
Abstract
Cyclodextrins (CDs) can enhance the stability and bioavailability of pharmaceutical compounds by encapsulating them within their cavities. This study utilized molecular dynamics simulations to investigate the interaction mechanisms between hydrocortisone (HC) and various methylated CD derivatives. The results reveal that the loading of HC into CD cavities follows different mechanisms depending on the degree and position of methylation. Loading into βCD and 6-MeβCD was more complete, with the hydroxyl groups of HC facing the primary hydroxyl rim (PHR) and the ketone side facing the secondary hydroxyl rim (SHR). In contrast, 2,3-D-MeβCD and 2,6-D-MeβCD showed a different loading mechanism, with the ketone side facing the PHR and the hydroxyl groups facing the SHR. The root mean square fluctuation (RMSF) analysis demonstrated that methylation increases the flexibility of CD heavy atoms, with 3-MeβCD and 2,3-D-MeβCD exhibiting the highest flexibility. However, upon inclusion of HC, 3-MeβCD, 2,3-D-MeβCD, 2-MeβCD, and 6-MeβCD showed a significant reduction in flexibility, suggesting a more rigid structure that effectively retains HC within their cavities. The radial distribution function revealed a significant reduction in the number of water molecules within the innermost layer of the methylated CD cavities, particularly in TMeβCD, indicating a decrease in polarity. The presence of HC led to the release of high-energy water molecules, creating more favorable conditions for HC loading. Conformational analysis showed that methylation caused a partial decrease in the area of the PHR, a significant decrease in the area of the middle rim, and a notable decrease in the area of the SHR. The loading of HC increased the area of the PHR in most derivatives, with the most pronounced increase observed in 2,6-D-MeβCD and 6-MeβCD. The analysis of interaction energies and binding free energies demonstrated that the binding of HC to methylated CD derivatives is thermodynamically more favorable than to βCD, with the strongest association observed for 6-MeβCD, 2-MeβCD, and 2,3-D-MeβCD.
Similar content being viewed by others


Introduction
Corticosteroids, a category of steroid hormones secreted by the adrenal cortex, include glucocorticoids and mineralocorticoids1. Glucocorticoids, of which hydrocortisone (HC) is a prime example, are important in the treatment of a variety of inflammatory and autoimmune diseases, including asthma, allergies, septic shock, rheumatoid arthritis, inflammatory bowel disease, and multiple sclerosis2. However, their clinical efficacy is hampered by adverse reactions associated with high doses (as seen in systemic vasculitis and SLE) and prolonged administration. These adverse effects include osteoporosis, skin atrophy, diabetes, abdominal obesity, glaucoma, cataracts, avascular necrosis, increased susceptibility to infection, growth retardation, and hypertension2,3. In addition, their therapeutic potential may be undermined by poor aqueous solubility, which affects their bioavailability and therapeutic efficacy4. To overcome these limitations, researchers have turned to Cyclodextrins (CDs) to form inclusion complexes with corticosteroids5,6.
CDs are cyclic oligosaccharides composed of glucose units arranged in a toroidal structure, creating a hydrophobic cavity capable of hosting hydrophobic drug molecules. Through host–guest interactions, CDs can encapsulate HC molecules, thereby improving their solubility, stability, and pharmacokinetic properties7,8.
Schönbeck et al.9 comprehensively analyzed the phase-solubility (PS) relationship of the HC/γ-cyclodextrin (γCD) system employing isothermal titration calorimetry (ITC), high-performance liquid chromatography (HPLC) for quantification, and precipitate composition analysis. ITC determined a 1:1 binding stoichiometry between HC and γCD with a precise binding constant (K11). The PS diagram exhibited a BS-type shape with three discernible regions. The initial solubility increase of HC was attributed to 1:1 complex formation, followed by a plateau due to the precipitation of a complex with approximately 1.5 γCDs per HC. Higher γCD concentrations led to a marked decrease in HC solubility due to the low solubility product (KS32) of the precipitating 3:2 γCD:HC complex.
Schwarz et al.10 investigated the solubilization of seven steroidal drugs (testosterone, estradiol, etc.) by six βCD derivatives. The anionic thioether heptakis-6-sulfoethylsulfanyl-6-deoxy-β-cyclodextrin (HSES) gave high steroid solubilities up to 19 mM, while the neutral thioethers heptakis-6-methylsulfanyl-6-deoxy-2-(2-(2-(2-methoxyethoxy)ethoxy)ethoxy)ethyl)]-βCD (HTMT) and heptakis-6-thioglyceryl-6-deoxy-βCD (HTG) exhibited selectivity for testosterone and estradiol, respectively. Solubilization occurred due to the inclusion of the steroid rings in the CD cavity. Further research could build upon these promising CD-steroid systems to enable improved delivery of hydrophobic steroidal drugs.
Kristmundsdóttir et al.11 developed a hydrocortisone mouthwash utilizing CDs to improve solubility and stability. The formulation demonstrated stability under storage and heating, effective antimicrobial preservation, and promising results in improving symptoms in 78% of 50 patients with oral inflammatory conditions. Further development of CD-enabled steroid formulations could provide convenient and effective treatments for oral diseases.
Hydrocortisone is frequently prescribed off-label for children, but appropriate pediatric formulations are lacking. As a result, hydrocortisone tablets are often manipulated by crushing or splitting to achieve lower doses, but this can lead to dosing errors or lack of bioequivalence compared to tablets12. To address this issue, Orlu-Gul et al.13 developed a reconstitutable oral solution with a 1:6 HC:hydroxypropyl-β-cyclodextrin (HPβCD) ratio for fast, complete solubilization. Key features included taste-masking with neotame and HPβCD for bitterness and stability, allowing flexible dosing with improved palatability and stability compared to compounded suspensions.
From an experimental viewpoint, guest-CD interactions are investigated using Isothermal Titration Calorimetry (ITC) for thermodynamic parameters, Thermogravimetric and Differential Thermal Analysis (TG/DTA) for thermal stability, Fourier Transform Infrared Spectroscopy (FTIR) for functional group identification, Proton Nuclear Magnetic Resonance (1H-NMR) for structural changes upon complexation14, and X-ray Crystallography for spatial arrangement and chemical bonding, all crucial for CD optimization in applications like drug delivery systems15. In this regard, computational methods such as molecular dynamics simulations offer significant advantages in studying drug-CD interactions and overall in drug delivery as well as other biological systems. They provide detailed, atomic-level insights into binding mechanisms, thermodynamic properties, and structural changes, enabling researchers to optimize drug formulations and predict biological behavior with increased accuracy and efficiency, ultimately advancing the field of personalized medicine16,17,18.
Main strategy
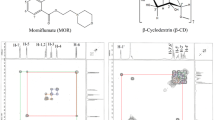
Methylated β-cyclodextrin (MeβCD) offers enhanced drug delivery capabilities compared to β-cyclodextrin (βCD), including improved solubility, bioavailability, reduced toxicity, targeted delivery, controlled release, and enhanced barrier penetration, rendering it advantageous for inclusion in drug delivery systems19,20,21. On the other side, the interaction between hydrocortisone and CDs, particularly methylated-CD, is significant due to its impact on the solubility, stability, and bioavailability of hydrocortisone. CDs form inclusion complexes with steroids like hydrocortisone, enhancing their water solubility and solution stability. For instance, hydroxypropyl-βCD (HPβCD) increases hydrocortisone bioavailability in aqueous humor and cornea5. MeβCDs form more soluble complexes with hydrocortisone compared to the native βCD22. MeβCDs decelerate the alkaline and acidic degradation of hydrocortisone, providing increased chemical stability23. The hydroxide ion-catalyzed rearrangement of hydrocortisone 17-butyrate into the 21-butyrate of lower biological activity was catalyzed by βCD and inhibited by dimethyl-βCD at pH 2–824. This study aimed to investigate the well-characterized structures of MeβCD from both thermodynamic and structural perspectives and to assess their interactions with HC (Log p = 1.61, water solubility 0.311 mg/mL)25,26,27 for potential use in drug delivery systems. Different polymerization states of MeβCD, which are well-known in experimental research, were considered based on their degree of polymerization28, as illustrated in Fig. 1.

Molecular structure of Methylated β-cyclodextrin derivatives and Hydrocortisone.
First, we simulated each MeβCD structure and βCD separately in water. Then, by placing an HC molecule 1.5 nm away from the secondary hydroxyl rim (SHR), we conducted simulations again to identify the potential of each of the polymerized states and to examine the interactions in the 1:1 stoichiometry without assuming any initial a priori inclusion, as shown in Fig. 2. This approach will illuminate the suitability of each structure for use in drug delivery systems, contributing to the development of more effective and targeted drug formulations.


Snapshots of the initial and final configurations of hydrocortisone toward βCD (a), 2-MeβCD (b), 3-MeβCD (c), 6-MeβCD (d), 2,3-D-MeβCD (e), 2,6-D-MeβCD (f), and TMeβCD (g).
Computational details
Molecular dynamics simulation
The molecular dynamics (MD) simulation was conducted using GROMACS 2022.2 software29,30,31, employing the GROMOS 54a7 force field32,33 to model a binary mixture of CDs and HC immersed in a solvent of 4500 water molecules. A validation related to the force field has been conducted by comparing some of the fundamental experimental parameters associated with βCD, as depicted in Figure S1 and Table S1. The water molecules were represented using the extended simple point charge model (SPC/E)34,35. The simulation employed periodic boundary conditions in all three dimensions (xyz) to mimic bulk behavior. To eliminate unfavorable atomic interactions, a steepest descent energy minimization was employed36, and the systems underwent equilibration processes within the NVT and NPT ensembles for 1 ns and 9 ns, respectively. After achieving equilibration, a total simulation time of 200 ns was considered in the NPT ensemble for each system. Integration of Newton's equations utilized the leap-frog algorithm37 with a time step of 2 fs. Coordinates, velocities, energies, and log data were saved every 2.0 ps, with compressed coordinates stored for efficient analysis. Bonded interactions were maintained using the LINCS algorithm38 with all bonds constrained, while van der Waals (vdW) interactions were truncated at a cutoff radius of 1.2 nm. Coulombic (Coul) interactions were calculated via the Particle Mesh Ewald (PME) method39 with a real-space cutoff of 1.2 nm, complemented by a long-range dispersion correction. Temperature was controlled using the V-rescale technique40 with a time constant of 0.1 ps and a reference temperature of 300 K for each coupling group. Pressure coupling utilized the Parrinello-Rahman method41 with an isotropic pressure coupling scheme, a time constant of 2.0 ps, and a reference pressure of 1.0 bar, considering the compressibility of water set to 4.6 × 10–5 bar-1.
Free energy computations
The solvation free energy of a solvated system was determined by evaluating the free energy difference in a transforming system as it moves between solvated and unsolvated states. This computational analysis involved the utilization of a coupling parameter (λ) in conjunction with the thermodynamic integration (TI) formula42,43.
The Hamiltonian (H) was quantified as a function of the coupling parameter λ. This parameter serves as an indicator of the extent of transformation occurring between specific states, such as the transition from solvated to unsolvated states or between states A and B. The TI method relies on the λ-dependence of the Hamiltonian for solute–solvent interactions, which gradually varies between full interactions (corresponding to λ = 0) and no interaction (corresponding to λ = 1). In other words, the solute molecule is gradually made to disappear from the solution using the coupling parameter λ. The Bennett acceptance ratio method (BAR)44 was employed to assess variations in free energy across different λ values in Hamiltonians. A thermodynamic cycle utilizing alchemical free energy computations45,46 was utilized to ascertain binding free energy. The computation involved gradually reducing the potential energy of interaction between HC and its surroundings using a λ parameter spanning 0 to 147,48. For calculating ΔGsolv, 25 λ points were employed. Initially, Coulombic solute–solvent interactions were disabled at higher λ values compared to vdW terms to prevent unstable Coulombic interactions, which could yield unreliable energies and configurations46,49. Soft-core potentials with specific parameters (α = 0.5, σ = 0.3, and p = 1) were integrated to prevent atom overlap between solute and solvent at low λ values46,47,48. Free energy calculations commenced from the final configuration of each simulated system, with each simulation lasting 5 ns.
Results and discussion
Interaction mechanism
CDs enhance the stability of pharmaceutical substances by protecting them from degradation, oxidation, or hydrolysis6,50. This protective effect is facilitated by the encapsulation of drugs within the cavity of CDs, effectively isolating them from external factors that could compromise their effectiveness or durability7,51. The interaction mechanism between CDs and pharmaceutical compounds is analyzed through the formation of inclusion complexes. This process leads to improvements in solubility, stability, release kinetics, and bioavailability, thereby enhancing the therapeutic effectiveness of the pharmaceutical agent52,53,54,55. The initial and final configurations of all HC-containing systems are illustrated in Fig. 2. The cavities of all CDs show the ability to host HC. Among these, HC is positioned in βCD, 2-MeβCD, 3-MeβCD, 6-MeβCD and TMeβCD with the hydroxyl groups facing the PHR and the ketone side (oxygen group attached to the aromatic ring) facing the SHR. It appears that the loading of HC into the cavities of βCD and 6-MeβCD is more complete, as a larger portion of the HC molecule is accommodated in their cavities. In contrast, loading of HC into the cavities of 2,3-D-MeβCD and 2,6-D-MeβCD follows a different mechanism, with the ketone side facing PHR and the hydroxyl groups facing SHR. In addition, it appears that loading into the cavity of 2,3-D-MeβCD is more complete than in 2,6-D-MeβCD. Although the final positioning of HC is clearly defined, understanding the time evolution of HC entry into the CDs cavities can provide insight into the kinetics of these interactions. To this end, the distances between the centre of geometry (COG) of the HC and the CDs are depicted in Fig. 3. As seen, loading of HC into the cavities of βCD, 6-MeβCD, 2,3-D-MeβCD, 2,6-D-MeβCD and TMeβCD occurs gradually, with entry times of approximately 43.6, 84.1, 55.1, 30.6 and 164.7 ns, respectively. In contrast, entry into the cavities of 2-MeβCD and 3-MeβCD is faster, with entry times of approximately 7.4 and 17.1 ns, respectively. Figure S2 illustrates the moment and mode of HC entry into the CDs cavities. It is evident that in βCD and 6-MeβCD, HC approaches the PHR from its ketone side, so that the efficient loading is facilitated. Conversely, in 2-MeβCD, 3-MeβCD, and TMeβCD, HC approaches the SHR from its hydroxyl groups side, leading to loading. Finally, in 2,3-D-MeβCD and 2,6-D-MeβCD, entry of HC occurs from the ketone side towards the SHR.

Time evaluation the distance between Hydrocortisone and the center of CD molecules.
Root mean square fluctuation (RMSF)
RMSF is crucial in MD simulations for understanding molecule flexibility, such as CDs in drug delivery. It reveals atom fluctuation, helping identify flexible or rigid parts. CD flexibility affects guest molecule binding, with RMSF analysis pinpointing conformational changes. RMSF also predicts encapsulation efficiency, vital for optimizing CD-based systems, improving drug carrier performance48,56,57,58. Here, we first consider the changes in the flexibility of the heavy atoms forming the main skeleton in all methylated derivatives as well as in βCD, followed by a discussion of the changes induced by the presence of HC. As shown in Fig. 4a, among the oxygen atoms in all structures, O6 shows the highest flexibility. This oxygen atom, located at the end of a short chain and distant from the ring, exhibits greater degrees of freedom and flexibility. Conversely, O1 and O5 show the least flexibility, probably due to their position within the solid ring structure. In terms of carbon atoms, C6 shows the highest flexibility, while the carbon atoms within the ring (C1, C3, C4, C5) show much less flexibility. After O6 and C6, the highest flexibility of all oxygen and carbon atoms is observed for O2 and C2. It seems that these two atoms within the ring have greater flexibility compared to others. As a result, methylation increases the flexibility of all heavy atoms, with 3-MeβCD and 2,3-D-MeβCD exhibiting the highest flexibility, distinguishing them from other structures. Remarkably, in the presence of HC (as shown in Fig. 4b), the heavy atoms within 3-MeβCD and 2,3-D-MeβCD, as well as 2-MeβCD and 6-MeβCD, undergo a significant reduction in flexibility. This observation suggests that these methylated derivatives have a more rigid structure compared to their counterparts. It appears that upon inclusion of HC, these four structures lose their flexibility and effectively retain HC within their cavities. Meanwhile, βCD exhibits the least flexibility as the parent structure in the absence of HC. The changes in the RMSF of its heavy atoms are not as severe in the presence of HC, and it remains as the most rigid structure.

Root mean square fluctuation (RMSF) of heavy atoms of CDs in different simulated system.
Water penetration into the cavity
In the absence of a guest molecule, it is common for water molecules to typically fill the CD cavity within an aqueous solution. X-ray and neutron diffraction analyses reveal that α-CD, β-CD, and γ-CD cavities typically contain 2.6, 6.5, and 8.8 water molecules, respectively, distributed within different layer of the cavities59,60,61,62,63,64. These confined water molecules exist in a high-energy state due to the lack of a complete hydrogen bond network. However, upon the introduction of a guest molecule into the cavity, these high-energy water molecules are released, facilitating the formation of a stable cavity-guest complex. This phenomenon indicates the thermodynamic favorability of cavity-guest complexation within CD cavities in aqueous solutions65,66,67,68. Hence, the probability of the presence of water molecules and their arrangement around CDs was determined using the Radial Distribution Function (RDF), as depicted in Fig. 5.

Radial distribution function (RDF) of water around CDs in different simulated systems.
As observed, βCD contains a significant number of water molecules at distances of less than 0.5 nm (the innermost layer). In this layer, the probability of the presence of water in the methylated derivatives is reduced, with a more pronounced reduction observed for 3-MeβCD, 2,6-D-MeβCD and TMeβCD, while the outflow of water is less pronounced for 2-MeβCD, 2,3-D-MeβCD and 6-MeβCD, with significant amounts of water still remaining. For a quantitative assessment, considering the location of the first minimum in the RDF at 0.5 nm, hypothetical spheres with different radii were considered in the central region of the CDs cavities and the number of water molecules present in these spheres was counted, as shown in Table S2. The average number of water molecules in this layer (0–0.5 nm) for βCD is 9.9, which is in good agreement with other simulation studies.
As a result of functionalization, there was a significant reduction in the number of water molecules in the innermost layer (0–0.5). The reduction in the number of water molecules in this layer ranges from a minimum of 42% (as observed for 2-MeβCD, with 5.74 water molecules) to a maximum of 97% (as observed for TMeβCD, with 0.25 water molecules). This can be attributed to the decrease in the polarity inside the cavity. Significant changes also occurred in the outermost layer (0.5–1.0), with reductions ranging from a minimum of 1.3% (as observed for 6-MeβCD, with 88.88 water molecules) to 15.4% (as observed for TMeβCD, with 76.15 water molecules). Upon HC loading, a significant release of high-energy water molecules from the innermost layer (0–0.5) of all CD cavities is observed, creating more favorable conditions for HC loading into the cavity. The second layer (0.5–1.0) also experienced a decrease in the number of water molecules due to the presence of HC. Although this decrease was noticeable, it was not as great as in the innermost layer.
Conformational changes
Understanding CDs conformational changes is vital for enhancing drug delivery systems, optimizing material properties, and designing molecular recognition systems. It influences host–guest interactions, nanostructure characteristics, and impacts stability, toxicity, and biological interactions. This knowledge is instrumental in creating advanced drug formulations and functional materials for diverse applications69,70,71. The most significant structural changes in various simulated systems of CDs have been summarized in Table 1. Instructions on computing both the area and volume of the cavity were provided in the Supplementary Information (page 4), and these methods have been confirmed in other simulation studies72,73. The functionalization of βCD and the subsequent formation of methyl derivatives leads to a partial decrease of the APHR in all methylated derivatives, with a notable reduction observed mainly in 2,3-D-MeβCD (1.00 nm2) and to a lesser extent in 3-MeβCD (1.02 nm2). The presence of HC increases the APHR in almost all derivatives. Particularly, for 2,6-D-MeβCD (1.28 nm2) and 6-MeβCD (1.27 nm2), a more pronounced increase is observed. This phenomenon can be attributed to the functionalization of position R6 in both structures, rendering this region more hydrophobic. The repulsive interaction between the hydrophilic oxygen of HC and these hydrophobic groups contributes to the increase in APHR. Conversely, the significant decrease in APHR observed in 2,3-D-MeβCD (0.97 nm2) can be explained by the mechanism of HC loading into its cavity. The attractive interaction between the hydroxyl groups present in this rim and the oxygen of HC, which penetrates deep into the cavity, leads to a significant decrease in APHR. The changes in AMID in methylated derivatives are much more severe, showing a significant decrease compared to βCD (0.83 nm2). It appears that methylation causes the internal part of the cavities of the derivatives to become highly non-polar, leading to intramolecular attraction in this region. Such an event has previously been observed in some βCD derivatives, accompanied by a significant outflow of water from the center of the cavity46. However, the loading of HC into the cavities leads to an increase in the AMID values, which are almost the same for all derivatives as for βCD. As can be seen, the changes in ASHR are also very significant in the methylated derivatives. The largest decrease is observed in 2,6-D-MeβCD, while the smallest decrease is observed in 2,3-D-MeβCD. It seems that the presence of two adjacent methyl groups at positions R2 and R3 causes some repulsion and prevents a further decrease in ASHR. However, upon HC loading, ASHR increases significantly in all derivatives, whereas it decreases in βCD. It seems that for HC loading, ASHR shows much greater structural desirability in methylated derivatives compared to βCD. In both the presence and absence of HC, βCD exhibits high circularity (ΩO1 ≈ 0.99). Despite the limited degree of freedom of the O1 atoms due to bonding with carbon atoms (C1 and C4) within the ring, functionalization of βCD and the formation of methyl structures lead to a decrease in Ω, especially notable in 3-MeβCD (0.92). Upon loading HC, increased steric repulsion interactions between its bulky atoms and the O1 atoms of the carriers appear to result in a more regular and nearly equidistant equilibrium position of the O1 atoms with respect to each other. Consequently, this increases ΩO1 across all carriers, yielding very similar ΩO1 values among them. The CDs heights (h12 and h16) also experience a significant reduction. Specifically, the values of 0.33 (h16) and 0.22 (h12) nm in βCD decreased to 0.22 and 0.12 nm in 2,6-D-MeβCD, and to 0.21 and 0.13 nm in TMeβCD. Other methylated derivatives also experience significant changes in this area, as shown in Table 1. Considering the defined equation for the determination of the volume of CDs cavities (page 4 of the SI), it is clear that what influences the volume of the cavities (VC) more than other structural changes is precisely the height and not the surfaces. Methylated derivatives of βCD seem to undergo substantial height changes after HC loading and are drawn along the \(\mathop{r}\limits^{\rightharpoonup}\) axis (Fig. 1). This consequently leads to a significant increase in VC, with the largest observed in 6-MeβCD and 2,6-D-MeβCD at 0.61 nm3 and 0.60 nm3, respectively. The reduction in VC of the methylated derivatives in comparison with βCD can be attributed to the increase in the non-polar nature of these cavities.
Relative shape anisotropy and surface-to-volume ratio
Relative shape anisotropy (RSA), also known as κ2, refers to the deviation of the shape of a polymer molecule from that of a perfect sphere or cylinder74,75. This characteristic is critical in drug delivery, as it affects the distribution and retention of drug carriers in the body, as well as their transport across biological barriers such as cell membranes and tissue extracellular matrix. Understanding the RSA of drug carriers is essential for improving the efficacy of drug delivery and minimizing side effects76,77. The calculation of RSA involves utilizing the principal moments of the radius of gyration (Rg), typically arranged as λ1 ≥ λ2 ≥ λ3, and summing them to obtain \({\text{R}}_{\text{g}}^{2}={\uplambda }_{1}+{\uplambda }_{2}+ {\uplambda }_{3}\), from which RSA can be derived78:
RSA can take values between 0 and 1, with a value of 0 corresponding to a highly symmetric polymer conformation and reaching 1 for an ideal linear chain. The examination of RSA for all CDs with and without HC was carried out, as shown in Fig. 6. In the absence of HC, βCD and TMeβCD demonstrated the highest symmetry, with RSA showing the highest probability around 0.1 (Fig. 6a). However, due to functionalization, all other methylated derivatives exhibited noticeable asymmetry compared to βCD, with a significant probability concentrated around RSA = 0.2. The methylated derivatives 3-MeβCD, 2-MeβCD, and 2,6-D-MeβCD displayed the greatest level of asymmetry, with probabilities around 4, 3, and 2, respectively. This points to a significant departure from symmetry for these methylated derivatives. Loading the HC altered everything (Fig. 6b), causing all CDs to have highly symmetric structures with the highest probability near RSA = 0.1. Such that a significant probability is not observed around RSA = 0.2, means that the symmetry increases sharply. Only in the case of 2,3-D-MeβCD, to some extent, a probability (~ 0.8) is still observable around RSA = 0.2.

The relative shape anisotropy parameter of CDs in different simulated systems.
The findings suggest that elongating CDs along the \(\mathop{r}\limits^{\rightharpoonup}\) axis (as illustrated in Fig. 1 and Table 1) leads to a more symmetrical structure in HC-containing systems. This symmetrical configuration may not only enhance dissolution and drug delivery but also offers additional benefits. The larger surface area of nanoparticles relative to their volume facilitates increased interaction with the environment, thereby enabling various applications such as drug delivery and catalysis79,80. Hence, we assessed the ratio of the solvent-accessible surface area (SASA) of each CD to its volume, as shown in Fig. 7.

The surface-to-volume ratio (SA/V) in different simulated systems.
As can be seen, in the presence of HC, the SA/V ratio of all CDs increases. The lowest changes correspond to βCD, followed by 2,3-D-MeβCD, and 2-MeβCD. The changes appear to be height-dependent. After functionalization and HC loading, 2,3-D-MeβCD and 2-MeβCD exhibit minimal height (h12 + h16) alterations in comparison to βCD, as shown in Table 1. Conversely, 3-MeβCD, 6-MeβCD, 2,6-D-MeβCD, and TMeβCD experience significant height changes following methylation and HC loading. Overall, the highest SA/V ratio in the presence of HC is associated with 2-MeβCD, βCD, and 2,6-D-MeβCD, respectively. It's important to note, however, that the circular polymer structure of CDs limits the potential expansion of surface area.
Analysis of interaction energies and binding free energy
The interaction energies between the solute and solvent determine the solubility and stability of a compound in a solution. These energies depend largely on Coulombic forces, vdW forces, and hydrogen bonding interactions. The magnitude and orientation of these interactions are affected by the chemical characteristics and shape of both the solute and solvent molecules. Understanding these interactions is essential for designing novel carriers, improving the conditions for reactions, and synthesizing new materials81,82,83.
The BAR method was employed to determine the solvation free energy (ΔGsolv) and binding free energy (ΔGbind) of HC to CDs, as described previously. The ΔGsolv of HC in water was found to be somewhat negative (-13.91 kJ/mol), as expected due to the abundance of hydroxyl groups, despite the hydrophobic nature of the main skeleton. It is evident that the binding of HC to all CDs was spontaneous and energetically favorable. Among these, the most exothermic processes, in order, are associated with the binding of HC to 6-MeβCD, 2-MeβCD, βCD, 2,3-D-MeβCD, 2,6-D-MeβCD, 3-MeβCD, and TMeβCD, as seen in Fig. 8a. Further insights into the forces involved in binding and solvation were gained by analyzing various types of interaction energies in simulated systems.

Energy Analysis among Various Components within Simulated Systems. (All results were obtained from the last 10% of the simulation time.).
Simultaneous investigation was conducted to examine the energy interactions between different components in various systems, as depicted in Fig. 8. The evidence suggests that in the interaction between HC and CDs, the vdW forces predominantly drive the thermodynamic favourability, while the Coulombic interactions have a minor effect (Fig. 8b). Among these interactions, HC showed more thermodynamic favorability with 6-MeβCD, 2,6-D-MeβCD and TMeβCD derivatives compared to other CDs, as indicated by more negative energy values. An interesting observation is that the thermodynamic interaction of HC with methylated derivatives appears to be more favourable than its interaction with βCD. This underlines the potential of these methylated derivatives as promising candidate carriers for HC. In the water/HC system, the energies of the vdW and Coulombic interactions between HC and water were measured to be -118.53 and -47.13 kJ/mol, respectively. In the presence of CDs, the contributions of Coulombic and vdW interactions are significantly reduced (Fig. 8c), ensuring that in none of the systems do the Coulombic and vdW interaction energies between HC and water (total energy) exceed the vdW interaction energy of HC and CDs. Consequently, the loading of HC in all CDs is assessed as thermodynamically favorable. Another important interaction that can determine the solubility of the inclusion complex is the interaction of CDs with water in the presence and absence of HC, Fig. 8d. As a result of methylation, the contribution of the Coulombic interaction between water and CDs is lower in all methylated derivatives than in βCD (energies become more positive), and a Coulombic repulsion due to methylation is observed. Conversely, the contribution of the vdW interaction remains almost unchanged (as seen in 3-MeβCD and 6-MeβCD) or increases in thermodynamic favorability (as seen in 2-MeβCD, 2,3-D-MeβCD, 2,6-D-MeβCD, TMeβCD), which is manifested by the negative shift of the energy of the vdW interaction between CDs and water. In the presence of HC, the Coulombic repulsion in 2,6-D-MeβCD and TMeβCD remains almost unchanged, while in 3-MeβCD and 6-MeβCD, it is partially accompanied by a positive shift. However, it can be seen that a Coulombic repulsion between the CDs and water occurs due to the presence of HC in βCD, 2-MeβCD, and 2,3-D-MeβCD. The same pattern and effect also occur for the contribution of the vdW interaction energies between CDs and water in the presence of HC. However, an interesting observation regarding the total interaction energies between CDs and water in the presence of HC shows that the energy of interaction of methylated derivatives with water in the presence of HC is more negative (in 2-MeβCD, 6-MeβCD, 2,6-D-MeβCD, TMeβCD) or almost equal to βCD (in 3-MeβCD and 2,3-D-MeβCD) as the parent structure, providing further evidence for the thermodynamic favorability of methylated structures as carriers of HC.
Hydrogen bonds
The significance of hydrogen bonding is essential in "host–guest" systems, as it is crucial for selective binding, stability, shape recognition, control of properties, reversibility, and can impact the pharmacokinetics of drugs84,85. Hence, the assessment of hydrogen bond formation ability among different components in simulation systems was carried out, with findings detailed in Table 2. HC in water (in the absence of any CD) forms 31.8 hydrogen bonds with water. Loading HC into CDs significantly reduces the number of this type of hydrogen bonds, with the greatest reduction resulting from loading into 3-MeβCD (1.11) and TMeβCD (0.97). The reason for this phenomenon could be elucidated by considering the mechanism of HC loading. In 3-MeβCD and TMeβCD, as discussed earlier, the loading of HC occurs in such a way that the hydroxyl groups of HC are completely covered by the cavity of the CDs (Fig. 2), making them inaccessible to water. As a result, the number of hydrogen bonds between HC and water is greatly reduced. In 2,3-D-MeβCD (2.37) and 2,6-D-MeβCD (1.96) the highest number of hydrogen bonds between HC and water is observed compared to the other derivatives. It is also evident that due to the loading mechanism of HC into the cavities of 2,3-D-MeβCD and 2,6-D-MeβCD, where only the ketone side of HC is completely covered within the cavities, the hydroxyl groups of HC are fully accessible to water molecules to form hydrogen bonds. Finally, the quality and persistence of hydrogen bonding (as indicated by the lifetime in ps) between HC and water is almost identical in all systems (~ 11 ps). A likely conclusion from these observations is that a higher number of hydrogen bonds between HC and water may lead to a faster release process. The number of hydrogen bonds between CDs and water also indicates that βCD is more capable of forming hydrogen bonds with water (19.41) than other CDs. The formation of methylated derivatives leads to a significant decrease in the number of hydrogen bonds. Among these, the most significant reduction is attributed to TMeβCD (9.73), while the least reduction is associated with 2-MeβCD (16.42) and 6-MeβCD (16.56). Therefore, methylation is considered thermodynamically undesirable for the solubility of CDs, although it is not the only thermodynamic determinant. HC forms hydrogen bonds with all CDs almost equally and imperceptibly, which certainly cannot be considered as the determining factor for HC loading. However, the most important observation is the significant decrease in the number of intramolecular hydrogen bonds of methylated derivatives compared to βCD. The presence of a full intramolecular hydrogen bond belt in βCD makes it rigid (between hydroxyl groups at O2 and O3 position), leading to lower water solubility than α-CD and γ-CD86. As observed, methylation leads to a significant disruption of the intramolecular hydrogen bonding network, resulting in a noticeable reduction in the number of intramolecular hydrogen bonds in methylated derivatives compared to βCD (5.94). The highest reduction is associated with TMeβCD (0.00) and the lowest reduction with 2-MeβCD (4.10). This observation can be thermodynamically evaluated as favorable for enhancing the solubility of CDs. It appears that the loading of HC increases this type of hydrogen bonding, which in turn reinforces the mentioned network and leads to the stiffening of the methylated derivatives, as evident in the "RMSF" (as discussed in Sect. "Root mean square fluctuation (RMSF)").
Conclusion
This comprehensive computational study has provided a detailed understanding of the interactions between hydrocortisone and various CD derivatives, including methylated βCDs. The results demonstrate the versatility of CDs as drug delivery carriers and highlight the importance of molecular-level investigations in optimizing these systems. The interaction mechanism between HC and the CDs was elucidated through the formation of inclusion complexes, which led to improvements in solubility, stability, and loading kinetics. The specific positioning and orientation of HC within the CD cavities were found to be crucial, with the hydroxyl groups of HC facing the primary hydroxyl rim and the ketone side facing the secondary hydroxyl rim in most cases. The kinetics of HC entry into the CD cavities varied significantly, with some derivatives exhibiting faster loading times compared to others. The analysis of structural flexibility, as measured by root mean square fluctuation (RMSF), revealed that methylation enhances the flexibility of the CD structures, but the presence of HC can lead to a reduction in flexibility, particularly for 3-MeβCD, 2,3-D-MeβCD, 2-MeβCD, and 6-MeβCD. This suggests that these derivatives may be more effective in retaining the guest molecule within their cavities. The investigation of water penetration into the CD cavities provided insights into the thermodynamic favorability of the cavity-guest complexation. The reduction in the number of high-energy water molecules within the innermost layer of the methylated CDs upon HC loading indicates the release of these water molecules, facilitating the formation of a stable inclusion complex. The conformational changes observed in the CD structures, such as the changes in the areas of the primary, middle, and secondary hydroxyl rims, as well as the cavity volume and circularity, further highlight the impact of functionalization and guest loading on the structural properties of these carriers. These changes can directly influence the solubility, stability, and drug release characteristics of the CD-based formulations. Overall, this study underscores the potential of CDs, particularly methylated derivatives, as effective drug delivery platforms for hydrophobic compounds like hydrocortisone. The insights gained from the computational analysis can guide the rational design and optimization of CD-based pharmaceutical formulations to enhance the therapeutic efficacy and bioavailability of challenging drug candidates.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Van Staa, T.-P. et al. Use of oral corticosteroids in the United Kingdom. QJM 93, 105–111 (2000).
Schäcke, H., Döcke, W.-D. & Asadullah, K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol. Ther. 96, 23–43 (2002).
Ramamoorthy, S. & Cidlowski, J. A. Corticosteroids: Mechanisms of action in health and disease. Rheum. Dis. Clin. 42, 15–31 (2016).
Mongioì, L. M., Condorelli, R. A., Barbagallo, F., La Vignera, S. & Calogero, A. E. Dual-release hydrocortisone for treatment of adrenal insufficiency: A systematic review. Endocrine 67, 507–515 (2020).
Fenyvesi, É., Puskás, I. & Szente, L. Applications of steroid drugs entrapped in cyclodextrins. Environ. Chem. Lett. 17, 375–391 (2019).
Singh, J. et al. Chronicle updates in cyclodextrin-based carriers for drug delivery. Bull. Natl. Res. Cent. 46, 202 (2022).
Dhiman, P. & Bhatia, M. Pharmaceutical applications of cyclodextrins and their derivatives. J. Incl. Phenom. Macrocycl. Chem. 98, 171–186 (2020).
Amiri, S. & Amiri, S. CDs: A potential candidate for improving water solubility and stability of hydrophobic guest molecules. Micro- Nano-Contain. Smart Appl. 327–357 (2022).
Schönbeck, C., Madsen, T. L., Peters, G. H., Holm, R. & Loftsson, T. Soluble 1: 1 Complexes and insoluble 3: 2 complexes–Understanding the phase-solubility diagram of hydrocortisone and γ-cyclodextrin. Int. J. Pharm. 531, 504–511 (2017).
Schwarz, D. H., Engelke, A. & Wenz, G. Solubilizing steroidal drugs by β-cyclodextrin derivatives. Int. J. Pharm. 531, 559–567 (2017).
Kristmundsdóttir, T., Loftsson, T. & Holbrook, W. P. Formulation and clinical evaluation of a hydrocortisone solution for the treatment of oral disease. Int. J. Pharm. 139, 63–68 (1996).
Boulos, N. Formulation matters: safe oral hydrocortisone use in children. Pharm. J. 307 (2021).
Orlu-Gul, M., Fisco, G., Parmar, D., Gill, H. & Tuleu, C. A new reconstitutable oral paediatric hydrocortisone solution containing hydroxypropyl-β-cyclodextrin. Drug Dev. Ind. Pharm. 39, 1028–1036 (2013).
D’Aria, F., Pagano, B. & Giancola, C. Thermodynamic properties of hydroxypropyl-β-cyclodextrin/guest interaction: A survey of recent studies. J. Therm. Anal. Calorim. 147, 4889–4897 (2022).
Wang, J.-W. et al. Structural insights into the host–guest complexation between β-cyclodextrin and bio-conjugatable adamantane derivatives. Molecules 26, 2412 (2021).
Mazurek, A. H., Szeleszczuk, Ł & Gubica, T. Application of molecular dynamics simulations in the analysis of cyclodextrin complexes. Int. J. Mol. Sci. 22, 9422 (2021).
Leonis, G., Ntountaniotis, D., Christodoulou, E., Mavromoustakos, T. Molecular dynamics protocols for the study of cyclodextrin drug delivery systems. Supramol. Drug Discov. Drug Deliv. Methods Protoc. 109–125 (2021).
Haimhoffer, Á. et al. Cyclodextrins in drug delivery systems and their effects on biological barriers. Sci. Pharm. 87, 33 (2019).
Haley, R. M., Gottardi, R., Langer, R. & Mitchell, M. J. Cyclodextrins in drug delivery: Applications in gene and combination therapy. Drug Deliv. Transl. Res. 10, 661–677 (2020).
Sheng, T. M. & Kumar, P. V. A new approach for β-cyclodextrin conjugated drug delivery system in cancer therapy. Curr. Drug Deliv. 19, 266–300 (2022).
Alshati, F. et al. Guest-host relationship of cyclodextrin and its pharmacological benefits. Curr. Pharm. Des. 29, 2853–2866 (2023).
Pitha, J., Harman, S. M. & Michel, M. E. Hydrophilic cyclodextrin derivatives enable effective oral administration of steroidal hormones. J. Pharm. Sci. 75, 165–167 (1986).
Andersen, F. M. & Bundgaard, H. Influence of β-cyclodextrin on the stability of hydrocortisone in aqueous solution. Arch. Pharm. Chem. Sci. Ed. 11, 61–66 (1983).
Chun, I. K. & Kim, B. Y. Influence of various cyclodextrins on the stability of hydrocortisone 17-butyrate in aqueous solution. Arch. Pharm. Res. 15, 176–183 (1992).
Lau, E. T. L. et al. Encapsulation of hydrocortisone and mesalazine in zein microparticles. Pharmaceutics 5, 277–293 (2013).
Ali, H. S. M. et al. Solubility of budesonide, hydrocortisone, and prednisolone in ethanol+ water mixtures at 298.2 K. J. Chem. Eng. Data 55, 578–582 (2010).
Zhao, Y. H. et al. Evaluation of human intestinal absorption data and subsequent derivation of a quantitative structure–activity relationship (QSAR) with the Abraham descriptors. J. Pharm. Sci. 90, 749–784 (2001).
Krait, S. et al. Complexation of daclatasvir by single isomer methylated β-cyclodextrins studied by capillary electrophoresis, NMR spectroscopy and mass spectrometry. Carbohydr. Polym. 273, 118486 (2021).
Bauer, P., Hess, B. & Lindahl, E. GROMACS 2022.2 Manual (2022). https://doi.org/10.5281/zenodo.6637572.
Bauer, P., Hess, B. & Lindahl, E. GROMACS 2022 Source code (2022). https://doi.org/10.5281/zenodo.6103835.
Abraham, M. J. et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1, 19–25 (2015).
Malde, A. K. et al. An automated force field topology builder (ATB) and repository: Version 1.0. J. Chem. Theory Comput. 7, 4026–4037 (2011).
Stroet, M. et al. Automated topology builder version 3.0: Prediction of solvation free enthalpies in water and hexane. J. Chem. Theory Comput. 14, 5834–5845 (2018).
Berendsen, H. J. C., Grigera, J. R. & Straatsma, T. P. The missing term in effective pair potentials. J. Phys. Chem. 91, 6269–6271 (1987).
Chatterjee, S., Debenedetti, P. G., Stillinger, F. H. & Lynden-Bell, R. M. A computational investigation of thermodynamics, structure, dynamics and solvation behavior in modified water models. J. Chem. Phys. 128 (2008).
JA, S. Practical mathematical optimization: an introduction to basic optimization theory and classical and new gradient-based algorithms, 2nd edn. Applied optimization, vol. 97, (2005).
Hockney, R. W., Goel, S. P. & Eastwood, J. W. Quiet high-resolution computer models of a plasma. J. Comput. Phys. 14, 148–158 (1974).
Hess, B., Bekker, H., Berendsen, H. J. C. & Fraaije, J. G. E. M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 (1997).
Darden, T., York, D. & Pedersen, L. Particle mesh Ewald: An N⋅ log (N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 (1993).
Bussi, G., Donadio, D. & Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 14101 (2007).
Parrinello, M. & Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 52, 7182–7190 (1981).
Shirts, M. R., Pitera, J. W., Swope, W. C. & Pande, V. S. Extremely precise free energy calculations of amino acid side chain analogs: Comparison of common molecular mechanics force fields for proteins. J. Chem. Phys. 119, 5740–5761 (2003).
Villa, A. & Mark, A. E. Calculation of the free energy of solvation for neutral analogs of amino acid side chains. J. Comput. Chem. 23, 548–553 (2002).
Bennett, C. H. Efficient estimation of free energy differences from Monte Carlo data. J. Comput. Phys. 22, 245–268 (1976).
Mobley, D. L., Chodera, J. D. & Dill, K. A. On the use of orientational restraints and symmetry corrections in alchemical free energy calculations. J. Chem. Phys. 125, 84902 (2006).
Ganjali Koli, M. & Fogolari, F. Exploring the role of cyclodextrins as a cholesterol scavenger: A molecular dynamics investigation of conformational changes and thermodynamics. Sci. Rep. 13, 21765. https://doi.org/10.1038/s41598-023-49217-8 (2023).
Mixcoha, E., Campos-Terán, J. & Piñeiro, A. Surface adsorption and bulk aggregation of cyclodextrins by computational molecular dynamics simulations as a function of temperature: α-CD vs β-CD. J. Phys. Chem. B. 118, 6999–7011 (2014).
Ganjali Koli, M., Eshaghi Malekshah, R. & Hajiabadi, H. Insights from molecular dynamics and DFT calculations into the interaction of 1,4-benzodiazepines with 2-hydroxypropyl-βCD in a theoretical study. Sci. Rep. 13, 9866. https://doi.org/10.1038/s41598-023-36385-w (2023).
Rizzi, A., Jensen, T., Slochower, D. R., Aldeghi, M., Gapsys, V., Ntekoumes, D., Bosisio, S., Papadourakis, M., Henriksen, N. M., De Groot, B. L. The SAMPL6 SAMPLing challenge: Assessing the reliability and efficiency of binding free energy calculations. J. Comput. Aided Mol. Des. 1–33 (2020).
Aiassa, V., Garnero, C., Zoppi, A. & Longhi, M. R. Cyclodextrins and their derivatives as drug stability modifiers. Pharmaceuticals 16, 1074 (2023).
Pitha, J., Szente, L. & Szejtli, J. Molecular encapsulation of drugs by cyclodextrins and congeners, in Control. Drug Deliv., 125–148 (CRC Press, 2019).
Pandey, A. Cyclodextrin-based nanoparticles for pharmaceutical applications: A review. Environ. Chem. Lett. 19, 4297–4310 (2021).
Kovacs, T. et al. Cyclodextrins: Only pharmaceutical excipients or full-fledged drug candidates?. Pharmaceutics 14, 2559 (2022).
Saokham, P., Muankaew, C., Jansook, P. & Loftsson, T. Solubility of cyclodextrins and drug/cyclodextrin complexes. Molecules 23, 1161 (2018).
Kim, D.-H., Lee, S.-E., Pyo, Y.-C., Tran, P. & Park, J.-S. Solubility enhancement and application of cyclodextrins in local drug delivery. J. Pharm. Investig. 50, 17–27 (2020).
Zhao, Y., Zeng, C. & Massiah, M. A. Molecular dynamics simulation reveals insights into the mechanism of unfolding by the A130T/V mutations within the MID1 zinc-binding Bbox1 domain. PLoS One 10, e0124377 (2015).
Martínez, L. Automatic identification of mobile and rigid substructures in molecular dynamics simulations and fractional structural fluctuation analysis. PLoS One 10, e0119264 (2015).
Mojdehi, M. F. P., Koli, M. G., Bolagh, M. D. O., Gardeh, M. G. & Hashemianzadeh, S. M. A detailed computational study on binding of kinase inhibitors into β-cyclodextrin: Inclusion complex formation. Mol. Syst. Des. Eng. 6, 80–92 (2021).
Chacko, K. K. & Saenger, W. Topography of cyclodextrin inclusion complexes 15. Crystal and molecular structure of the cyclohexaamylose-7.57 water complex, form III. Four-and six-membered circular hydrogen bonds. J. Am. Chem. Soc. 103, 1708–1715 (1981).
Saenger, W. Cyclodextrin inclusion compounds in research and industry. Angew. Chem. Int. Ed. Engl. 19, 344–362 (1980).
Saenger, W. et al. Structures of the common cyclodextrins and their larger analogues beyond the doughnut. Chem. Rev. 98, 1787–1802 (1998).
Betzel, C., Saenger, W., Hingerty, B. E. & Brown, G. M. Topography of cyclodextrin inclusion complexes, part 20. Circular and flip-flop hydrogen bonding in. beta.-cyclodextrin undecahydrate: A neutron diffraction study. J. Am. Chem. Soc. 106, 7545–7557 (1984).
Lindner, K. & Saenger, W. β-Cyclodextrin dodecahydrate: crowding of water molecules within a hydrophobic cavity. Angew. Chem. Int. Ed. Engl. 17, 694–695 (1978).
Manor, P. C. & Saenger, W. Topography of cyclodextrin inclusion complexes. III. Crystal and molecular structure of cyclohexaamylose hexahydrate, the water dimer inclusion complex. J. Am. Chem. Soc. 96, 3630–3639 (1974).
Biedermann, F., Nau, W. M. & Schneider, H. The hydrophobic effect revisited—Studies with supramolecular complexes imply high-energy water as a noncovalent driving force. Angew. Chem. Int. Ed. 53, 11158–11171 (2014).
Setny, P., Baron, R. & McCammon, J. A. How can hydrophobic association be enthalpy driven?. J. Chem. Theory Comput. 6, 2866–2871 (2010).
Liu, L. & Guo, Q.-X. The driving forces in the inclusion complexation of cyclodextrins. J. Incl. Phenom. Macrocycl. Chem. 42, 1–14 (2002).
Connors, K. A. The stability of cyclodextrin complexes in solution. Chem. Rev. 97, 1325–1358 (1997).
Szente, L. & Fenyvesi, É. Cyclodextrin-lipid complexes: Cavity size matters. Struct. Chem. 28, 479–492 (2017).
Izawa, H. et al. β-Cyclodextrin-crosslinked alginate gel for patient-controlled drug delivery systems: Regulation of host–guest interactions with mechanical stimuli. J. Mater. Chem. B. 1, 2155–2161 (2013).
Poulson, B. G. et al. Cyclodextrins: Structural, chemical, and physical properties, and applications. Polysaccharides 3, 1–31 (2021).
Khuntawee, W., Wolschann, P., Rungrotmongkol, T., Wong-Ekkabut, J. & Hannongbua, S. Molecular dynamics simulations of the interaction of beta cyclodextrin with a lipid bilayer. J. Chem. Inf. Model. 55, 1894–1902 (2015).
Sandilya, A. A., Natarajan, U. & Priya, M. H. Molecular view into the cyclodextrin cavity: Structure and hydration. ACS Omega 5, 25655–25667 (2020).
Blavatska, V. & Janke, W. Polymer adsorption on a fractal substrate: Numerical study. J. Chem. Phys. 136, 104907 (2012).
Blavatska, V. & Janke, W. Shape anisotropy of polymers in disordered environment. J. Chem. Phys. 133, 184903 (2010).
Champion, J. A. & Mitragotri, S. Role of target geometry in phagocytosis. Proc. Natl. Acad. Sci. 103, 4930–4934 (2006).
Simone, E. A., Dziubla, T. D. & Muzykantov, V. R. Polymeric carriers: role of geometry in drug delivery. Expert Opin. Drug Deliv. 5, 1283–1300 (2008).
Arkın, H. & Janke, W. Gyration tensor based analysis of the shapes of polymer chains in an attractive spherical cage. J. Chem. Phys. 138, 54904 (2013).
Khan, I., Saeed, K. & Khan, I. Nanoparticles: Properties, applications and toxicities. Arab. J. Chem. 12, 908–931 (2019).
Weng, Z. & Zaera, F. Increase in activity and selectivity in catalysis via surface modification with self-assembled monolayers. J. Phys. Chem. C. 118, 3672–3679 (2014).
Dill, K. & Bromberg, S. Molecular driving forces: Statistical thermodynamics in biology, chemistry, physics, and nanoscience. Garland Sci. https://doi.org/10.4324/9780203809075 (2010).
Boroushaki, T., Koli, M. G., Malekshah, R. E. & Dekamin, M. G. Elucidating anticancer drugs release from UiO-66 as a carrier through the computational approaches. RSC Adv. 13, 31897–31907 (2023).
Roosta, S., Majid Hashemianzadeh, S. & Ganjali Koli, M. Investigation of glutathione as a natural antioxidant and multitarget inhibitor for Alzheimer’s disease: Insights from Molecular Simulations. J. Mol. Liq. https://doi.org/10.1016/j.molliq.2021.117960 (2021).
Liu, Z., Dai, X., Sun, Y. & Liu, Y. Organic supramolecular aggregates based on water-soluble cyclodextrins and calixarenes. Aggregate 1, 31–44 (2020).
Wankar, J. et al. Recent advances in host–guest self-assembled cyclodextrin carriers: Implications for responsive drug delivery and biomedical engineering. Adv. Funct. Mater. 30, 1909049 (2020).
Szejtli, J. Introduction and general overview of cyclodextrin chemistry. Chem. Rev. 98, 1743–1754 (1998).
Author information
Authors and Affiliations
Contributions
R. G.: investigation, data curation, data analysis, design, and conceptualization. K. A.: writing, investigation, validation, data curation, data analysis, design, supervised, and conceptualization. M. G. K.: writing, investigation, validation, data curation, data analysis, design, and conceptualization.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gholami, R., Azizi, K. & Ganjali Koli, M. Unveiling the dynamic and thermodynamic interactions of hydrocortisone with β-cyclodextrin and its methylated derivatives through insights from molecular dynamics simulations. Sci Rep 14, 12495 (2024). https://doi.org/10.1038/s41598-024-63034-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-63034-7
- Springer Nature Limited