
Abstract
The Itaipu Hydroelectric Power Plant is the second largest in the world in power generation. The artificial barrier created by its dam imposes an obstacle for fish migration. Thus, in 2002, a fish pass system, named Piracema Channel, was built to allow fish to access areas upstream of the reservoir. We tested the potential of environmental DNA metabarcoding to monitor the impact of both the dam and associated fish pass system in the Paraná River fish communities and to compare it with traditional monitoring methods. Using a fragment of the 12S gene, we characterized richness and community composition based on amplicon sequence variants, operational taxonomic units, and zero-radius OTUs. We combined GenBank and in-house data for taxonomic assignment. We found that different bioinformatics approaches showed similar results. Also, we found a decrease in fish diversity from 2019 to 2020 probably due to the recent extreme drought experienced in southeastern Brazil. The highest alpha diversity was recorded in the mouth of the fish pass system, located in a protected valley with the highest environmental heterogeneity. Despite the clear indication that the reference databases need to be continuously improved, our results demonstrate the analytical efficiency of the metabarcoding to monitor fish species.
Similar content being viewed by others
Introduction
The Itaipu Hydroelectric Power Plant, built at the border between Brazil and Paraguay, is the second largest in the world in power generation1, second only to the Three Gorges Power Plant in China. With the formation and filling of its reservoir, in 19822, the natural barrier to the migration of fishes of the middle section of the Paraná River (Sete Quedas falls) was replaced by the artificial barrier of the Itaipu dam, located 170 km downstream. This artificial barrier (196 m high) caused impacts on the adjacent fish assemblages, such as the reduction in reproductive activity in the first kilometers downstream of the dam3. To allow for fish migration and mitigate the environmental impact of the dam, a fish passage system known as the Piracema Channel was created in 2002, linking the Paraná River to Itaipu's Reservoir4. However, the real contribution to the reproductive success of the long-distance migratory species is still under investigation, and this channel also allowed for the dispersal of species originally restricted to the lower Paraná River upstream and species originally restricted to the upper Paraná River downstream5. These potential impacts are continuous and can interact with natural disturbance, such as several droughts as which happened in 2020. In this context, monitoring the impact of both dam and fish pass system in the Paraná River fish communities is essential.
Fish diversity estimates in Brazilian freshwater are still imprecise due to the scarcity of complete inventories5,6,7. Many species are described every year and several groups are in need of taxonomic revision5,8. Furthermore, traditional assessment methods for fish diversity surveys are costly and time consuming, given that they depend on capture (e.g. netting, trawling) or observation9,10 and expertise for taxonomic identification11. In this sense, designing methods for cost-effective monitoring fish diversity and community composition is an urgent task. Most sampling efforts in Brazil have historically been primarily funded by the hydroelectric sector, focusing particularly on rivers where power dams were built12. The areas of the dam construction have some of the most comprehensive knowledge of fish assemblage composition in comparison with other Brazilian regions and therefore offer an ideal opportunity to compare taxonomic surveys with molecular approaches.
A promising alternative to traditional taxonomic surveys and biomonitoring methods is the use of environmental DNA (eDNA), combined with a high-throughput sequencing approach, as in the case of metabarcoding13. This technique has the advantage of obtaining DNA from environmental samples, such as water, without first isolating the target organism and therefore can sample entire communities14. Metabarcoding is a powerful tool for biodiversity assessment that has been widely used for several purposes and different taxonomic groups15,16,17, and is considered a transformative technology for the entire field18. However, some limitations, such as the relative scarcity of DNA sequences for several species, which is even more problematic in highly diverse regions such as the Neotropics19, may create constraints that hamper its full application20,21.
The absence of a comprehensive DNA reference database may lead to a misidentification of several species. Therefore, putting together a curated and complete DNA reference database is fundamental for species identification through a metabarcoding approach7. But, even with an incomplete DNA reference database, the use of molecular units, such taxonomic units clustered by similarity (operational taxonomic units—OTUs22) or unique sequences (e.g. amplicon sequence variants—ASVs23, or zero-radius OTUs—ZOTUs24) allows for diversity monitoring in the context of biodiversity assessment in megadiverse biomes. Such estimates without comprehensive species identification limit ecological conclusions but allowed for monitoring of natural and artificial impacts16,25. The metabarcoding approach has been successfully used for molecular identification of several vertebrate groups in temperate regions26,27, monitoring of endangered species such as freshwater fish in Australia28 and turtles in the United States29, and to describe biodiversity even with limited taxonomic identification30.
In this context, our goal here is to describe an effective survey protocol for detecting fish assemblages through eDNA metabarcoding in an ecologically complex and highly diverse freshwater system, the Piracema channel, that connects the Paraná River with Itaipu Reservoir. For this, we used an in-house molecular database of fishes occurring in the channel complemented with GenBank sequences. We also describe fish alpha diversity and community structure in the Piracema channel system. Additionally, we compare our metabarcoding results with the traditional sampling campaigns made between 2017 and 2021.
Materials and methods
Study area
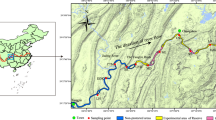
Our study was conducted at the Piracema channel (Fig. 1), a fish pass system connecting the Paraná River with the Upper Paraná River floodplain (main reservoir). For the traditional taxonomic survey, we sampled three points at mouth of channel at Paraná River (Fig. 1, blue square), the main lake at the Piracema channel (Fig. 1, red circle), and the reservoir near the water intake to the Channel (Fig. 1, green triangle) between 2017 to 2021. Fish were collected monthly during the fish reproductive period (October to March), and once during the winter (July or August), employing active and passive methods (Table 1).

Sampling location. The map shows the sampling location of each collection point. We sampled one point at mouth of channel at Paraná River (blue square), four points along the Piracema Channel (circles; Bela Vista River 1 = purple, Bela Vista 2 = yellow, Brasilia stream = gray, and lake = red), and one point at Itaipu’s reservoir (green triangle). Up at figure is possible to visualize the Itaipus’ dam that created the reservoir. Inset panel shows the location of Itaipu’s dam in relation to South America. Map was created in QGIS v.3.6.2 software88.
For each point, gill nets and longlines were set out in the afternoon (16:00 h) and inspected every 4 h during a 24 h cycle; cast nets were operated 3 times each mesh, after every gear inspection. Boarded electrofishing was operated two times in each point, at dawn and at dusk, covering 100 m of the environment margin each time. Fish were euthanized by immersion in benzocaine solution, following current legislation31, and identified accordingly Britski et al.32, Ota et al.33 and Neris et al.34. Fragments of muscle were collected with a scalpel, placed into 2 ml tubes filled with 99.8% ethanol and stored at 4 °C until processing. Voucher specimens are housed in the Nupelia-UEM fish collection.
For metabarcoding, we sampled one site at the mouth of channel at Paraná River (Fig. 1, blue square), four sites along the Piracema Channel (Fig. 1, circles), and one site at Itaipu Reservoir (Fig. 1, green triangle). Each sampling point was collected in sextuplicate. All six sites were sampled in 2019 and three sites (mouth of channel at Paraná River, lake at Piracema channel [red circle], and the reservoir) were sampled again in 2020, totaling 54 samples. All sampling sites were provided with GPS coordinates.
Sampling design for molecular analysis
We collected water by partially submerging a one litter polypropylene bottle. The objective was to sample water at the air/water interface. After water collection, bottles were closed and cleaned with a 10% sodium hypochlorite solution, following by rinsing with distilled water. We used gloves which were changed in between each new sampling replicate to reduce the risk of cross-sample contamination.
After the collection and cleaning steps, the bottles were stored in polystyrene boxes containing artificial ice to maintain the temperature of the samples at 4 to 10 °C. The samples were filtered, on the same day of collection, using nitrocellulose membranes (0.45 µm pore) with the aid of a vacuum pump. Filters were kept in 100% ethanol under refrigerated conditions until molecular analysis was performed. All filters were processed at the ATGC laboratory at the Universidade Federal do Paraná (UFPR).
DNA extraction
For total DNA extraction, we kept the collected filters at room temperature to allow the residual ethanol to dry completely. After dried we extract the DNA using magnetic beads (microspheres surrounded by magnetite and carboxyl), which bind to DNA (carboxyl bond—DNA) by the process of Solid Phase Reversible Immobilization (SPRI). The DNA extract was stored at − 20 °C until the amplification. The extraction and quantification processes were carried out in separate rooms, as suggested by Pie et al.35. We checked the DNA concentration using both a spectroscope (Nanodrop, Thermo, USA) and a fluorimeter (Qubit, Invitrogen, USA).
PCR amplification
We targeted the 12S rRNA gene using the MiFish forward (5′-GTCGGTAAAACTCGTGCCAGC-3′) and reverse (5′-CATAGTGGGGTATCTAATCCCAGTTTG-3′) primers designed by Miya et al.36 to yield 163–185 bases long fragments. Amplification was performed in a total volume of 20 μl in GoTaqG2 system (Promega, USA), 500 nM of forward and reverse primers, and 20 ng of DNA template. The PCR conditions consisted of an initial denaturation step of 2 min at 95 °C and then 25 cycles of denaturation at 94 °C for 30 s, hybridization at 55 °C for 45 s, and elongation at 72 °C for 30 s, followed by a final elongation at 72 °C for 5 min and finishing at 4 °C. To avoid PCR inhibition BSA (0.5 μg/μl) was added to the reaction as suggested by Boeger et al.37. The quality of amplification was verified on a 1.5% agarose gel in TBE buffer (9 mM TRIS, 9 mM boric acid, 1 mM EDTA), stained with SYBR Safe DNA Gel Stain (ThermoFisher Scientific, Country). All replicates from each sampling point were amplified to increase the chance of detecting rare species. The PCR product was then diluted (20×) and used as a template for the addition of adapters in the second PCR. Indexing was performed for Illumina MiSeq sequencing (Illumina, USA), using the above PCR system with Nextera indexes (Illumina) in a total volume of 10 μl. PCR conditions were an initial step of 95 °C for 3 min, following by 12 cycles of denaturation at 94 °C for 30 s, hybridization at 55 °C for 45 s, and elongation at 72 °C for 30 s, followed by a final elongation at 72 °C for 5 min and finishing at 4 °C. We checked the DNA concentration in a Qubit® fluorimeter (Invitrogen, USA), normalized and pooled the PCR products following the Illumina protocol. The samples were sequenced at GoGenetic (Curitiba, Brazil) using Illumina MiSeq Reagent 600V3 (PE 300b). Three negative controls (distilled water) were used as control for extraction, amplification, and sequencing. The raw sequences are deposited in GenBank under Bioproject PRJNA750895 (biosamples SAMN20500524–SAMN20500577).
Sequence analyses and taxonomic assessment
For the amplicon sequence variants (ASVs) approach, we used the Cutadapt package38 in Python v.3.339 to remove primers. We then used the DADA2 package23 in R v. 4.0.240 to quality filter reads, merge sequences, remove chimeras, and to infer ASVs. We excluded reads with ambiguous bases (maxN = 0). Based on the quality scores of the forward and reverse sequences, each read was required to have < 3 or < 5 errors, respectively (maxEE = c (3,5), truncQ = 2). Therefore, ASVs were inferred for forward and reverse reads for each sample using the run-specific error rates. To assemble paired-end reads, we considered a minimum of 12 base pairs of overlap and excluded reads with mismatches in the overlapping region. Chimeras were removed using the consensus method of "removeBimeraDenovo" implemented in DADA2.
For operational taxonomic units (OTUs) and zero-radios OTU (ZOTUs) analyses, we used the USEARCH/UPARSE v.11.0.667 Illumina paired reads pipeline41 to primer remove, quality filtering, dereplicate and sort reads by abundance, to infer OTUs and ZOTUs, and to remove singletons. We filtered the sequences to discard chimeras and clustered sequences into OTUs at a minimum similarity of 97% using a ‘greedy’ algorithm that performs chimera filtering and OTU clustering simultaneously and the UNOISE algorithm to denoised sequence as zero-radios OTUs to create or ZOTUs table41,42.
We build a reference dataset of DNA sequences for the 205 fish taxa that have been historically recorded in the Itaipu system using the following steps. First, we looked for 12S sequences of these species in GenBank by searching for their corresponding names. We were able to find sequences for 126 species in our reference database. Additionally, we created an in-house database which included sequences for 42 additional species to the 79 species previously identified as present on Itaipu system but not available on GenBank. Sequences for the in-house database were obtained via Sanger sequencing of tissue samples and were uploaded to GenBank (accession numbers MZ778813-MZ778856). We manually blasted all sequences against the NCBI GenBank database to verify misidentification or problematic sequences (e.g. blasted in the different family). In total, our reference database included 168 (82%) sequences from the 205 taxa recorded in the Itaipu system. Finally, we blasted the ASVs, OTUs, and ZOTUs sequences with our reference database to verify the taxonomic composition using the “Blastn” function of the program Blast+43 with an e-value of 0.001. We kept ASVs, OTUs, and ZOTUs that have matched with a fish species at minimum level of 75% similarity (as these sequences are probably fishes species not present in our reference database), and considered identified species just ASVs, OTUs, and ZOTUs that matched in a minimum level of 97% similarity. We summed reads for each ASVs, ZOTUs, and OTUs present in the three negative controls and divided by the total reads of each ASVs, ZOTUs, and OTUs. All ASVs, ZOTUs and OTUs with a proportion > 0.01% of reads in negative controls were discarded (13 ASVs, 2 ZOTU, and 7 OTUs).
Statistical analysis
We conducted all analyses in R using RStudio44. We used the tidyverse package v. 1.3.045 for data curation and ggplot2 v. 3.3.246, ggfortify v. 0.4.1147, gridExtra v. 2.348, and ggpubr v. 0.4.049 for data visualization (scripts in Appendix 1).
For analysis of alpha and beta diversity with metabarcoding data, we made the analysis at ASVs, OTUs, and ZOTUs level. Since the number of observed ASVs, ZOTUs, and OTUs is dependent on the number of reads, we rarefied all samples to the lowest number of reads obtained from any one plot (157 for ASVs, 147 for ZOTUs, and 219 for OTUs; Supplementary Fig. S1) using the “rarefy” function with the vegan v.2.5.750 R package. Because in the ZOTUs table the minimum reads of a plot was nine, we used the second lower value to avoid having to downsize the other samples to such a low number of reads51. Because rarefying of counts is considered inappropriate for detection of differentially abundant species51, even more with so different sampling depth as in our case, we also calculated true effective number of ASVs, ZOTUs, and OTUs of order q = 1, which is equivalent to the exponential of the Shannon entropy52, using the function “AlphaDiversity” of the Entropart v.1.6.753 R package. The effective number is more robust against biases arising from uneven sampling depth than the simple counts of ASVs, ZOTUs, and OTUs51,54. Additionally, for alpha diversity, we also calculated the ASV, OTU, and ZOTU richness (the number of ASV, OTU, and ZOTU per point), Chao1, and Fisher’s alpha diversity (i.e., the relationship between the number of ASV, OTU, and ZOTU in any given point and the number of reads of each ASV, OTU, and ZOTU) using the phyloseq v.1.34.055 R package.
For beta diversity, we also used rarefaction (with “rrarefy” function of vegan package) and hill number (with “varianceStabilizingTransformation” function in DESeq2 v.1.28.156 R package) to normalize our data. While rarefaction normalizes data by random subsampling without replacement, the hill number transformation normalizes the count data with respect to sample size (number of reads in each sample) and variances, based on fitted dispersion-mean relationships56. We then constructed two-dimensional Principal Coordination Analysis (PCoA) ordinations of the abundance (reads) and presence/absence data for both rarefied and hill numbered data. We used the ‘cmdscale’ function and Bray–Curtis distances in the vegan package to assess community dissimilarity among all samples in the PCoA. We used the “envfit” method in vegan to fit sampling localities and sample year onto the PCoA ordination as a measure of the correlation among the sampling localities with the PCoA axes.
For traditional survey data, we calculated the alpha diversity using the observed richness, Chao1, and ACE with the function “estimate”, and Shannon index with the function “diversity” both with vegan package. We also constructed two-dimensional PCoA ordinations of the abundance (reads) and presence/absence data, and used the “envfit” to fit sampling localities and sample year onto the PCoA ordination.
Ethics statement
We confirm that all methods were carried out in accordance with relevant guidelines and regulations and in compliance with the ARRIVE guidelines. We confirm that all experimental protocols were in accordance with the precepts of Law nº 11.794, of 8 October 2008, of Decree nº 6.899, of 15 July 2009, and with the edited rules from Conselho Nacional de Controle da Experimentação Animal (CONCEA), and it was approved by the ANIMAL USE ETHICS COMMITTEE OF THE AGRICULTURAL SCIENCES CAMPUS OF THE FEDERAL UNIVERSITY OF PARANA, BRAZIL, with degree 1 of invasiveness, on March 22th, 2021.
Results
For the traditional surveys, 4447 fishes were collected, for a total 122 species. Most specimens were collected at the mouth of channel at Paraná River, with 2240 (51%) fishes belonging to 105 species. The reservoir showed the lowest number of collected specimens: 1,034 (23%) and a total of 64 species. Of these, 29 species (24%) were identified with metabarcoding approach, while 93 species (76%) were only identified with traditional surveys (Table 2). Other 27 species were identified at > 97% of similarity only with the metabarcoding approach (Table 2).
For metabarcoding data, we obtained a total of 17,616,032 reads. After all cleaning steps, we kept a total of 2,280,447 sequences belonging to 7096 ASVs. Of these, 1,015,157 (44% of the total) sequences belonging to 190 ASVs were classified into species corresponding to our reference database in the level of 75% similarity. Of these, 7,591 reads (0.75% of the total) were recorded in the sum of the three negative controls (Appendix 3). A total of 13 ASVs with a proportion of reads > 0.01% of the total reads were removed from the analysis. A total of 121 ASVs (64%) were classified in 35 species matches at a level > 97% of similarity (Table 2), which is certainly an underestimation of the real number of species, since the other 69 ASVs should belong to species do not present in our database.
For the OTU and ZOTU analyses, after all cleaning steps, we obtained 1,157,738 and 1,145,129 reads belonging to 796 OTUs and 207 ZOTUs, respectively. Of these, 1,002,883 (87%) and 1,002,335 (87%) reads belonging to 136 OTUs and 94 ZOTUs, respectively, were classified into species corresponding to our reference database at the level of 75% similarity. Of these, 7,493 and 7,494 reads (0.75% of total) were registered in the sum of the three negative controls in the OTUs and ZOTUs tables, respectively (Appendix 4 for OTUs and Appendix 5 for ZOTUs) and 2 ZOTU and 7 OTUs with a proportion of reads > 0.01% of the total reads were removed from the analysis. As the OTUs analysis already classified the sequences by 97% similarity, the 131 OTUs (all matched with fishes less the 5 present in the negative controls) probably correspond to the number of species present in our samples (more than all species sampled in five years with traditional surveys). Yet, only 37 species belonging to 42 OTUs and 34 species belonging to 46 ZOTUs at > 97% similarity were assigned in both analyses. Eighty-one (66%) OTUs and 41 (47%) ZOTUs were identified as a fish species with a similarity lower than 97%, representing species not present in our reference database.
All the alpha diversity measures of ASVs, OTUs, and ZOTUs per sampling point varied, with the point at mouth of the channel at Paraná River, in 2019, showing the highest diversity for all molecular units and the lake at Piracema Channel in 2020 the lowest (Fig. 2). For the traditional surveys, the variation was more random, but the mouth of channel at Paraná River also had the highest diversity (Supplementary Fig. S2).

Alpha diversity estimation for (A) ASVs, (B) ZOTUs, (C) OTUS. Alpha diversity varied by location and by sampling year. Each point is one of the replicates sampled. Colors and symbols represent collection points (mouth of channel at Paraná River = blue square, Itaipu’s reservoir = green triangle, and Piracema Channel = circles [Bela Vista River 1 = purple, Bela Vista 2 = yellow, Brasilia stream = gray, and lake = red]), and fill represent year of collection (filled = 2019, empty = 2020).
Fish communities varied among sampled sites. For the abundance based in hill numbers, the first axis of the PCoA separated the samples by year (envfit: R2 = 0.30 [ZOTUs], 0.34 [ASVs], and 0.36 [OTUs], p < 0.001), except for the Itaipu's reservoir, with the positive values associated with 2019 and the negative values associated with 2020 (Fig. 3). The second axis separated the samples by locality with some overlap (envfit: R2 = 0.95 [ZOTUs]–0.96 [ASVs and OTUs], p < 0.001; Fig. 3). For the presence/absence data also based in hill numbers, the overlap was higher but yet the separation by year (envfit: R2 = 0.30 [ZOTUs], 0.32 [OTUs], and 0.41 [ASVs], p < 0.001) and locality (envfit: R2 = 0.91 [ZOTUs]–0.92 [ASVs and OTUs], p < 0.001) was similar to the abundance data (Fig. 3). The results of rarefied data were similar with more overlap among sampling points (Appendix 1, Supplementary Fig. S3). For traditional surveys, the points are clustered by localities (envfit: R2 = 0.60 for abundance and 0.87 for presence/absence data, p < 0.001), but not by year (p > 0.05, Supplementary Fig. S4).

Principal Coordinates Analysis (PCoA) of fishes’ communities from Itaipu based in hill numbers for (A) ASVs abundance, (B) ASVs presence/absence, (C) ZOTUs abundance, (D) ZOTUs presence/absence, (E) OTUs abundance, and (F) OTUs presence/absence. The axis 1 separated mainly the samples by year, while the axis 2 separated samples mainly by locality. Each point is one of the replicates sampled. Colors and symbols represent collection points (mouth of channel at Paraná River = blue square, Itaipu’s reservoir = green triangle, and Piracema Channel = circles [Bela Vista River 1 = purple, Bela Vista 2 = yellow, Brasilia stream = gray, and lake = red]), and filled represent year of collection (fill = 2019, empty = 2020).
Discussion
Our results support mounting evidence that eDNA analysis provides a cost-effective alternative to characterize fish biodiversity. We also demonstrate that different bioinformatic approaches show similar results in terms of alpha and beta diversity, supporting the use of molecular approaches to monitor biodiversity even with incomplete taxonomic identification. However, a serious caveat for using these molecular methods for biodiversity assessments is the scarcity of comprehensive taxonomic reference databases, especially for the tropical regions of the globe. Here, we also highlight these caveats for the Neotropical fish database, which are taxonomically limited, limiting the identification of several species. With a complete reference database, eDNA could detect mostly fish community and also fish species that are poorly or non-represented by conventional methods, as suggested by our results.
We identified 35 species with ASVs, 37 with OTUs, and 34 with ZOTUs approaches at > 97% similarity. However, many other ASVs, OTUs, and ZOTUs were identified at < 97% similarity, representing species not present in our database. Considering that 76 species sampled with traditional survey had no available sequences, many of these species may be present in our metabarcoding data but blasted as another species. We produced our reference database based on the historical taxonomic survey of Piracema Channel that may prevent identification of species that had not been recorded by conventional fish survey methods. However, the use of a database without curatorship can include spurious species identifications, such as species unlikely to be physically present at sampling sites10. That occurs because when the database does not contain the sequence of a certain species, the sequences will match with the closest species in that database, which can occur in a completely different environment (e.g. marine), beyond other factors that also contribute to registering spurious species, such as misannotated sequences57 or low variability in the target sequenced region10 that will sign any species with such similar sequence. For instance, our sequence for Prochilodus lineatus is identical to other Prochilodus species, such as P. harttii and P. costatus. Furthermore, there are many species undescribed, making it impossible to identify them. A recent compilation to list Paraná state fish species included 42 undescribed species5, and this number may be underestimated due to the presence of crypt species and sampling biases.
Even with the previously mentioned limitations, the use of molecular units such as ASVs58, OTUs22, and ZOTUs24 allows for assessing of genetic diversity and enables comparison among multiple sites59, space–time dynamics16 and evaluate natural and anthropogenic impacts60. For instance, vertebrate populations from freshwater ecosystems are declining at alarming rates (83% decline since 1970)61, and their conservation and management are a priority for global biodiversity62. The Neotropical region harbors one of the largest freshwater biodiversity, with an estimated 9000 described fish species (around 30% of total freshwater species)11. The increasing construction of dams is threatening fish populations over the entire planet63,64,65 but specially in Neotropical countries such as Brazil5,66,67, and effective ways to monitor fish biodiversity to understand its impact is essential.
As observed with the use of conventional ichthyofauna monitoring methods68, the number of species, ASVs, OTUs, ZOTUs, or 12S gene sequence readouts identified in our study showed a variation between the two sampling occasions (2019 and 2020). Such variations in fish assemblages can be related to a series of factors, both biotic (ecological characteristics of the species, for example) and abiotic (variations in water quality, and other environmental factors). In addition, physical characteristics of the environment such as total water volume and hydrological characteristics can also play a key role in the ecology and occurrence of fish species68. For instance, the recent extreme drought experienced in southeastern Brazil69 may have impacted fish assemblages. Our results showed a decrease of alpha diversity in 2020 in both mouth of channel at Paraná River (blue squares) and the lake (red circles; Fig. 2). In addition to the direct effects caused by this type of climatic phenomenon, such as the reduction in the volume of water, indirect effects such as reduced oxygen concentration in the water and food availability can cause severe impacts on fish’s communities68,70,71. Such effects were more evident at the mouth of channel at Paraná River, where the water level dropped 7 m from 2019 to 2020. At the reservoir, alpha diversity did not vary as water level fluctuation was less evident as a result of a stable environment due to the large size of this water body (green triangles; Fig. 2). However, the traditional survey in Piracema Channel was unable to significantly detect the diversity variation throughout the period of the study (Supplementary Fig. S2), highlighting the high sensibility of eDNA metabarcoding for monitoring.
Among sampling points, the highest alpha diversity was recorded in those collected in mouth of channel at Paraná River, while the lowest alpha diversity was registered in the lake (Fig. 2). Habitat heterogeneity is recognized as a main factor supporting functional and phylogenetic diversity, which is often reflected in the taxonomic richness of the fish communities72. Mouth of channel at Paraná River, the entrance of the Piracema Channel, is in a protected valley, where the riparian vegetation is conserved, allowing the colonization by a diversified flora and fauna. Besides this, the confluence with the Paraná River produces adjacent lotic and lentic microhabitats, supporting a higher alpha diversity when compared to the main lake or the water intake of the Channel, which are lentic and uniform environments. Such pattern of fish diversity/limnologic gradients meets the patterns previously assessed for the reservoir tributaries73.
The beta diversity showed that in 2020, with the event of the extreme drought, a homogenization of fish assemblage happened (Fig. 3). Both samples from the mouth of channel at Paraná River (blue squares) and the lake (red circles) cluster together with the reservoir in both years. The Itaipu’s Reservoir was filled in 1982 and the Piracema Channel (a fish pass), connecting the region just downstream from Itaipu Dam to the Itaipu Reservoir, was opened 38 years later. Both events allowed the dispersion of species (including non-native species) in both directions promoting the homogenization of communities from upper and lower Paraná River5,74,75. Our results show the importance of the closest rivers and streams for system diversity and resilience, as the mostly community variation was found in the Boa Vista River and Brasilia Stream (Fig. 3).
Although eDNA metabarcoding is a powerful tool for biodiversity, as it has been widely used for different purposes and different taxonomic groups, including identification and quantification of Neotropical ichthyofauna16,76,77, many issues can hamper the metabarcoding results7,10,78,79. Shaw et al.10 drew attention to methodological considerations related to the eDNA sampling process for freshwater fishes. According to them, the number of replicates is extremely important to obtain accurate data. Specifically, they demonstrated that the collection of two eDNA replicates per point were insufficient to detect less abundant taxa; however, adopting five replicates must have a 100% detection rate. In addition, sampling water column was more effective in detecting fish communities than sampling sediment10. Here, we collected six replicates per sampling point on the water surface. Furthermore, the rarefaction curves clearly show that many individual samples have a very low sequencing depth, but considering the replicates all our sampled localities reach the asymptote (Supplementary Fig. S1), although samples from the lake in 2020 just reach the asymptote considering OTUs analyses and the reservoir in 2020 had not reached the asymptote considering ZOTUs analyses (Supplementary Fig. S1).
The bioinformatic methodological choices can also affect the metabarcoding results. Here, we used three pipelines that showed the best results compared with other approaches80. We used both OTU-level clustering at 97% level, with UPARSE41, and the unique sequences with zero-radius ZOTU-level denoising, with UNOISE324, and ASV-level Divisive Amplicon Denoising Algorithm 2, with DADA223. Both the OTUs and the ZOTUs are created using in USEARCH81. The initial steps as merging, filtering, and deduplicating are the same for both approaches, with just the last step been different. The third approach generated ASVs through a parametric model, based in Q-scores to calculate a substitution model, estimating a probability for each possible base substitution, to infer true biological sequences from reads as implemented in DADA223. Although we recorded some variation in the number of reads and "species" registered in each pipeline, the results are very similar, highlighting their robustness.
Another potential bias in the results is data treatment. Here we used several data normalizations for both alpha and beta diversity. Although historically more used, rarefied data is biased to detect differentially abundant species51 and the hill numbers are considered the best approach for metabarcoding data54. Also, due to PCR biases, variation in the copy number of 12S genes per cell/genome, as well as differences in size and biomass across the targeted organisms can compromise a straightforward interpretation of OTU reads as an abundance measure82,83,84. However, rare (low abundant) ASVs, ZOTUs and OTUs are more likely to be an artefact (both erroneous sequence or because of cross-talk85) and the true sequences are more stochastically distributed due to the intrinsic low occurrence and detection probability86,87. Therefore, analyses that weight more the most abundant molecular units could be preferable. As each method has its own biases, we present here both approaches.
Finally, it is important to highlight that, in general, molecular data derived from “environmental sequencing” should be seen as complementary to, rather than as competing with, traditional taxonomic studies. Indeed, a confluence of both lines of evidence is highly warranted, as it will be necessary to overcome their respective shortcomings. For instance, we have shown here that many species occurring in the Itaipu fish pass system have no genetic data to allow their identification. Even so, other 47 species with sequences available were only identified with traditional surveys. This difference can be related to species density but also to primers biases. To improve the species detection with metabarcoding it is crucial to enhance the genetic reference database through traditional and to test if the primers used in metabarcoding studies are able to amplify species present in the studied system. Indeed, the metabarcoding approach is an intricate web of feedback loops with the species taxonomy and ecology.
Conclusion
Despite the clear indication that the reference databases need to be continuously fed with additional information on species that occur in the region, our results demonstrate the analytical efficiency of the metabarcoding approach for monitoring fish species in the Itaipu’s fish pass system. In addition, the methodology allowed, even when the specific identity of the ASVs, OTUs, and ZOTUs were below 97% similarity with the species in our database, to carry out estimates of species alpha and beta diversity. The use of such a methodology enables the monitoring of the fish community with sufficient sensitivity to detect changes due to some natural or anthropogenic event.
References
de Souza Dias, V., Pereira da Luz, M., Medero, G. M. & Tarley Ferreira Nascimento, D. An overview of hydropower reservoirs in Brazil: Current situation, future perspectives and impacts of climate change. Water 10, 592 (2018).
Patias, J., Zuquette, L. V. & Rodrigues-Carvalho, J. A. Piezometric variations in the basaltic massif beneath the Itaipu hydroelectric plant (Brazil/Paraguay border): Right Buttress Dam. Bull. Eng. Geol. Environ. 74, 207–231 (2015).
Agostinho, A. A. Pesquisas, monitoramento e manejo da fauna aquática em empreendimentos hidrelétricos. In Seminário Sobre Fauna Aquática E O Setor Elétrico Brasileiro 38–59 (Brasil, 1994).
Makrakis, S., Gomes, L. C., Makrakis, M. C., Fernandez, D. R. & Pavanelli, C. S. The Canal da Piracema at Itaipu Dam as a fish pass system. Neotrop. Ichthyol. 5, 185–195 (2007).
Dos Reis, R. B., Frota, A., Depra, G. D. C., Ota, R. R. & Da Graca, W. J. Freshwater fishes from Paraná State, Brazil: An annotated list, with comments on biogeographic patterns, threats, and future perspectives. Zootaxa 4868, 451–494 (2020).
Becker, R. A., Sales, N. G., Santos, G. M., Santos, G. B. & Carvalho, D. C. DNA barcoding and morphological identification of neotropical ichthyoplankton from the Upper Paraná and São Francisco. J. Fish Biol. 87, 159–168 (2015).
Milan, D. T. et al. New 12S metabarcoding primers for enhanced Neotropical freshwater fish biodiversity assessment. Sci. Rep. 10, 1–12 (2020).
Agostinho, A. A., Pelicice, F. M. & Gomes, L. C. Dams and the fish fauna of the Neotropical region: Impacts and management related to diversity and fisheries. Braz. J. Biol. 68, 1119–1132 (2008).
Bonar, S. A., Hubert, W. A. & Willis, D. W. Standard methods for sampling North American freshwater fishes. American Fisheries Society, Bethesda, (USA, 2009).
Shaw, J. L. A. et al. Comparison of environmental DNA metabarcoding and conventional fish survey methods in a river system. Biol. Conserv. 197, 131–138 (2016).
Reis, R. E. et al. Fish biodiversity and conservation in South America. J. Fish Biol. 89, 12–47 (2016).
Baumgartner, G. et al. Peixes do baixo rio Iguaçu. (Eduem, 2012).
Taberlet, P., Bonin, A., Coissac, E. & Zinger, L. Environmental DNA: For Biodiversity Research and Monitoring (Oxford University Press, 2018).
Taberlet, P., Coissac, E., Pompanon, F., Christian, B. & Willerslev, E. Towards next-generation biodiversity assessment using DNA metabarcoding. Mol Ecol 33, 2045–2050 (2012).
Ritter, C. D. et al. The pitfalls of biodiversity proxies: Differences in richness patterns of birds, trees and understudied diversity across Amazonia. Sci. Rep. 9, 1–3 (2019).
Sales, N. G. et al. Space-time dynamics in monitoring neotropical fish communities using eDNA metabarcoding. Sci. Total Environ. 754, 142096 (2021).
Zinger, L. et al. Body size determines soil community assembly in a tropical forest. Mol. Ecol. 28, 528–543 (2019).
Baird, D. J. & Hajibabaei, M. Biomonitoring 2.0: A new paradigm in ecosystem assessment made possible by next-generation DNA sequencing. Mol. Ecol. 21, 2039–2044 (2012).
Zinger, L. et al. Advances and prospects of environmental DNA in neotropical rainforests. Adv. Ecol. Res. 62, 331–373 (2020).
Cilleros, K. et al. Unlocking biodiversity and conservation studies in high-diversity environments using environmental DNA (eDNA): A test with Guianese freshwater fishes. Mol. Ecol. Resour. 19, 27–46 (2019).
Sales, N. G., Wangensteen, O. S., Carvalho, D. C. & Mariani, S. Influence of preservation methods, sample medium and sampling time on eDNA recovery in a neotropical river. Environ. DNA 119–130. https://doi.org/10.1002/edn3.14 (2020).
Blaxter, M. et al. Defining operational taxonomic units using DNA barcode data. Philos. Trans. R. Soc. B Biol. Sci. 360(1462), 1935–1943. https://doi.org/10.1098/rstb.2005.1725 (2005).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016).
Edgar, R. C. UNOISE2: Improved error-correction for Illumina 16S and ITS amplicon sequencing. BioRxiv 81257 (2016).
Muha, T. P., Rodriguez-Barreto, D., O’Rorke, R., Garcia de Leaniz, C. & Consuegra, S. Using eDNA metabarcoding to monitor changes in fish community composition after barrier removal. Front. Ecol. Evol. 9, 28 (2021).
Kitano, T., Umetsu, K., Tian, W. & Osawa, M. Two universal primer sets for species identification among vertebrates. Int. J. Legal Med. 121, 423–427 (2007).
Stoeckle, M. Y., Soboleva, L. & Charlop-Powers, Z. Aquatic environmental DNA detects seasonal fish abundance and habitat preference in an urban estuary. PLoS One 12, e0175186 (2017).
Bylemans, J. et al. An environmental DNA-based method for monitoring spawning activity: A case study, using the endangered Macquarie perch (Macquaria australasica). Methods Ecol. Evol. 8, 646–655 (2017).
De Souza, L. S., Godwin, J. C., Renshaw, M. A. & Larson, E. Environmental DNA (eDNA) detection probability is influenced by seasonal activity of organisms. PLoS One 11, e0165273 (2016).
Ritter, C. D. et al. Locality or habitat? Exploring predictors of biodiversity in Amazonia. Ecography (Cop.) 42, 321–333 (2019).
CFMV-Resolução no 1000 de 11 de maio de 2012—Dispõe sobre procedimentos e métodos de eutanásia em animais e dá outras providências. (2012).
Britski, H. A., de Silimon, K. Z. S. & Lopes, B. S. Peixes do Pantanal: manual de identificação, ampl. Brasília, DF, Embrapa Informação Tecnológica (2007).
Ota, R. R., Deprá, G. de C., Graça, W. J. da & Pavanelli, C. S. Peixes da planície de inundação do alto rio Paraná e áreas adjacentes: revised, annotated and updated. Neotrop. Ichthyol. 16(2). https://www.scielo.br/j/ni/a/tScwvm8JLhKnbxKjtBQLPBx/abstract/?lang=en (2018).
Neris, N., Villalba, F., Kamada, D. & Viré, S. Guía de peces del Paraguay/Guide of fishes of Paraguay. Zamphiropolos, (Paraguay, 2010).
Pie, M. R. et al. Development of a real-time PCR assay for the detection of the golden mussel (Limnoperna fortunei, Mytilidae) in environmental samples. An. Acad. Bras. Cienc. 89, 1041–1045 (2017).
Miya, M. et al. MiFish, a set of universal PCR primers for metabarcoding environmental DNA from fishes: Detection of more than 230 subtropical marine species. R. Soc. Open Sci. 2, 150088 (2015).
Boeger, W. A. et al. Testing a molecular protocol to monitor the presence of golden mussel larvae (Limnoperna fortunei) in plankton samples. J. Plankton Res. 29, 1015–1019 (2007).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10–12 (2011).
Van Rossum, G. & Drake, F. L. Python 3 References Manual. Scotts Valley CA: CreateSpace. (2009).
R Core Team. R: the R project for statistical computing. 2019. https://www.r-project.org/ (accessed 30 Mar 2020).
Edgar, R. C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998 (2013).
Edgar, R. C. & Flyvbjerg, H. Error filtering, pair assembly and error correction for next-generation sequencing reads. Bioinformatics 31, 3476–3482 (2015).
Camacho, C. et al. BLAST+: Architecture and applications. BMC Bioinform. 10, 421 (2009).
Team, Rs. RStudio: integrated development for R. RStudio, Inc., Boston, MA https://www.rstudio.com42, 84 (2015).
Wickham, H. tidyverse: Easily Install and Load “Tidyverse” Packages (Version R package version 1.1. 1). (2017).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Springer, 2016).
Tang, Y., Horikoshi, M. & Li, W. ggfortify: Unified interface to visualize statistical results of popular R packages. R J. 8, 474 (2016).
Auguie, B. & Antonov, A. gridExtra: Miscellaneous functions for “grid” graphics (Version 2.2. 1)[Computer software]. (2016).
Kassambara, A. & Kassambara, M. A. Package ‘ggpubr’. (2020).
Oksanen, J. et al. Vegan: Community ecology package. R package version 1.17-4. https://cran.r-project.org. Acesso em 23, 2010 (2010).
McMurdie, P. J. & Holmes, S. Waste not, want not: Why rarefying microbiome data is inadmissible. PLoS Comput. Biol. 10, e1003531 (2014).
Jost, L. Entropy and diversity. Oikos 113, 363–375 (2006).
Marcon, E., Herault, B. & Marcon, M. E. Package ‘entropart’. (2021).
Mächler, E., Walser, J.-C. & Altermatt, F. Decision making and best practices for taxonomy-free eDNA metabarcoding in biomonitoring using Hill numbers. BioRxiv (2020).
McMurdie, P. J. & Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8, e61217 (2013).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
León, A., Reyes, J., Burriel, V. & Valverde, F. Data quality problems when integrating genomic information. In International Conference on Conceptual Modeling 173–182 (Springer, 2016).
Callahan, B. J., McMurdie, P. J. & Holmes, S. P. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 11, 2639–2643 (2017).
Stahlhut, J. K. et al. DNA barcoding reveals diversity of hymenoptera and the dominance of parasitoids in a sub-arctic environment. BMC Ecol. 13, 2 (2013).
Gillet, B. et al. Direct fishing and eDNA metabarcoding for biomonitoring during a 3-year survey significantly improves number of fish detected around a South East Asian reservoir. PLoS One 13, e0208592 (2018).
Barrett, M. et al. Living planet report 2018: Aiming higher. WWF. Available at: https://www.globallandscapesforum.org/publication/living-planet-report-2018-aiming-higher/ (2018).
Díaz, S. M. et al. The global assessment report on biodiversity and ecosystem services: Summary for policy makers. Intergovernmental Science-Policy Platform on Biodiversity and Ecosystem Services. 56, (2019).
Dudgeon, D. Asian river fishes in the Anthropocene: Threats and conservation challenges in an era of rapid environmental change. J. Fish Biol. 79, 1487–1524 (2011).
Dudgeon, D. Multiple threats imperil freshwater biodiversity in the Anthropocene. Curr. Biol. 29, R960–R967 (2019).
He, F. et al. Disappearing giants: A review of threats to freshwater megafauna. Wiley Interdiscip. Rev. Water 4, e1208 (2017).
Agostinho, A. A., Thomaz, S. M. & Gomes, L. C. Threats for biodiversity in the floodplain of the Upper Paraná River: Effects of hydrological regulation by dams. (2018). Int. J. Ecohydrol. Hydrobiol Warsaw. 4(3), 267–280 (2004).
Santana, M. L., Carvalho, F. R. & Teresa, F. B. Broad and fine-scale threats on threatened Brazilian freshwater fish: Variability across hydrographic regions and taxonomic groups. Biota Neotrop. 21 (2). https://www.scielo.br/j/bn/a/YqFbWSy5vbfHy3QK9kNpdKp/?format=html&lang=en (2021).
Matthews, W. J. Patterns in Freshwater Fish Ecology. (Springer Science & Business Media, 2012).
de Oliveira Bueno, E., Alves, G. J. & Mello, C. R. Hydroelectricity water footprint in Parana hydrograph region, Brazil. Renew. Energy 162, 596–612 (2020).
Camacho Guerreiro, A. I., Amadio, S. A., Fabre, N. N. & da Silva Batista, V. Exploring the effect of strong hydrological droughts and floods on populational parameters of Semaprochilodus insignis (Actinopterygii: Prochilodontidae) from the Central Amazonia. Environ. Dev. Sustain. 23, 3338–3348 (2021).
Jespersen, H., Rasmussen, G. & Pedersen, S. Severity of summer drought as predictor for smolt recruitment in migratory brown trout (Salmo trutta). Ecol. Freshw. Fish 30, 115–124 (2021).
Pool, T. K., Grenouillet, G. & Villéger, S. Species contribute differently to the taxonomic, functional, and phylogenetic alpha and beta diversity of freshwater fish communities. Divers. Distrib. 20, 1235–1244 (2014).
de Oliveira, E. F., Goulart, E. & Minte-Vera, C. V. Fish diversity along spatial gradients in the Itaipu Reservoir, Paraná, Brazil. Braz. J. Biol. 64, 447–458 (2004).
Daga, V. S. et al. Homogenization dynamics of the fish assemblages in Neotropical reservoirs: Comparing the roles of introduced species and their vectors. Hydrobiologia 746, 327–347 (2015).
Vitule, J. R. S. Introdução de peixes em ecossistemas continentais brasileiros: revisão, comentários e sugestões de ações contra o inimigo quase invisível. Neotrop. Biol. Conserv. 4, 111–122 (2009).
Mariac, C. et al. Species‐level ichthyoplankton dynamics for 97 fishes in two major river basins of the Amazon using quantitative metabarcoding. Mol. Ecol. https://onlinelibrary.wiley.com/action/showCitFormats?doi=10.1111%2Fmec.15944 (2021).
Jackman, J. M. et al. eDNA in a bottleneck: Obstacles to fish metabarcoding studies in megadiverse freshwater systems. Environ. DNA 3, 837–849 (2021).
Bessey, C. et al. Maximizing fish detection with eDNA metabarcoding. Environ. DNA 2, 493–504 (2020).
Evans, N. T. et al. Fish community assessment with eDNA metabarcoding: Effects of sampling design and bioinformatic filtering. Can. J. Fish. Aquat. Sci. 74, 1362–1374 (2017).
Prodan, A. et al. Comparing bioinformatic pipelines for microbial 16S rRNA amplicon sequencing. PLoS One 15, e0227434 (2020).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010).
Elbrecht, V. & Leese, F. Can DNA-based ecosystem assessments quantify species abundance? Testing primer bias and biomass—sequence relationships with an innovative metabarcoding protocol. PLoS ONE 10, e0130324 (2015).
Pawluczyk, M. et al. Quantitative evaluation of bias in PCR amplification and next-generation sequencing derived from metabarcoding samples. Anal. Bioanal. Chem. 407, 1841–1848 (2015).
Holman, L. E., Chng, Y. & Rius, M. How does eDNA decay affect metabarcoding experiments? Environ. DNA https://onlinelibrary.wiley.com/action/showCitFormats?doi=10.1002%2Fedn3.201 (2021).
Edgar, R. C. UNCROSS2: identification of cross-talk in 16S rRNA OTU tables. BioRxiv 400762 (2018).
MacArthur, R. H. Geographical Ecology: Patterns in the Distribution of Species. (Princeton University Press, 1984).
Leray, M. & Knowlton, N. Random sampling causes the low reproducibility of rare eukaryotic OTUs in Illumina COI metabarcoding. PeerJ 5, e3006 (2017).
Team, Q. D. QGIS geographic information system. Open Source Geospatial Found. Proj. Versão 2, (2015).
Acknowledgements
Funding was provided by Itaipu Binacional to AON (Grant #4500049847). We thank Itaipu Binacional for providing postdoctoral fellow grants to GDP and NC and the Alexander van Humboldt foundation for providing a postdoctoral fellow grant to CDR. We also thank Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) for awarding AO and MRP with research fellowship grants (Grants #304633/2017-8 and #302904/2020-4, respectively).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
G.D.P, A.H, N.C., O.S.M.N., A.O., and M.P. study conceptualization and experimental design. A.O.A, P.V.S., and E.B. molecular analysis. C.H. taxonomic sampling and identification. C.D.R. bioinformatics and statistical analysis. A.O. an M.P. fund-raising. A.O. make the map figure. C.D.R. and G.D.P. wrote the first version with the collaboration of all authors. All authors read and approved the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dal Pont, G., Duarte Ritter, C., Agostinis, A.O. et al. Monitoring fish communities through environmental DNA metabarcoding in the fish pass system of the second largest hydropower plant in the world. Sci Rep 11, 23167 (2021). https://doi.org/10.1038/s41598-021-02593-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-02593-5
- Springer Nature Limited