
Abstract
A key in species conservation is understanding the amount and distribution of genetic diversity and how environmental changes that occurred in the recent past may have influenced current patterns of population structure. Commerson’s dolphin, Cephalorhynchus commersonii, has two subspecies, one of which is endemic to South America (C. commersonii commersonii) and little is known about its population genetics. Our objective was to investigate the population genetics of this subspecies throughout its distribution. Using 70 skin samples and information available in GenBank, 308 mitochondrial DNA sequences and 28 species-specific microsatellites were analyzed. The species presented low genetic diversity when compared to other dolphin species, but was consistent with other species within the genus. Strong population structure based on mitochondrial DNA was exhibited throughout its entire distribution, a pattern consistent with female philopatry. However, this pattern was not detected when using microsatellites, suggesting male-mediated gene flow. Demographic tests suggested a population expansion beginning approximately 15,000 years ago, after the Last Glacial Maximum. In a climate change scenario, we recommended considering each sampling location as an independent population management unit in order to evaluate the impact of possible environmental changes on the distribution of genetic information within the species.
Similar content being viewed by others

Introduction
Understanding population structure is vital for studying the ecology of endemic species and planning or designing conservation strategies1. As we improve our understanding of the structuring of natural populations, it becomes essential that researchers also focus on understanding the processes underlying these patterns2. Most species are divided into populations, even in apparently continuous environments such as the marine environment, where barriers to gene flow are not always clear and can be associated with currents, temperature, salinity or primary productivity3,4,5,6.
Despite their broad range and their dispersal capabilities, marine mammals usually exhibit strong population structure at some geographic scale2,7,8,9,10. Populations may respond differently to a common threat11. Their susceptibility to changes will largely depend on genetic variability, i.e. their capacity to cope with environmental changes12,13. The largest threats to marine mammals currently include global warming14, and other anthropogenic impacts, especially fishery bycatch, resource overfishing, oil exploration, and pollution15,16. Predicting how populations will respond to these threats is complex and may depend on factors that shape their distribution patterns (such as prey availability) and their genetic diversity17. However, distribution of current genetic diversity also reflects the influence of long-term historical processes. By examining environmental changes that took place over the course of the Pleistocene and Holocene (including climate change), we gain insight into how similar factors will impact contemporary populations18. The Last Glacial Maximum (LGM) at the end of the Pleistocene is one of the largest global environmental changes and had significant impact on the genetic diversity of many natural populations of top predators19. After the LGM, many populations of marine mammals showed demographic expansion both in the northern and southern hemispheres, such as narwhals Monodon monoceros20, belugas Delphinapterus leucas21, bowhead whales Balaena mysticetus22, South American fur seal Arctocephalus australis23, South American sea lion Otaria flavescens24,25, southern right whale Eubalaena australis26, dusky dolphin Lagenorhynchus obscurus27, and spectacled porpoise Phocoena dioptrica28. These historical climatic fluctuations, characterized by periods of contraction and retraction, have been suggested to be the primary driving factor behind the origin and radiation of some species from the southern hemisphere, e.g., those that are involved in the genus Cephalorhynchus29.
The genus Cephalorhynchus includes four dolphin species widely distributed in cool temperate waters of the Southern Hemisphere, with each species endemic to a different region30. Among them, Commerson’s dolphin (Cephalorhynchus commersonii), has the largest distribution and comprises two subspecies: C. commersonii commersonii (South America) and C. commersonii kerguelenssi (Kerguelen Islands), which differ from each other both morphologically and genetically29,31. Along the southeastern coast of South America, the species is distributed between 40° and 56° S, including Strait of Magellan and the Falkland (Malvinas) Islands32,33. It is found frequently in shelf waters (< 200 m deep) and near shore (< 60 km from the Argentine coast and < 25 km from Falkland (Malvinas) Islands), where the continental shelf is wide and flat, there are large tidal cycles, and the waters are relatively cold as they are influenced largely by the Malvinas Current33,34,35,36,37,38,39. This species is present in different coastal habitats, with the highest densities in areas in close proximity to river mouths40.
The population structure of Commerson’s dolphins is poorly known, with existing information primarily focused on the southern range of the species. There, some degree of genetic differentiation at regional scale was detected using mitochondrial markers suggesting site fidelity by females41,42. However, there is still a gap of information regarding their northern distribution and contemporary genetic variation. Increasing the sampling effort will lead to a better understanding of how genetic diversity is distributed throughout the species range. Also, the information obtained here is crucial for the development of effective and sustainable management plans for the species43. To achieve these goals, we used mitochondrial and microsatellite DNA variability to investigate the population genetic diversity and structure of Commerson’s dolphin throughout their distribution in South America.
Materials and methods
Study area, sample collection, and DNA extraction
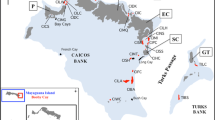
Tissue samples from 70 Cephalorhynchus commersonii were collected between 1999 and 2019 from five localities along the Patagonian coast: Playa Unión (43° 16′ S, 65° 02′W), Bahía Camarones (45° 2′ S, 65° 34′ W), Caleta Olivia (46° 26′S, 67° 30′W), Golfo San Jorge (46° 03′ S, 65° 59′ W), and Puerto Deseado (47° 47′ S , 65° 48′ W; Fig. 1). Of these, eight samples were from stranded individuals, eight from incidental capture, and the remaining fifty four were obtained via biopsy skin sampling44. All methods are reported in accordance with ARRIVE guidelines and were carried out in agreement with relevant guidelines and regulations. Also, sampling was approved under permits Nº41/2009, Nº002/2009, Nº23/07 N°001/10, Nº178/07, Nº001/19, Nº93/15, Disposición Nº8 and Nº16 awarded by Dirección de Fauna y Flora Silvestre, Subsecretaria de Recursos Naturales, Ministerio de Industria, Agricultura y Ganadería, Subsecretaría de Conservación y Áreas Protegidas, Secretaría de Turismo, Dirección de Fauna Silvestre, Consejo Agrario Provincial and Administración de Parques Naturales. To cover the entire distribution of the species in South America, 253 mitochondrial DNA (mtDNA) control region sequences available in GenBank (accession numbers are presented in Supplementary 1 Table S1, see Pimper et al.42 and Cipriano et al.41) were included for further analysis, increasing the sampling location to the south of our sampling area (Fig. 1).

Study area showing Cephalorhynchus commersonii distribution in red shadow. Sampling sites underlined correspond to present study, whereas those with asterisk (*) indicate the localities for the mitochondrial DNA control region sequences from Cipriano et al.41 and Pimper et al.42. PU = Playa Unión, BC = Bahía Camarones, CO = Caleta Olivia, GSJ = Golfo San José, PD = Puerto Deseado, SJ = San Julián, RG = Río Gallegos, SM = Strait of Magellan, TdF = Tierra del Fuego (N = North, C = Center, S = South). Pie charts represent the respective frequency of mitochondrial DNA control region haplotypes from each sampling location. The map was created in QGIS 3.12 (https://www.qgis.org/es/site/), whereas the final figure edition was generated using Photopea online graphics editor (https://www.photopea.com/).
Total genomic DNA was extracted following a glass milk protocol modified from Elphinstone et al.45 using a Perkin Elmer Multiprobe II Plus Liquid Handling System (Perkin Elmer, Waltham, MA). Before extraction, tissue samples were digested at 55ºC for 2 days, adding 4 μl of Proteinase K (20 mg/ml, New England BioLabs, NEB) approximately every 8 h. DNA quality and quantity were evaluated by electrophoresis on 1% agarose gel. Sex of the individuals sampled via skin biopsy was identified molecularly following Bérubé and Palsbøll46 method for the amplification of ZFX and ZFY regions.
Molecular protocol
Mitochondrial DNA
The mtDNA control region was amplified using primers MTCRf (5′-TTCCCCGGTCTTGTAAACC) and MTCRr (5′-TTTTCAGTGTCTTGCTTT)47. The reaction was performed in a 20 μl final volume including: 0.5 units of TSG; 2.5 mM MgCl2, 200 μM dNTPs, 10 mM buffer (Tris–HCl, pH 8.4), 0.3 μM of each primer and 1 μl of DNA template. The PCR profile was: 3 min at 93 °C; then 30 cycles of 1 min at 92 °C, 1 min at 48 °C and 1 min at 72 °C; then a final extension phase of 5 min at 72 °C. Each PCR product was purified with enzymatic PCR cleanup at 37 °C for 30 min and then at 80 °C for 20 min, prior reaction using: 0.025 μl of Exonuclease I (NEB), 0.25 μl Antarctic Phosphatase (NEB), 2.5 μl Antarctic Phosphatase buffer (NEB), and 7.225 μl of H2O. For all samples, a target fragment of ~ 900 bp was sequenced at MacrogenUSA (Rockville, MD), followed by visual checks and alignment on MEGA748.
Microsatellites
A single sample with the highest quality and quantity DNA was selected for shotgun sequencing and microsatellite design by means of visualization on 1% agarose gel stained with GelGreen (BioTium Fremont, CA). Our sequencing library was developed using 1 ng of genomic DNA following the Illumina Nextera XT Sample Preparation Kit protocol (Illumina, San Diego, California)49. Briefly, the protocol consisted of tagmentation genomic DNA, followed by PCR amplification. Purification of the PCR products was conducted using in-house made speedbeads follow Faircloth, et al.50. DNA library sequencing was performed using a MiSeq Benchtop Sequencer (Illumina, San Diego, California). Forward read output data from the MiSeq were imputed into Msatcommander 1.0.8-beta51 for the detection of microsatellite loci, with final product size, including primers and flanks, between 50 and 120 bp. A total of 34,973 microsatellites and 5017 sets of primers were identified following this criterion, and 70 were chosen at random for amplification/testing. Amplification was carried out in two multiplex PCR reaction (35 microsatellites each one) per individual. Further details on the molecular protocol to primer amplification are available in Ruzzante et al.49 and references therein. Summarized, this step consisted of diluting the multiplex PCR products and indexing them via a second PCR. PCR products were then pooled in equal proportion and subsequently cleaned via speedbeads. After quantification, the library was diluted to 15 pM and sequenced in a single direction using an Illumina MiSeq Benchtop Sequencer (Illumina, San Diego, CA). Post-sequencing data were automatically genotyped using Megasat52 followed by manual verification.
Analyses
Mitochondrial DNA
Haplotype and nucleotide diversity53 were estimated with DNAsp v554, considering each sampling location as a separate population. The analysis of molecular variance (AMOVA) based on FST (using haplotypes frequencies) and ΦST (using genetic distances with Kimura 2-parameter algorithm) was performed in Arlequin55,56, and significance was tested using 10,000 permutations. The substitution model (Kimura 2-parameter) was selected under Akaike Information Criterion (AIC) generated with jModelTest v2.1.1057. These analyses were tested under two scenarios: using all datasets and a data subset that includes sampling sites with microsatellite information (PU, BC, CO, PD) avoiding related individuals (see next section: “Microsatellites”). Gene flow among populations was estimated using genetic structure measures (ΦST) in Arlequin58. A haplotype network applying the median-joining algorithm and default parameters was built in Network 5.0.1.059. Demographic tests were explored through the Fu neutrality test60 and mismatch distribution analysis61 using Arlequin 3.5.2.258. A Mantel test was performed in Genalex 662 to examine isolation by distance through the use of linearized Φst63. BEAUTi v1.8.3 and BEAST v1.8.364 were used for Bayesian skyline plot reconstruction, assuming a piecewise-linear Bayesian skyline tree prior. The substitution model selected was Hasegawa–Kishino–Yano (HKY) based on AIC generated with jModelTest v2.1.1057. In addition, we used a lognormal relaxed clock rate (uncorrelated) with a mutation rate for the control region of 6.3 × 10–7 s/s/year derived for Lagenorhynchus obscurus65. Ninety million iterations of the MCMC (Markov Chain Monte Carlo) were applied, discarding the first 9,000,000 steps as “burn in” and sampling every 1000 iterations. Finally, convergence was checked through the effective sample size (ESS > 200) and a Bayesian skyline plot was built, both using Tracer v1.666.
Microsatellites
Potential null alleles were assessed using Microchecker 2.2.367. Tests for Hardy–Weinberg equilibrium (HWE) and linkage disequilibrium (LD) between pairs of loci were conducted in Arlequin 3.5.2.258, applying 1,000,000 Markov chain steps (MCMC) with 100,000 dememorization steps and 30,000 permutations, respectively; p values were adjusted using false Discovery rate adjustment68. Duplicates among the samples were tested using Genalex 662, removing one individual from each resampled pair. Allelic richness (AR) for each location were estimated with FSTAT 2.9.369, and Arlequin 3.5.2.258 was used to estimate observed (Ho) and expected heterozygosity (He). Effective population size was estimated with LDNe70. Population structure was estimated through AMOVA using Arlequin 3.5.2.258. Also, hierarchical population structure analysis was performed in Structure 2.3.471 under the admixture model, using 5,000,000 MCMC, 1,500,000 burn-in steps, and 10 replicates for each genetic cluster (K = 1–8). The Evanno method72 implemented in Structure Harvester73 was used to estimate the most likely K. Both genetic differentiation tests were conducted using two datasets: first using all samples and, second a data subset without related individuals. To account for filial relatedness, Queller and Goodnight 1989 estimator74 was applied using Genalex 662. Those individuals with a relatedness coefficient ≥ 0.5 (first-order relationship, such as parents and offspring or full-siblings) were removed. Contemporary gene flow among sampling locations was estimated using BayesAss Version 375, with a run setting of 150,000,000 iterations, 10% burn-in, and the rest of parameters as default. The model convergence was checked through the effective sample size (ESS > 200) using Tracer v1.666. Finally, a Mantel test63 was performed in Genalex 662 to examine isolation by distance among sampling sites using linearized FST values.
Results
mtDNA
We amplified the mtDNA control region in 55 of 70 individuals sampled, with the per individual sequence length ranging from 326 to 698 bp; the shortest sequences belong to the oldest samples. Three duplicate individuals (identified with microsatellite loci) were removed from the analysis. We used a 423 bp consensus sequence to align all sequences available in GenBank. From a total of 308 sequences used for population analyses, we found 26 polymorphic sites, defining 27 distinct haplotypes distributed throughout the species distribution (Fig. 1). Five of these haplotypes are novel for the species (Table 1, Supplementary 1 Table S1). Overall haplotype diversity was high (h = 0.801 ± 0.014), while nucleotide diversity was moderate (π = 0.00477 ± 0.00298) (Table 1).
AMOVA showed significant genetic differentiation among all sampling areas (populations defined a priori), FST = 0.139 and ΦST = 0.194, suggesting female philopatry (Table 2). Differences among populations are significant and represent almost 20% of variability. Furthermore, most of the pairwise ΦST values were significant (Table 3) and migration rates estimated using this genetic structure measure were mostly less than five effective migrants (Supplementary 1 Table S2). Data subset without related individuals (n = 42), also, showed significant differentiation based on both genetic structure measures (Supplementary 2 Table S1, S2). Finally, the Mantel test indicated a significant isolation by distance model (R2 = 0.14; p = 0.03; Supplementary 1 Fig. S1).
The minimum spanning network reflects the geographic distribution of the different haplotypes with a star-like configuration suggesting demographic expansion (Fig. 2). Haplotypes H5 and H4 were the most frequent (H5 = 32.48%, H4 = 25.72%). Only four haplotypes were new, singletons and unique to the four northern populations (Playa Unión, Bahía Camarones, Caleta Olivia, Golfo San Jorge), while 23 were shared among populations.

Haplotype network of Cephalorhynchus commersonii mitochondrial DNA control region sequences built using the median-joining algorithm (N = 308, 423 bp). The size of the circles is proportional to haplotype frequency, and the dashes between them represent one mutational step away from the next. PU = Playa Unión, BC = Bahía Camarones, CO = Caleta Olivia, GSJ = Golfo San José, PD = Puerto Deseado, SJ = San Julián, RG = Río Gallegos, SM = Strait of Magellan, TdF = Tierra del Fuego.
Neutrality tests were performed with all population samples combined. Fu’s Fs-values were large, negative, and significant for the entire population (Fu’s Fs = − 13,622, p = 0.002, α = 0.02), suggesting a population expansion. Also, the mismatch distribution showed a unimodal distribution suggesting a recent population expansion event (SSD = 0.004, p = 0.515; Harpending's raggedness index r = 0.025, p = 0.693). Furthermore, Bayesian skyline reconstruction also revealed a recent demographic expansion around 15,000 years ago (Fig. 3).

(a)—Mismatch distribution analysis according to a population expansion scenario of Commerson’s dolphin (Cephalorhynchus commersonii). The observed number of differences are given as a red dashed line, and those expected under a population expansion model are given as a black solid line. (b)—Bayesian Skyline plot representing historical demographic trends of Commerson’s dolphin population. Estimates of effective population size (Ne) means are joined by a solid line; whereas the colored area corresponds to the credibility interval based on 95% highest posterior density interval.
Microsatellites
From the 70 microsatellites/primer sets tested, 30 polymorphic loci were amplified successfully in 50 individuals from four sampling locations only (Playa Unión = 17; Bahía Camarones = 13; Caleta Olivia = 6; Puerto Deseado = 14). No evidence of linkage disequilibrium was detected, nor were null alleles detected. Two loci showed significant departure from Hardy–Weinberg equilibrium after false Discovery rate adjustment and were removed from the analysis. Moreover, three individuals were eliminated following their identification as duplicates. Summary statistics for the remaining 28 microsatellites loci in the four populations from north-central Patagonia are presented in Supplementary 1 Table S3, whereas genetic diversity within populations is summarized in Table 4. A total of 100 alleles were detected across all loci, 16 of those alleles were private and all four populations exhibited private alleles. Microsatellite locus CCo8 was the most variable with 15 alleles, followed by CCo52 with 8 alleles. Allelic richness (AR) per locus, which is independent of sample size, ranged from 1.132 to 8.003 and the highest mean over loci AR was found in Caleta Olivia (3.053). Average Ho and He ranged from 0.315 and 0.320 to 0.348 and 0.341, respectively. No significant difference was found in AR, Ho and He among populations (Kruskal–Wallis test, df = 3, p > 0.05).
Although AMOVA analysis showed evidence of genetic structure (FST = 0.032, p = 0.023); this result is based in only two small significant FST values between the two most geographically distanced populations: Playa Unión (43° S) and Bahía Camarones (45° S) with Puerto Deseado (47°S) (Table 3). Evanno et al.72 method indicated K = 2 as the most likely scenario to Structure assignment test, however, high variance, low Delta K values, and the fact that no individual was 100% assigned to a single genetic cluster, suggest only one panmictic population (Fig. 4, Supplementary 1 Table S4, S5 and Fig. S2). Both genetic differentiation analyses using the data subset without related individuals (n = 39) showed a single population (AMOVA: FST = 0.012, p = 0.316; Supplementary 2 Table S3, S4 and Fig. S1, S2). Using a Mantel test, the correlation was significant, supporting the presence of isolation by distance between regions (R2 = 0.75; p = 0.04; Supplementary 1 Fig. S3). Finally, contemporary migration rates were less than 0.1 between populations (Supplementary 1 Table S6).

Hierarchical STRUCTURE analysis of Commerson’s dolphin (Cephalorhynchus commersonii) from the four sampling regions based on 28 microsatellite loci and 50 individuals. Each individual is represented by a vertical line, which is partitioned into K colored segments, where each colored segment length is proportional to the individual's estimate membership coefficient.
Discussion
In marine mammal population genetic studies, a common assumption is that the species under study represent a single genetic population, with likely some degree of gene flow between predefined "populations"76. However, coastal odontocetes (like the species under study) are prone to population fragmentation and isolation, even in the short-term and along the contiguous coastline without physical barriers, which is probably due to natal fidelity77,78. Through the use of mtDNA and species-specific microsatellites we analyzed the population genetics of Commerson’s dolphins to understand how its genetic information is distributed along their range in southern South America. Our results showed that Commerson's dolphin represents a single panmictic population, with a strong female philopatry along the entire distribution based on mtDNA data and gene flow mediated by males at least in the northern of its distribution where microsatellite data could be analyzed.
Genetic structure analysis based in mtDNA and nuclear DNA markers exhibited contrasting patterns. Considering the mtDNA control region analysis a strong genetic structure among Commerson’s dolphins sampling locations was detected, suggesting a strong female philopatry which is not biased by kinship relationships as when kin pairs were detected one member of the pair was eliminated from the analysis. Female dispersal was low (nM < 5), which is consistent with significant breaks in dispersal. In contrast, no structure among northern populations was detected using microsatellite data, supporting the idea of male-mediated gene flow at least in the northern region of Patagonian continental shelf. In genetic populations studies of mammals, is common to assume the existence of sex-biased dispersal behavior, which is based on female philopatric behavior due to the requirements needed to breed and parental care18,79. Also, males dispersal is expected to improve reproductive success and avoid inbreeding depression18. Although it is best known in terrestrial mammals79, several studies with marine mammals, especially with cetaceans, support this hypothesis showing male-mediated gene flow18,80,81,82. However, it does not appear to be a common pattern for the species in the genus Cephalorhynchus. Based on mitochondrial markers, Heaviside’s dolphin, C. heavisidii, did not show evidence of population structure83, whereas Chilean dolphin, C. eutropia, showed genetic differentiation only through microsatellite analysis84. In contrast, Hector’s dolphin, C. hectori, displayed significant genetic differentiation and limited gene flow among populations using mtDNA and microsatellites85,86. Particularly with Commerson’s dolphin from South America, previous genetic analysis of mtDNA population structure found high levels of genetic differentiation indicating a strong site fidelity by females41,42. This was also confirmed in the present study, which covers the entire Commerson’s dolphin distribution in South America and extensive survey of microsatellite data, not reported before.
The genetic variability estimated with both molecular markers is distributed homogeneously suggesting, also, a single genetic population of Commerson’s dolphin. Furthermore, those estimated through mtDNA is similar to previously reported for the species in the south of its distribution (between 47 and 56° S) and for the genus41,42,78,87.
A combination of high h (> 0.5) and low π (< 0.005) is characteristic of species/populations that suffered a population bottleneck followed by a rapid demographic expansion and the consequent accumulation of mutations88. In this context, the Commerson´s dolphin population of the coast of Argentina might have experienced periods of population expansion and contraction coupled with glaciation events that occurred during the Pleistocene. Thus, population growth and geographic expansion occurred, likely with rapid diversification of haplotypes with minimum nucleotide differences. The haplotype network in a star-shape coincides with the results of the paired differences curve (mismatch distributions) and Fu values, suggesting recent population expansion for the species.
Other marine mammals from the region also exhibited recent demographic expansion23,24,26,27,28 likely associated with the LGM and sea ice retreat, around 15,000 years ago (as it is reported here based on Bayesian skyline reconstruction analysis). Habitat changes including changes in the distribution and abundance of prey are likely to have taken place during this period. These changes in primary productivity and relative abundance of prey played a key role in the history of some coastal dolphin populations. An example is dusky dolphin historic demography which was coupled with fluctuations in anchovy populations, its main prey17. Marine mammals were favored by these changes, leading to a sudden demographic expansion along the coast of the Southwestern Atlantic Ocean29,89.
The hypothesis of Cephalorhynchus radiation, based on the west-wind drift mechanism, suggests that Commerson’s dolphin is one of the latest species to establish its current distribution, after setting up the New Zealand population29. Once established in South America, the species appears to have expanded northward across the south-western Atlantic Ocean coasts. As can be visualized in the haplotype network (Fig. 2) and in the haplotype composition of each sampling site (Fig. 1), the majority of haplotypes were concentrated in the south of Argentina but many of them were found throughout almost the entire distribution. Among these haplotypes, the ancestral haplotypes (H4 and H5)41,42 are geographically spread throughout the entire distribution along southwestern Atlantic ocean. Therefore, historical changes in the habitat added to the recent radiation of the species with a rapid demographic expansion, is likely to have led to the absence of population genetic structure.
Population expansion from the southern to the northern ranges, with strong female philopatry and contemporary male-mediated gene flow, can produce the isolation-by-distance pattern (IBD) observed in our data90. However, while it is possible that the IBD mechanism is not the most appropriate model to represent the complexities of natural populations, it is a useful hypothesis for testing different scenarios. Commerson´s dolphin migration rates differed between marker type. The low sample size used with microsatellites is likely a factor contributing to the failure to identify recent migrants with BAYESASS91. Longer movements among far away locations may also occur more frequently than between nearby locations, e.g., between the northern and southern populations, where the southern populations act as a source (where the density of individuals is the highest) and the northern as a sink (with the lowest density)39. Although the species' range has not been estimated, it was reported individual movements of up to 250 km in distance34. This range of movement is higher than the home range estimated for other species of the genus (< 60 km)30, and it may explain the lack of a clear pattern of population structure, such as "stepping-stone model" suggested for Hector's dolphins population85. Consequently, the Commerson´s dolphin population on the coast of Argentina might respond to a meta-population model. Therefore, it is necessary to increase the analysis with nuclear markers analysis using samples from southern localities to have a better understanding of the population structure.
The condition of panmixia does not necessarily mean that the species should be considered as a single management unit. If indeed the species is structured according to a metapopulation, any impact at the local level could disrupt the dynamic of the populations and generate isolation between them. Taking into account the effects caused by past climatic fluctuations and in a current climate change scenario, alterations in sea surface temperature could produce changes in the habitat and affect the population structure of the species. To assess whether the pattern of genetic differentiation in the species conforms to a metapopulation model, an increased sampling effort in terms of both sample size and geographic coverage is recommended. The analysis of molecular markers in the present study is geographically uneven, with the analysis of autosomal markers restricted to the northern part of the species distribution. Hence, based on the precautionary principle, we recommend that the sampling areas analyzed here should be considered as single management units, where future evaluations and technical reports consider them independently.
Data availability
All the data generated or analyzed during this study are included in this published article (and its Supplementary Information file). Molecular markers information is available in Genbank (https://www.ncbi.nlm.nih.gov/genbank/) under accession numbers ON776962-ON777036. The dataset analyzed in the study is available upon request by contacting the corresponding author.
References
Waples, R. S. & Gaggiotti, O. INVITED REVIEW: What is a population? An empirical evaluation of some genetic methods for identifying the number of gene pools and their degree of connectivity. Mol. Ecol. 15, 1419–1439 (2006).
Mendez, M., Rosenbaum, H. C., Subramaniam, A., Yackulic, C. & Bordino, P. Isolation by environmental distance in mobile marine species: Molecular ecology of franciscana dolphins at their southern range. Mol. Ecol. 19, 2212–2228 (2010).
De Meeûs, T. et al. Population genetics and molecular epidemiology or how to “débusquer la bête”. Infect. Genet. Evol. 7, 308–332 (2007).
Durigan, M. et al. Population genetic analysis of Giardia duodenalis: Genetic diversity and haplotype sharing between clinical and environmental sources. MicrobiologyOpen 6, e00424 (2017).
Amaral, A. R. et al. Seascape genetics of a globally distributed, highly mobile marine mammal: The short-beaked common dolphin (genus Delphinus). PLoS ONE 7, e31482 (2012).
Mendez, M. et al. Molecular ecology meets remote sensing: Environmental drivers to population structure of humpback dolphins in the Western Indian Ocean. Heredity 107, 349–361 (2011).
de los Angeles Bayas-Rea, R., Félix, F. & Montufar, R. Genetic divergence and fine scale population structure of the common bottlenose dolphin (Tursiops truncatus, Montagu) found in the Gulf of Guayaquil. Ecuador. PeerJ 6, e4589 (2018).
Natoli, A., Peddemors, V. M. & Rus Hoelzel, A. Population structure and speciation in the genus Tursiops based on microsatellite and mitochondrial DNA analyses. J. Evol. Biol. 17, 363–375 (2004).
Oliveira, L. R., Loizaga De Castro, R., Cárdenas-Alayza, S. & Bonatto, S. L. Conservation genetics of South American aquatic mammals: An overview of gene diversity, population structure, phylogeography, non-invasive methods and forensics. Mammal Rev. 42, 275–303 (2012).
Vollmer, N. L. & Rosel, P. E. Fine-scale population structure of common bottlenose dolphins (Tursiops truncatus) in offshore and coastal waters of the US Gulf of Mexico. Mar. Biol. 164, 1–15 (2017).
MacLeod, C. D. Global climate change, range changes and potential implications for the conservation of marine cetaceans: A review and synthesis. Endanger. Species Res. 7, 125–136 (2009).
Hartl, D. L., Clark, A. G. & Clark, A. G. Principles of Population Genetics, Vol. 116 (Sinauer associates Sunderland, 1997).
Thomas, C. D. et al. Extinction risk from climate change. Nature 427, 145 (2004).
Reeves, R. R., Smith, B. D., Crespo, E. A. & Notarbartolo di Sciara, G. Dolphins, whales and porpoises: 2002–2010 conservation action plan for the world's cetaceans, Vol. 58 (IUCN, 2003).
Crespo, E. A. & Hall, M. A. In Marine Mammals, 463–490 (Springer, 2002).
Crespo, E. A. et al. Direct and indirect effects of highseas fisheries on the marine mammal populations in the northern and central Patagonian coast. J. Northwest Atl. Fish. Sci. 22, 189–207 (1997).
Harlin-Cognato, A. D., Markowitz, T., Würsig, B. & Honeycutt, R. L. Multi-locus phylogeography of the dusky dolphin (Lagenorhynchus obscurus): Passive dispersal via the west-wind drift or response to prey species and climate change?. BMC Evol. Biol. 7, 1–17 (2007).
Hoelzel, A. Evolution of population genetic structure in marine mammal species. In Population genetics for animal conservation, 294–318 (Cambridge University Press, Cambridge, 2009).
Fraser, C. I., Nikula, R., Ruzzante, D. E. & Waters, J. M. Poleward bound: Biological impacts of Southern Hemisphere glaciation. Trends Ecol. Evol. 27, 462–471 (2012).
Louis, M. et al. Influence of past climate change on phylogeography and demographic history of narwhals, Monodon monoceros. Proc. R. Soc. B 287, 20192964 (2020).
Skovrind, M. et al. Circumpolar phylogeography and demographic history of beluga whales reflect past climatic fluctuations. Mol. Ecol. 30, 2543–2559 (2021).
Foote, A. D. et al. Ancient DNA reveals that bowhead whale lineages survived Late Pleistocene climate change and habitat shifts. Nat. Commun. 4, 1–7 (2013).
Crespo, E. A. et al. Status, population trend and genetic structure of South American fur seals, Arctocephalus australis, in southwestern Atlantic waters. Mar. Mamm. Sci. 31, 866–890 (2015).
Feijoo, M., Lessa, E. P., De Castro, R. L. & Crespo, E. A. Mitochondrial and microsatellite assessment of population structure of South American sea lion (Otaria flavescens) in the Southwestern Atlantic Ocean. Mar. Biol. 158, 1857–1867 (2011).
Túnez, J. I., Cappozzo, H. L., Nardelli, M. & Cassini, M. H. Population genetic structure and historical population dynamics of the South American sea lion, Otaria flavescens, in north-central Patagonia. Genetica 138, 831–841 (2010).
Oliveira, L., Ott, P. H., Grazziotin, F. G., White, B. & Bonatto, S. In Paper (SC/S11/RW26) presented to the Southern Right Whale Assessment Workshop (Commission International Whaling).
Loizaga de Castro, R., Dans, S. L. & Crespo, E. A. Spatial genetic structure of dusky dolphin, Lagenorhynchus obscurus, along the argentine coast: Preserve what scale?. Aquat. Conserv. Mar. Freshw. Ecosyst. 26, 173–183 (2016).
Pimper, L. E., Goodall, R. N. P. & Remis, M. I. First mitochondrial DNA analysis of the spectacled porpoise (Phocoena dioptrica) from Tierra del Fuego, Argentina. Mamm. Biol. 77, 459–462 (2012).
Pichler, F. B. et al. Origin and radiation of Southern Hemisphere coastal dolphins (genus Cephalorhynchus). Mol. Ecol. 10, 2215–2223 (2001).
Dawson, S. M. In Encyclopedia of Marine Mammals, 166–172 (Elsevier, 2018).
Robineau, D., Goodall, R. N. P., Pichler, F. & Baker, C. S. Description of a new subspecies of Commerson’s dolphin, Cephalorhynchus commersonii (Lacépède, 1804), inhabiting the coastal waters of the Kerguelen Islands. Mammalia 71, 172–180 (2007).
Crespo, E. A. et al. Cephalorhynchus commersonii, Commerson's Dolphin. IUCN; The IUCN Red List of Threatened Species; 10-2017; 1-14 (2017).
Goodall, R. Commerson’s dolphin Cephalorhynchus commersonii (Lacépède 1804). Handb. Mar. Mamm. 5, 241–267 (1994).
Coscarella, M. A. Ecologıa, comportamiento y evaluación del impacto de embarcaciones sobre manadas de tonina overa Cephalorhynchus commersonii en Bahıa Engano, Chubut (Universidad de Buenos Aires, Buenos Aires, 2005).
Dellabianca, N. A. et al. Spatial models of abundance and habitat preferences of commerson’s and peale’s dolphin in southern patagonian waters. PLoS ONE 11, e0163441 (2016).
Goodall, R. et al. Studies of Commerson’s dolphins, Cephalorhynchus commersonii, off Tierra del Fuego, 1976–1984. Report of the International Whaling Commission (Special Issue 9), 143–160 (1988).
White, R. The Distribution of Seabirds and Marine Mammals in Falkland Islands Waters (Joint Nature Conservation Committee, 2002).
Loizaga de Castro, R., Dans, S. L., Coscarella, M. A. & Crespo, E. A. Living in an estuary: Commerson’s dolphin (Cephalorhynchus commersonii (Lacépède, 1804)), habitat use and behavioural pattern at the Santa Cruz River, Patagonia, Argentina. Latin Am. J. Aquat. Res. 41, 985–991 (2013).
Pedraza, S. Ecología poblacional de la tonina overa, Cephalorhynchus commersonii, (Lacépède, 1804) en el litoral patagónico. Unpublished PhD thesis, Universidad de Buenos Aires, Buenos Aires, Argentina (2008).
Garaffo, G. V. et al. Modeling habitat use for dusky dolphin and Commerson’s dolphin in Patagonia. Mar. Ecol. Prog. Ser. 421, 217–227 (2011).
Cipriano, F., Hevia, M. & Iñíguez, M. Genetic divergence over small geographic scales and conservation implications for Commerson’s dolphins (Cephalorhynchus commersonii) in southern Argentina. Mar. Mamm. Sci. 27, 701–718 (2011).
Pimper, L. E., Baker, C. S., Goodall, R. N. P., Olavarría, C. & Remis, M. I. Mitochondrial DNA variation and population structure of Commerson’s dolphins (Cephalorhynchus commersonii) in their southernmost distribution. Conserv. Genet. 11, 2157–2168 (2010).
O’Brien, S. J. A role for molecular genetics in biological conservation. Proc. Natl. Acad. Sci. 91, 5748–5755 (1994).
Loizaga de Castro, R., Hoelzel, A. & Crespo, E. Behavioural responses of Argentine coastal dusky dolphins (Lagenorhynchus obscurus) to a biopsy pole system. Anim. Welf. 22, 13–23 (2013).
Elphinstone, M. S., Hinten, G. N., Anderson, M. J. & Nock, C. J. An inexpensive and high-throughput procedure to extract and purify total genomic DNA for population studies. Mol. Ecol. Notes 3, 317–320 (2003).
Bérubé, M. & Palsbøll, P. Identification of sex in cetaceans by multiplexing with three ZFX and ZFY specific primers. Mol. Ecol. 5, 283–287 (1996).
Hoelzel, A., Hancock, J. & Dover, G. Evolution of the cetacean mitochondrial D-loop region. Mol. Biol. Evol. 8, 475–493 (1991).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 (2016).
Ruzzante, D. E. et al. Validation of close-kin mark–recapture (CKMR) methods for estimating population abundance. Methods Ecol. Evol. 10, 1445–1453 (2019).
Faircloth, B. C., Branstetter, M. G., White, N. D. & Brady, S. G. Target enrichment of ultraconserved elements from arthropods provides a genomic perspective on relationships among H ymenoptera. Mol. Ecol. Resour. 15, 489–501 (2015).
Faircloth, B. C. MSATCOMMANDER: Detection of microsatellite repeat arrays and automated, locus-specific primer design. Mol. Ecol. Resour. 8, 92–94 (2008).
Zhan, L. et al. MEGASAT: Automated inference of microsatellite genotypes from sequence data. Mol. Ecol. Resour. 17, 247–256 (2017).
Nei, M. Molecular Evolutionary Genetics (Columbia University Press, 1987).
Librado, P. & Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452 (2009).
Schneider, S., Roessli, D. & Excoffier, L. Arlequin: A software for population genetics data analysis, version 2.000. Genetics Biometry Laboratory, Department of Anthropology, University of Geneva, Switzerland (2000).
Excoffier, L., Smouse, P. E. & Quattro, J. M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 131, 479–491 (1992).
Darriba, D., Taboada, G. L., Doallo, R. & Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 9, 772 (2012).
Excoffier, L. & Lischer, H. E. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour 10, 564–567 (2010).
Bandelt, H.-J., Forster, P. & Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 16, 37–48 (1999).
Fu, Y.-X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147, 915–925 (1997).
Rogers, A. R. & Harpending, H. Population growth makes waves in the distribution of pairwise genetic differences. Mol. Biol. Evol. 9, 552–569 (1992).
Peakall, R. & Smouse, P. E. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 6, 288–295 (2006).
Mantel, N. The detection of disease clustering and a generalized regression approach. Can. Res. 27, 209–220 (1967).
Drummond, A. J. & Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7, 214 (2007).
Harlin, A. D., Markowitz, T., Baker, C. S., Würsig, B. & Honeycutt, R. L. Genetic structure, diversity, and historical demography of New Zealand’s dusky dolphin (Lagenorhynchus obscurus). J. Mammal. 84, 702–717 (2003).
Rambaut, A., Suchard, M., Xie, D. & Drummond, A. Tracer v1. 6. http://beast.bio.ed.ac.uk/Tracer (2014).
Van Oosterhout, C., Hutchinson, W. F., Wills, D. P. & Shipley, P. MICRO-CHECKER: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 4, 535–538 (2004).
Rice, W. R. Analyzing tables of statistical tests. Evolution 43, 223–225 (1989).
Goudet, J. FSTAT, a program to estimate and test gene diversities and fixation indices, version 2.9. 3. http://www2.unil.ch/popgen/softwares/fstat.htm (2001).
Waples, R. S. & Do, C. LDNE: A program for estimating effective population size from data on linkage disequilibrium. Mol. Ecol. Resour. 8, 753–756 (2008).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 14, 2611–2620 (2005).
Earl, D. A. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361 (2012).
Queller, D. C. & Goodnight, K. F. Estimating relatedness using genetic markers. Evolution 43, 258–275 (1989).
Wilson, G. A. & Rannala, B. Bayesian inference of recent migration rates using multilocus genotypes. Genetics 163, 1177–1191 (2003).
Milinkovitch, M. C., Leduc, R., Tiedemann, R. & Dizon, A. In Marine Mammals: Biology and Conservation (ed Evans, P. G. H. & Raga, J. A.) 325–359 (Springer, 2002).
Pichler, F. Population structure and genetic variation in Hector’s dolphin (Cephalorhynchus hectori), ResearchSpace@ Auckland (2001).
Pichler, F. & Baker, C. Loss of genetic diversity in the endemic Hector’s dolphin due to fisheries-related mortality. Proc. R. Soc. Lond. Ser. B Biol. Sci. 267, 97–102 (2000).
Greenwood, P. J. Mating systems, philopatry and dispersal in birds and mammals. Anim. Behav. 28, 1140–1162 (1980).
Chilvers, B. L. & Wilkinson, I. S. Philopatry and site fidelity of New Zealand sea lions (Phocarctos hookeri). Wildl. Res. 35, 463–470 (2008).
Engelhaupt, D. et al. Female philopatry in coastal basins and male dispersion across the North Atlantic in a highly mobile marine species, the sperm whale (Physeter macrocephalus). Mol. Ecol. 18, 4193–4205 (2009).
Möller, L. M. & Beheregaray, L. B. Genetic evidence for sex-biased dispersal in resident bottlenose dolphins (Tursiops aduncus). Mol. Ecol. 13, 1607–1612 (2004).
Jansen van Vuuren, B., Best, P., Roux, J. P. & Robinson, T. Phylogeographic population structure in the Heaviside’s dolphin (Cephalorhynchus heavisidii): Conservation implications. Anim. Conserv. 5, 303–307 (2002).
Pérez-Alvarez, M. J. et al. Microsatellite markers reveal strong genetic structure in the endemic Chilean dolphin. PLoS ONE 10, e0123956 (2015).
Hamner, R. M., Pichler, F. B., Heimeier, D., Constantine, R. & Baker, C. S. Genetic differentiation and limited gene flow among fragmented populations of New Zealand endemic Hector’s and Maui’s dolphins. Conserv. Genet. 13, 987–1002 (2012).
Pichler, F., Dawson, S., Slooten, E. & Baker, C. Geographic isolation of Hector’s dolphin populations described by mitochondrial DNA sequences. Conserv. Biol. 12, 676–682 (1998).
Kraft, S. et al. From settlers to subspecies: Genetic differentiation in commerson’s Dolphins between South America and the Kerguelen Islands. Front. Mar. Sci. 8, 782512 (2021).
Grant, W. & Bowen, B. W. Shallow population histories in deep evolutionary lineages of marine fishes: Insights from sardines and anchovies and lessons for conservation. J. Hered. 89, 415–426 (1998).
Ponce, J. F., Rabassa, J., Coronato, A. & Borromei, A. M. Palaeogeographical evolution of the Atlantic coast of Pampa and Patagonia from the last glacial maximum to the Middle Holocene. Biol. J. Lin. Soc. 103, 363–379 (2011).
Wright, S. Isolation by distance. Genetics 28, 114 (1943).
Meirmans, P. G. Nonconvergence in B ayesian estimation of migration rates. Mol. Ecol. Resour. 14, 726–733 (2014).
Acknowledgements
We would like to thank colleagues at the Laboratorio de Mamíferos Marinos (LAMAMA—CESIMAR—CONICET) and at the Ruzzante Lab (Department of Biology—Dalhousie University, Canada) for logistic and institutional support. C.A.D. acknowledges a scholarship (2019–2020) from: (1) The Emerging Leaders in the Americas Program (ELAP), Government of Canada and (2) funds from the Small Grants in Aid of Research program of the Society for Marine Mammalogy (3) Funds for the collections were awarded to E.A.C. Consultancy by the International Fund for Animal Welfare; Mohamed bin Zayed Species Conservation Fund, Fundación BBVA (BIOCON 04, PNUD ARG-02/018), by the Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET-PIP 0742/98), the Agencia Nacional de Promoción Científica y Tecnológica (01-04030 A and 11679), the) and the GEF Project CNP-BB 27. C.A.D. was supported by a Doctoral Fellowship from the National Research Council of Argentina (CONICET).
Author information
Authors and Affiliations
Contributions
C.A.D., R.L., and E.A.C. conceived the study and collected the samples; C.A.D. and G.R.M. generated the molecular data and conducted analysis along with D.E.R and R.L.; C.A.D., R.L., D.E.R. and E.A.C. wrote the paper. All authors contributed critically to the drafts and gave final approval for publication.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Durante, C.A., Loizaga, R., McCracken, G.R. et al. Commerson’s dolphin population structure: evidence for female phylopatry and male dispersal. Sci Rep 12, 22219 (2022). https://doi.org/10.1038/s41598-022-26192-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-26192-0
- Springer Nature Limited