days when cellular confluence reached 70–90%.
Transient gene expression assays
Transient gene expression assays to assess COUP-TFII function were performed in 96-well plates (#0030730119, Eppendorf) using HEK293-T cells, FuGENE 6 transfection reagent (#E231A, Promega), and Dual Luciferase Reporter Assay system (#E1910, Promega) with pCMV-RL Renilla luciferase (pRL Renilla (#E2231, Promega)) expression as a marker of transfection efficiency.
For each well of 96-well, 25,000 HEK293-T cells were seeded and transfected with FuGENE 6 and plasmid DNA at the carrier (µL):DNA (µg) ratio of 3:1. The transfection protocol was optimized from previous studies on Lhb and INSL3 promoters33,45 (Supplementary Figure S6, Supplementary Tables S2 and S3). A total of 50 ng of wild-type or mutant NR2F2 expression vector were co-transfected with or without pCMX-NR5A1 (5 ng) into HEK293-T cells with rat Lhb or human LHB-reporter reporter (50 ng) and pRL Renilla (5 ng). For assays using the INSL3-reporter, 6.25 ng of wild-type or mutant NR2F2 expression vector were co-transfected with or without pCMX-NR5A1 (6.25 ng) into HEK293-T cells with INSL3-reporter (100 ng) and pRL Renilla (5 ng). For all transfections, the total concertation of DNA was kept uniform in each well, with the empty control vectors making up the difference.
Cells were lysed 48 h after transfection. The promoter activity was measured by the Dual-Luciferase Reporter Assay system (#E1910, Promega) using the Glomax Multi + Detection System (Promega). All data were standardized for Renilla activity. Results were plotted as the mean ± standard error of the mean (SEM) of three independent experiments, each with 12 replicates. Statistical analysis was performed using RStudio (Boston, MA, USA). Outliers were determined by the interquartile range method, and removed. Pairwise comparisons were performed using the approximative two-sample Fisher-Pitman permutation test with stratification of the three independent experiments (Rstudio, coin package, one-way test). A P-value of less than 0.05 was considered statistically significant.
Immunofluorescence
To study the subcellular localization of the COUP-TFII variants, 20,000 HEK293-T cells were seeded in each well of LabTek chamber slides (#055071, Dutscher) and were transfected using FuGENE 6 with 100 ng of plasmid encoding whether WT or p.Arg246His COUP-TFII. After 48 h, the medium was removed. Cells were rinsed with 1X phosphate-buffered saline (PBS) (#D8537, SIGMA) and fixed with 4% paraformaldehyde for 10 min. After three washes with 1X PBS (5 min each), cells were permeabilized with PBS-0.1% Triton X-100 (#0694-1L, Amresco), and saturated with PBS-5% Bovine Serum Albumin (BSA) (#A3059-50G, SIGMA), for 30 min each. Cells were incubated overnight with rabbit anti-COUP-TFII antibody (1:200, #ab42672, Abcam) diluted in PBS-3% BSA at 4 °C. The following day, cells were washed thrice, with 1X PBS (5 min each) and incubated at room temperature for 45 min with secondary antibody: Alexa488 goat anti-rabbit (1:1000, #ab205718, Abcam) diluted in PBS-3% BSA. After counterstaining with 4′,6-diamidino-2-phenylindole (DAPI, 1:5,000, #62248, Thermo Scientific), slides were mounted using ProLong Gold Antifade Mountant with DAPI (#P39941, ThermoFisher Scientific). Images were obtained using Zeiss LSM900 confocal microscope at 40X magnification and analyzed using ImageJ61.
Duolink proximity ligation assay (PLA)
To study the interaction of the COUP-TFII variants with NR5A1, 20,000 HEK-293T cells were seeded in each well of a LabTek chamber slide (#055071, Nunc). Cells were co-transfected using FuGENE 6 with 100 ng of plasmid encoding whether WT or p.Arg246His COUP-TFII and 100 ng of pCMX-NR5A1. After 48 h of culture, cells were fixed as described in the immunofluorescence section. The Duolink PLA was performed following the manufacturer’s instructions. In brief, cells were washed with PBS and permeabilized with PBS-0.1% Triton X-100 for 30 min. They were then incubated with the blocking solution (supplied in the kit) for an hour. Subsequently, overnight incubation at 4 °C with both mouse anti-NR5A1 (1:200, #sc-393592, Santa-Cruz Biotechnologies) and rabbit anti-COUP-TFII (1:200, #ab42672, Abcam) primary antibodies was performed. After 16 h, slides were rinsed and incubated successively with PLUS (anti-Rabbit) (#DUO92002-100RXN, SIGMA) and MINUS (anti-Mouse) (#DUO92004-100RXN, SIGMA) PLA probes, ligation solution and amplification solution (#DUO92014-100RXN (Green), SIGMA) at 37 °C. Following several washes, slides were mounted with Duolink In situ Mounting Medium with DAPI (#DUO082040, SIGMA). Images were obtained using Zeiss LSM900 confocal microscope at 63X magnification and analyzed with ImageJ software61. For each condition, the number of interactions was counted in at least 50 individual cells and the median (interquartile range) of interaction numbers was compared using Mann–Whitney test. A P-value of less than 0.05 was considered statistically significant.
Protein preparation
A total of 350,000 HEK293-T cells were seeded in each well of 6-well plates (#EP0030720113, Eppendorf). Transfection was performed after 24 h at 40–50% cell confluence, using 3 µL of FuGENE 6 and 1 µg of plasmid encoding whether WT or p.Arg246His COUP-TFII in each well. After 48 h of culture, cells were rinsed with 1X PBS and lysed with IP lysis buffer (#87788, ThermoFisher Scientific) supplemented with 100X Halt Protease Inhibitor Cocktail (#78440, ThermoFisher Scientific) and 100X EDTA (#78440, ThermoFisher Scientific) for 30 min. The lysates were centrifuged for 15 min at 20,000 g and the supernatants were retrieved. Protein quantification was performed using the Pierce Detergent Compatible Bradford Assay kit (#23,246, ThermoFisher Scientific) according to the manufacturer’s instructions.
Western blot
10 µg of protein samples were incubated for denaturation, with 4X XT loading buffer (#1610791, Bio-Rad) at 95 °C for 5 min. Proteins were separated on Criterion XT 10% polyacrylamide gel (#3450112, Bio-Rad) and transferred to PVDF membrane (#T831.1, Merk Millipore). Membranes were blocked in Tris-buffered saline containing 0.1% TWEEN 20 (#27949, SIGMA) (TBS-T) and 5% non-fat powdered milk for an hour at room temperature. After overnight incubation at 4 °C with rabbit anti-NR2F2 (1:2000, #ab42672, Abcam) diluted in TBS-T-5% BSA, membranes were washed thrice (15 min each) with 1X TBS-T. Following the wash, the membranes were incubated with anti-rabbit (#ab205718, Abcam) IgG antibody coupled to the Horse Radish Peroxidase (HRP) for 45 min. The revelation was performed using Pierce ECL Western blotting substrate (#32132, ThermoFisher Scientific) and X-ray films. The detection of several proteins on the same blot was attained by treating the membrane with Antibody Stripping solution (#L7710A, Interchim) followed by probing with new primary antibodies. β-Actin (1:2500, #A2228, SIGMA) was used for normalization. Band intensity was quantified using ImageJ software61, and results were plotted as the mean ± SEM of three independent experiments. The statistical comparison of the means was performed using the student t-test. A P-value of less than 0.05 was considered statistically significant.
Ethics approval
This study was approved by the local French ethical committee (2014/18NICB; registration no. IRB00003835) and the Independent Ethical Committee at Hospital de Pediatria Garrahan (2016/971). An informed consent to genetic testing was obtained from the parents for this study.
Data availability
The data presented in the study are deposited in the ClinVar repository (https://www.ncbi.nlm.nih.gov/clinvar/), accession number VCV002683753.1.
References
Capel, B. Vertebrate sex determination: evolutionary plasticity of a fundamental switch. Nat. Rev. Genet. 18, 675–689 (2017).
Nicol, B., Estermann, M. A., Yao, H.H.-C. & Mellouk, N. Becoming female: Ovarian differentiation from an evolutionary perspective. Front. Cell Dev. Biol. 10, 944776 (2022).
Nicol, B. et al. RUNX1 maintains the identity of the fetal ovary through an interplay with FOXL2. Nat. Commun. 10, 5116 (2019).
Ostrer, H. Disorders of sex development (DSDs): An update. J. Clin. Endocrinol. Metab. 99, 1503–1509 (2014).
Ahmed, S. F. et al. Society for endocrinology UK guidance on the initial evaluation of a suspected difference or disorder of sex development (Revised 2021). Clin. Endocrinol. (Oxf.) 95, 818–840 (2021).
Hughes, I. A. et al. Consensus statement on management of intersex disorders. Arch. Dis. Child. 91, 554–563 (2006).
Cools, M. et al. Caring for individuals with a difference of sex development (DSD): A consensus statement. Nat. Rev. Endocrinol. 14, 415–429 (2018).
Knarston, I., Ayers, K. & Sinclair, A. Molecular mechanisms associated with 46,XX disorders of sex development. Clin. Sci. 130, 421–432 (2016).
Baxter, R. M. et al. Exome sequencing for the diagnosis of 46,XY disorders of sex development. J. Clin. Endocrinol. Metab. 100, E333-344 (2015).
Eggers, S. et al. Disorders of sex development: insights from targeted gene sequencing of a large international patient cohort. Genome Biol. 17, 243 (2016).
Hughes, L. A. et al. Next generation sequencing (NGS) to improve the diagnosis and management of patients with disorders of sex development (DSD). Endocr. Connect. 8, 100–110 (2019).
Achermann, J. C., Ito, M., Ito, M., Hindmarsh, P. C. & Jameson, J. L. A mutation in the gene encoding steroidogenic factor-1 causes XY sex reversal and adrenal failure in humans. Nat. Genet. 22, 125–126 (1999).
Bashamboo, A. et al. Human male infertility associated with mutations in NR5A1 encoding steroidogenic factor 1. Am. J. Hum. Genet. 87, 505–512 (2010).
Lourenço, D. et al. Mutations in NR5A1 associated with ovarian insufficiency. N. Engl. J. Med. 360, 1200–1210 (2009).
Bashamboo, A. et al. A recurrent p.Arg92Trp variant in steroidogenic factor-1 (NR5A1) can act as a molecular switch in human sex development. Hum. Mol. Genet. 25, 3446–3453 (2016).
Igarashi, M. et al. Identical NR5A1 missense mutations in two unrelated 46,XX individuals with testicular tissues. Hum. Mutat. 38, 4 (2017).
Baetens, D. et al. NR5A1 is a novel disease gene for 46,XX testicular and ovotesticular disorders of sex development. Genet. Med. 19, 367–376 (2017).
Swartz, J. M. et al. A 46,XX ovotesticular disorder of sex development likely caused by a steroidogenic factor-1 (NR5A1) variant. Horm. Res. Paediatr. 87, 191–195 (2017).
Frasier, S. D., Bashore, R. A. & Mosier, H. D. Gonadoblastoma associated with pure gonadal dysgenesis in monozygous twins. J. Pediatr. 64, 740–745 (1964).
Miller, R. W., Fraumeni, J. F. & Manning, M. D. Association of Wilms’s tumor with Aniridia, hemihypertrophy and other congenital malformations. N. Engl. J. Med. 270, 922–927 (1964).
Drash, A., Sherman, F., Hartmann, W. H. & Blizzard, R. M. A syndrome of pseudohermaphroditism, Wilms’ tumor, hypertension, and degenerative renal disease. J. Pediatr. 76, 585–593 (1970).
Pelletier, J. et al. Germline mutations in the Wilms’ tumor suppressor gene are associated with abnormal urogenital development in Denys-Drash syndrome. Cell 67, 437–447 (1991).
Eozenou, C. et al. Testis formation in XX individuals resulting from novel pathogenic variants in Wilms’ tumor 1 ( WT1) gene. Proc. Natl. Acad. Sci. 117, 13680–13688 (2020).
Polvani, S., Pepe, S., Milani, S. & Galli, A. COUP-TFII in health and disease. Cells 9, 101 (2019).
Lin, F.-J., Qin, J., Tang, K., Tsai, S. Y. & Tsai, M.-J. Coup d’Etat: An orphan takes control. Endocr. Rev. 32, 404–421 (2011).
Bashamboo, A. et al. Loss of function of the nuclear receptor NR2F2, encoding COUP-TF2, causes testis development and cardiac defects in 46,XX Children. Am. J. Hum. Genet. 102, 487–493 (2018).
Rastetter, R. H. et al. Marker genes identify three somatic cell types in the fetal mouse ovary. Dev. Biol. 394, 242–252 (2014).
Sato, Y. et al. Immunolocalization of nuclear transcription factors, DAX-1 and COUP-TF II, in the normal human ovary: Correlation with adrenal 4 binding protein/ steroidogenic factor-1 immunolocalization during the menstrual cycle. J. Clin. Endocrinol. Metab. 88, 3415–3420 (2003).
Lottrup, G. et al. Expression patterns of DLK1 and INSL3 identify stages of Leydig cell differentiation during normal development and in testicular pathologies, including testicular cancer and Klinefelter syndrome. Hum. Reprod. Oxf. Engl. 29, 1637–1650 (2014).
Nef, S. & Parada, L. F. Cryptorchidism in mice mutant for Insl3. Nat. Genet. 22, 295–299 (1999).
Zimmermann, S. et al. Targeted disruption of the Insl3 gene causes bilateral cryptorchidism. Mol. Endocrinol. Baltim. Md. 13, 681–691 (1999).
Di-Luoffo, M., Pierre, K. J., Robert, N. M., Girard, M.-J. & Tremblay, J. J. The nuclear receptors SF1 and COUP-TFII cooperate on the Insl3 promoter in Leydig cells. Reprod. Camb. Engl. 164, 31–40 (2022).
Zheng, W., Horton, C. D., Kim, J. & Halvorson, L. M. The orphan nuclear receptors COUP-TFI and COUP-TFII regulate expression of the gonadotropin LHβ gene. Mol. Cell. Endocrinol. 330, 59–71 (2010).
Carvalheira, G. et al. The natural history of a man with ovotesticular 46,XX DSD caused by a novel 3-Mb 15q26.2 deletion containing NR2F2 gene. J. Endocr. Soc. 3, 2107–2113 (2019).
Arsov, T. et al. Expanding the clinical spectrum of pathogenic variation in NR2F2: Asplenia. Eur. J. Med. Genet. 64, 104347 (2021).
Ganapathi, M. et al. Heterozygous rare variants in NR2F2 cause a recognizable multiple congenital anomaly syndrome with developmental delays. Eur. J. Hum. Genet. https://doi.org/10.1038/s41431-023-01434-5 (2023).
Zidoune, H. et al. Novel genomic variants, atypical phenotypes and evidence of a digenic/oligogenic contribution to disorders/differences of sex development in a large North African Cohort. Front. Genet. 13, 900574 (2022).
Baetens, D., Verdin, H., De Baere, E. & Cools, M. Update on the genetics of differences of sex development (DSD). Best Pract. Res. Clin. Endocrinol. Metab. 33, 101271 (2019).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 17, 405–424 (2015).
Venselaar, H., te Beek, T. A., Kuipers, R. K., Hekkelman, M. L. & Vriend, G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinf. 11, 548 (2010).
Kruse, S. W. et al. Identification of COUP-TFII orphan nuclear receptor as a retinoic acid-activated receptor. PLoS Biol. 6, e227 (2008).
van den Driesche, S. et al. Proposed role for COUP-TFII in regulating fetal Leydig cell steroidogenesis, perturbation of which leads to masculinization disorders in rodents. PloS One 7, e37064 (2012).
Qin, J., Tsai, M.-J. & Tsai, S. Y. Essential roles of COUP-TFII in Leydig cell differentiation and male fertility. PloS One 3, e3285 (2008).
Mendoza-Villarroel, R. E., Robert, N. M., Martin, L. J., Brousseau, C. & Tremblay, J. J. The nuclear receptor NR2F2 activates star expression and steroidogenesis in mouse MA-10 and MLTC-1 Leydig cells. Biol. Reprod. 91, 26 (2014).
Mehanovic, S. et al. Identification of novel genes and pathways regulated by the orphan nuclear receptor COUP-TFII in mouse MA-10 Leydig cells†. Biol. Reprod. 105, 1283–1306 (2021).
Mäkelä, J.-A., Koskenniemi, J. J., Virtanen, H. E. & Toppari, J. Testis development. Endocr. Rev. 40, 857–905 (2019).
Reyes, F. I., Boroditsky, R. S., Winter, J. S. & Faiman, C. Studies on human sexual development. II. Fetal and maternal serum gonadotropin and sex steroid concentrations. J. Clin. Endocrinol. Metab. 38, 612–617 (1974).
Young, J. et al. Clinical management of congenital hypogonadotropic hypogonadism. Endocr. Rev. 40, 669–710 (2019).
Lucas-Herald, A. K., Rodie, M. E. & Ahmed, S. F. Update on the management of a newborn with a suspected difference of sex development. Arch. Dis. Child. https://doi.org/10.1136/archdischild-2020-320872 (2021).
Wang, Y., Gong, C., Qin, M., Liu, Y. & Tian, Y. Clinical and genetic features of 64 young male paediatric patients with congenital hypogonadotropic hypogonadism. Clin. Endocrinol. (Oxf.) 87, 757–766 (2017).
Maione, L. et al. Reproductive phenotypes in men with acquired or congenital hypogonadotropic hypogonadism: A comparative study. J. Clin. Endocrinol. Metab. 107, e2812–e2824 (2022).
Butler, M. G. et al. Two patients with ring chromosome 15 syndrome. Am. J. Med. Genet. 29, 149–154 (1988).
Klaassens, M. et al. Congenital diaphragmatic hernia and chromosome 15q26: Determination of a candidate region by use of fluorescent in situ hybridization and array-based comparative genomic hybridization. Am. J. Hum. Genet. 76, 877–882 (2005).
You, L.-R. et al. Mouse lacking COUP-TFII as an animal model of Bochdalek-type congenital diaphragmatic hernia. Proc. Natl. Acad. Sci. 102, 16351–16356 (2005).
Baptista, J. et al. Breakpoint mapping and array CGH in translocations: Comparison of a phenotypically normal and an abnormal cohort. Am. J. Hum. Genet. 82, 927–936 (2008).
Rump, P. et al. Drayer’s syndrome of mental retardation, microcephaly, short stature and absent phalanges is caused by a recurrent deletion of chromosome 15(q26.2→qter). Clin. Genet. 74, 455–462 (2008).
Choi, J.-H. et al. Clinical and functional characteristics of a novel heterozygous mutation of the IGF1R gene and IGF1R haploinsufficiency due to terminal 15q26.2->qter deletion in patients with intrauterine growth retardation and postnatal catch-up growth failure. J. Clin. Endocrinol. Metab. 96, E130–E134 (2011).
Li, Y., Lambert, M. H. & Xu, H. E. Activation of nuclear receptors: A perspective from structural genomics. Struct. Lond. Engl. 1993(11), 741–746 (2003).
Mendoza-Villarroel, R. E. et al. The INSL3 gene is a direct target for the orphan nuclear receptor, COUP-TFII, in Leydig cells. J. Mol. Endocrinol. 53, 43–55 (2014).
Achermann, J. C. et al. Gonadal determination and adrenal development are regulated by the orphan nuclear receptor steroidogenic factor-1, in a dose-dependent manner. J. Clin. Endocrinol. Metab. 87(4), 1829–1833 (2002).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012).
Bertelloni, S., Russo, G. & Baroncelli, G. I. Human chorionic gonadotropin test: Old uncertainties, new perspectives, and value in 46,XY disorders of sex development. Sex. Dev. 12, 41–49 (2018).
Acknowledgements
We are grateful to the patient and his parents for their participation in this study. We thank Prof. Jacques J. Tremblay (Université Laval, Québec, Canada) for providing the Luciferase reporter plasmid with the human INSL3 promoter.
Funding
This work is funded in part by a research grant from the European Society of Pediatric Endocrinology (to ABa) and by the Agence Nationale de la Recherche (ANR; ANR-10-LABX-73 REVIVE, ANR-17-CE14-0038-01, ANR 20 CE14 0007, ANR-23-CE14-0061 to KM and ANR-19-CE14-0022, ANR-19-CE14-0012, ANR-23-CE14-0068 to ABa). SW is supported by the Faculty of Medicine Ramathibodi Hospital, Mahidol University. The presented work resulted from collaboration made possible through the ESPE-sponsored program “ESPE Visiting Professorship”. In the interest of open-access publication, the author has applied a CC-BY open-access license to any manuscript accepted for publication (AAM) resulting from this submission.
Author information
Authors and Affiliations
Contributions
S.W., K.M., A.B. and M.E. conceived the experiments; S.W., H.Z., L.F., J.B-T., L.S. and M.E. performed the experiments. H.Z., A.B. and N.N. provided primary patient care. S.W., H.Z., D.H., L.F., K.M., A.B. and M.E. collated the experimental data. The manuscript was written by S.W., K.M., A.B. and M.E. All the authors read and agreed with the data being presented in the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wankanit, S., Zidoune, H., Bignon-Topalovic, J. et al. Evidence for NR2F2/COUP-TFII involvement in human testis development. Sci Rep 14, 17869 (2024). https://doi.org/10.1038/s41598-024-68860-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-68860-3
- Springer Nature Limited