
Abstract
The reduction of bumblebee populations has been reported in the last decades, and the microsporidian parasite Nosema bombi is considered as one of the factors contributing to such reduction. Although the decline of bee populations affects both wild plants and human food supply, the effects of Nosema spp. infections are not known because it is difficult to obtain infective spores from wild bees due to their low prevalence. Microscopical observation of fecal samples or midgut homogenates and/or PCR are generally used for N. bombi detection. However, the germination rate of microsporidian spore declines if they are kept at 4 °C for a long time or frozen. It is therefore crucial to minimize the diagnosis and isolation time of infective spores from field-collected samples. Therefore, we performed a loop-mediated isothermal amplification (LAMP) assay for the direct detection of N. bombi in bumblebee midgut homogenates. Using this method, we could detect N. bombi from individuals from which it was visible under the microscope and directly from wild individuals.
Similar content being viewed by others


Introduction
Pollinators play a crucial role in agricultural productivity and in the genetic diversity of wild plants1,2. Bumblebees are wild pollinators throughout temperate ecosystems, and their recent domestication has increased their economic importance3. Recently, the decrease of wild bumblebee populations has been reported worldwide4,5,6,7,8,9, and four species have become locally extinct in 11 European countries over the last 60 years10. Several factors have been proposed to contribute to this phenomenon, including the decline or fragmentation of their habitats, use of pesticides, and native and invasive pathogens. Within this scenario, the invasive microsporidian pathogen Nosema bombi has attracted attention11.
Microsporidia are obligatory intracellular parasites, belonging to kingdom fungi12. The host range of microsporidia species is broad as these can infect almost all vertebrate and invertebrate animals, with insects considered as one of the main host categories13. Microsporidia are generally opportunistic parasites, but they can cause severe symptoms to certain insect species14,15. Nosema apis and N. ceranae are considered parasites of the western honeybee Apis mellifera16 and eastern honeybee A. cerana17, respectively. Recently, N. ceranae was found to infect A. mellifera leading to the reduction of the survival rate of workers and collapse of colonies, suggesting that the host switch between A. mellifera and A. cerana contributed to the reduction of A. mellifera populations18,19.
Since the first report on N. bombi infecting bumblebees (Bombus spp.) in 191420, several pathological studies have been conducted on this bumblebee/honeybee-parasite system, from parasite transmission21 and tissue tropism22, to the reduced fitness of hosts due to decreasing sperm production and reduced survival rate of workers23, and male longevity and colony size24. In the United States, N. bombi is considered as one of the factors causing the wild population decline of bumblebees because N. bombi prevalence was higher in the declining bumblebee species than in others9,11. However, little evidence has been reported on the negative effects of N. bombi infection on bumblebee species. To elucidate on this issue, a bioassay using a substantial amount of infectious N. bombi spores should be performed. However, it is difficult to isolate active spores from field samples. Firstly, the prevalence of N. bombi is often low in wild bumblebees. In the United States, the overall prevalence in nine different bumblebee species was 2.9% (N = 9909), and the highest prevalence was 37.2% (N = 172)25. In Spain, the mean prevalence was 1.2% (N = 83) in Bombus terrestris26, and in Japan, it was 7.5% (N = 120) and 0% (N = 100) in B. terrestris and native species, respectively27. Secondly, although the microsporidia prevalence can be high at the colony level, for example 61.8% of the commercial colonies were infected in Ireland28, wild nests of bumblebees are hardly found in the field because they are established under ground or beneath grasses. Finally, even if some active spores are isolated from wild bumblebees, the low stability of these spores often hinders further bioassays. Because the long-time storage and freezing of N. apis and N. ceranae spores reduce their infectivity29, this is also expected for N. bombi spores. Moreover, N. bombi is usually detected by microscopy of fecal samples or PCR assays, which are time-consuming methodologies, especially when many samples are examined.
Loop-mediated isothermal amplification (LAMP) is an isothermal gene amplification method of target DNA, which is inexpensive, easy, and rapid30. This method can be performed under isothermal conditions due to the strand displacement activity of Bst DNA polymerase, and it shows high specificity because the primer set used (F3, B3, FIP, and BIP) binds six regions of the target DNA. Two additional “loop-primers”, LF and LB, contribute to increase the specificity of the LAMP reaction and to accelerate it31. In this method, the amplification can be detected without electrophoresis using magnesium pyrophosphate as a by-product32,33,34. Another important feature of LAMP is that the Bst DNA polymerase is hardly affected by the most common inhibitors35, which enables amplification of a target sequence without DNA extraction36,37. In the present study, we performed LAMP for N. bombi detection and compared its sensitivity to that of conventional PCR. We also tested LAMP as a method for detecting N. bombi from midgut homogenates without DNA extraction, which improves the versatility of LAMP.
Results
Optimal LAMP reaction temperature
Our LAMP primer set was designed based on the SSU rRNA nucleotide sequence of N. bombi (GenBank Accession No. AY008373; Fig. 1). To optimize the reaction temperatures of the LAMP assay for detecting N. bombi, we conducted a LAMP assay from 61 °C to 64 °C using the DNA extracted from the spore suspension of N. bombi (c. 1 ng/μl). The fluorescence started to increase most rapidly at 63 °C, at about 15 min of reaction time (Fig. 2). Experiments were conducted three times with similar results. It should be noted that fluorescence started to increase within 20 min for all temperatures, indicating that the LAMP assay developed in this study had tolerance to slight temperature changes. In the following experiments, we performed LAMP at 63 °C.

The LAMP primer set used in the present study. Right arrows indicate forward primers and left arrows indicate reverse primers. Terminals nucleotides are shown in capital letters. Numbers refer to nucleotide positions in the sequence of Nosema bombi SSU rRNA retrieved from GenBank (Accession No. AY008373).

LAMP assay using different reaction temperatures. The reaction temperatures corresponding to each curve are indicated. This LAMP assay was performed for 40 min using 1 ng of DNA extracted from N. bombi spores. The vertical axis indicates the fluorescence signal and the horizontal axis the reaction time.
Sensitivity of PCR and LAMP assays
We compared the sensitivity of the LAMP assay to that of the PCR assay using 1 μl of each 10-fold serial dilution of the DNA template (1 ng/μl to 1 fg/μl) from N. bombi spore suspension and the primers for N. bombi SSU rRNA designed by Klee et al.38. Both PCR and LAMP assays could detect N. bombi using 10 pg of template DNA (Fig. 3). Therefore, the PCR assay using the N. bombi-specific primers and our LAMP assay showed the same sensitivity.

Sensitivity of the PCR and LAMP assays. (a) PCR assay; Numbers 1 to 7 indicate the samples resulting from the 10-fold serial dilutions with DNA concentrations ranging from 1 ng/μl to 1 fg/μl. M and N are the DNA marker and the negative control, respectively. (b) LAMP assay; the vertical axis indicates the fluorescence signal and the horizontal axis the reaction time. The LAMP was performed at 63 °C for 40 min. 1 μl each of DNA was used in both PCR and LAMP. Full-length gel is presented in Supplementary Fig. S1.
Specificity test of LAMP primer
In bumblebees, the infections of N. ceranae has been reported in Uruguay39 and South America40, and thus the specificity of our lamp primer set was examined using both N. bombi and N. ceranae DNA (the DNA concentrations were 1.0 and 1.7 ng/μl, respectively). Our LAMP primer set could detect not only N. bombi but also N.ceranae (Fig. 4).

Specificity of our LAMP primer. The LAMP assay was performed at 63 °C for 40 min using N. bombi and N. ceranae DNA extracted from their spore suspensions. The DNA concentrations were 1.0 and 1.7 ng/μl, respectively. 1 μl of DNA was used in the LAMP assay. The vertical axis indicates the fluorescence signal and the horizontal axis indicates the reaction time.
Detection of N. bombi by the direct LAMP assay and microscopy
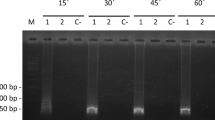
To apply LAMP for direct N. bombi detection without DNA extraction, we tested if LAMP could detect N. bombi from midgut homogenates and compared its result with that of microscopical observation. In this experiment, the artificially-infected midgut homogenates with spore concentrations ranging from 4.57 × 104 spores/μl to 4.57 spores/μl were prepared, and then 1 μl of them were used for microscopic observations. In the direct LAMP assay, the samples were added to reaction reagent using toothpicks. This is because in the direct LAMP assay, no signal was observed when 1 μl of midgut homogenate was used (data not shown). The direct LAMP assay detected N. bombi from homogenates at concentration as low as 4.57 × 101 spores/μl (Fig. 5a). On the other hand, using microscopy, N. bombi was detected from homogenates containing 4.57 × 102 spores/μl, but not from homogenates with lower concentrations (Fig. 5b). Noteworthy, it took more than 5 min to find at least one spore even in the midgut homogenate with 4.57 × 102 spores/μl by microscopy. This suggested that we were not able to find spores in midgut homogenates with approximately 100 spores/μl by microscopical observation, whereas LAMP could reliably detect N. bombi spores in homogenates with concentrations lower than this. Considering these results, direct LAMP can detect N. bombi in samples in which N. bombi spores could be observed under a microscope. On the contrary, in conventional PCR, amplifications did not occur even when homogenate with the highest spore concentration, which was added using a toothpick, was used, probably due to impurities.

Detection limit of the direct LAMP assay using midgut homogenates as templates. (a) Direct LAMP assay using midgut homogenates. The vertical axis indicates the fluorescence signal and the horizontal axis the reaction time. Numbers indicate spore concentrations (spores/μl). LAMP was performed at 63 °C for 40 min. (b) Images of the artificially-infected midgut homogenates obtained by phase contrast microscopy (×400). Numbers indicate spore concentrations (spores/μl). Arrowheads indicate N. bombi spores. Scale bar = 20 μm. In direct LAMP assay, midgut homogenates were added using toothpicks, while in microscopic observation, 1 μl each of midgut homogenate was observed.
Direct detection of N. bombi from wild bumblebees
To confirm that direct LAMP could detect N. bombi-infected individuals collected in the field, we used the midgut homogenates of eight B. hypocrita sapporoensis and eight B. terrestris collected from the field for direct LAMP. To validate the results obtained by direct LAMP, we performed a PCR assay using 5 to 10 ng/μl each of template DNA extracted from each midgut sample, as well as normal LAMP also using template DNA. The PCR assay detected N. bombi from two out of the 16 individuals, H2 and H8 (Fig. 6a), and N. bombi infection was verified by direct sequencing analysis. Nosema bombi was also detected in these two individuals in the LAMP assay using template DNA by both Genie®II and block incubator (Fig. 6b,c). Moreover, similar results were obtained for midgut homogenates using a block incubator and Genie®II (Fig. 6d,e), indicating that LAMP can directly detect N. bombi from wild individuals. Under the microscope, N. bombi spores were observed in the midgut homogenates of H2 and H8 with spore concentrations of 6.25 × 105 spores/μl and 3.00 × 105 spores/μl, respectively. On the other hand, in H3 in Fig. 6c, no spores were found by microscopic observation; the PCR and the other LAMP results (Fig. 6a,b,d,e) were also negative. This LAMP assay using extracted DNAs was repeated once more; H3 was negative but T7 became positive, while H2 and H8 showed positive (Supplementary Fig. S3a). To further confirm the conclusion that only H2 and H8 were infected by N. bombi, we conducted an additional LAMP assay using these four samples (H2, H3, H8, and T7), two randomly selected samples (T1 and T5), a positive control (the same as for the PCR assays in this paper), and a negative control (ultra-pure water) (Supplementary Fig. S3b). In this assay, N. bombi was not detected from H3 and T7. Additionally, to examine whether these four samples, H2, H3, H8, and T7, were infected with N. ceranae, we conducted PCR for detecting N. ceranae using the primer targeting N. ceranae SSU rRNA gene (Table 1), which assured that no sample was infected (Supplementary Fig. S4). The results found in H3 and T7 were sometimes observed in LAMP assays37, which could be the unreproducible amplifications, and such false positive results occurred more frequently when a template homogenate was used without dilution.

Detection of N. bombi by PCR and LAMP assays using wild individuals. (a) PCR assay using extracted DNA (from 5 to 10 ng/μl). (b) LAMP assay using extracted DNA and a block incubator. (c) LAMP assay using extracted DNA and the Genie®ІІ. (d) Direct LAMP assay using midgut homogenates and a block incubator. (e) Direct LAMP assay using midgut homogenates and the Genie®ІІ. Each LAMP assay was performed at 63 °C for 40 min. Sample names starting with “H” correspond to B. hypocrita sapporoensis samples and with “T” to B. terrestris sample; M: DNA marker, P: positive control, N: negative control. 1 μl each of DNA was used in (a–c), while the midgut homogenates were added using toothpicks in (d,e). Full-length gel is presented in Supplementary Fig. S2.
To confirm the result of our direct LAMP assay with the conventional PCR and the microscopic observation, we performed additional direct LAMP assays using another 73 samples collected from the field. Twelve out of 73 samples showed positive results in the direct LAMP assay and spores were observed under the microscopy from 3 out of these 12 samples, while the other 9 samples were negative in both the microscopy and PCR (Supplementary Table S1). According to these results, there were no samples which were positive under the microscopy but negative in the direct LAMP assay; that is, our direct LAMP assay could perfectly detect N. bombi infected samples from which some spores were visible under microscopy. We also confirmed that the nine samples that were negative in both microscopy and PCR were not infected by N. ceranae (Supplementary Fig. S4).
Discussion
In this study, a direct LAMP assay for detecting Nosema parasites from midgut homogenates of bees was established for the first time. Nosema spp. are considered as possible agents of the current decline observed in bees worldwide. We could reliably detect N. bombi from infected individuals by suspending their midguts in PBS and performing LAMP using this suspension as the template. Using LAMP, N. bombi-infected bumblebees can be easily and rapidly (in less than 1 h) detected. Moreover, LAMP sensitivity is identical to that of conventional PCR and higher than that of microscopical observation. However, our LAMP primer set could also detect not only N. bombi but also N. ceranae (Fig. 4). Moreover, unreproducible amplification occurred sometimes. In the future, a specific primer set for N. bombi detection targeting other N. bombi gene should be prepared and solutions that could reduce unreproducible amplification should be identified.
A previous study41 also used LAMP for detecting N. apis and N. ceranae infecting honeybees, but the template material was the total DNA extracted from infected bees. Thus, its results cannot be directly applied to a practical field diagnosis. Microsporidian spores generally have a thick chitinous cell wall42, which protects them from severe environmental conditions, such as inadequate temperature and relative humidity43,44,45. For this reason, the extraction of the large amount of DNA from Nosema spp. spores involves physical crushing via glass beads or liquid nitrogen, which is time-consuming and requires special equipment11,46,47,48. On the other hand, the direct LAMP method established here could amplify the target DNA without these extraction steps.
We also showed that LAMP can be performed using a simple incubator, which allows us to examine many samples whereas in Genie®II we can only examine 16 samples simultaneously. Moreover, the reaction stability of the LAMP reaction at several temperatures and the stability of LAMP reagents at room temperature49 might allow to diagnose the Nosema spp. infections using non-specific equipment, as in a water bath at 60 °C. Using the LAMP method developed in this study, we will be able to obtain infective N. bombi spores by screening wild bumblebees, without DNA extraction in the field, and examine the influence of Nosema spp. infections on bumblebee populations. Furthermore, the reaction time of our LAMP method could be shortened to 30 min because when the midgut homogenate from which we could detect N. bombi spores by microscopy was used as a template, amplification occurred within 30 min. This direct detection method can also be applied for screening N. apis and N. ceranae in honeybees using the previously published LAMP primer sets41. The results obtained here suggest that direct LAMP can also be applied to other pollinating bee and Nosema species and thus contribute to examine the causes underlying the decline in wild bee populations.
It remains unclear how LAMP can detect N. bombi DNA directly from midgut homogenates. One possible reason is the existence of a small amount of “naked” N. bombi DNA (uncovered by cell membranes) leaking into the midgut of bees from spores that either died from succumbing to host immunity or were in mid-reproduction.
Although LAMP-based diagnoses have been applied to several human and plant diseases, to plant quarantine, and to food inspection50,51, and direct LAMP has been able to detect not only viruses or fungi of plants but also fungal virus37, such methods are not necessarily applied to pathogen detection in the field. Thus, to apply LAMP in on-site surveys, future studies on the development of simplified, direct diagnosis methods are needed.
Materials and Methods
Preparation of the Nosema bombi and Nosema ceranae spore suspension
N. bombi spores used for the experiments were isolated from four queens of Bombus hypocrita hypocrita collected in Yamanashi prefecture, Japan, in September 2016, and N. ceranae spores were isolated from A. cerana workers collected from a wild colony in Tokyo, Japan, in April 2016, after confirming N. bombi and N. ceranae infection as described below. Firstly, each midgut was homogenized in 100 μl of phosphate-buffered saline (PBS; 1370 mM NaCl, 27 mM KCl, 81 mM Na2HPO4 12H2O, and 14.7 mM KH2PO4; pH 7.4) (hereafter referred to as midgut homogenate) and filtered through a lens paper into a 15-ml tube. The filtrate was centrifuged at 3000 × g for 10 min at 20 °C and the resulting supernatant was removed. The precipitate was washed by resuspension with 1 ml PBS and further centrifuged under the same conditions until the pellet became white. Finally, the pellet was resuspended in 100 μl PBS (hereafter referred to as the spore suspension) and N. bombi spore concentration calculated using a hemocytometer was 4.57 × 104 spores/μl.
After DNA extraction and PCR using this spore suspension, direct sequencing analysis was conducted as described below. The obtained sequence was aligned using MEGA 6.052 and compared with the N. bombi and N. ceranae SSU rRNA sequence deposited in GenBank (http://www.ncbi.nlm.nih.gov/genbank/) under accession number AY008373 and XR002966746, respectively. These extracted DNA were used as a positive control in all PCR assays.
DNA extraction
A sample of midgut homogenate (50 μl) or spore suspension (25 μl) was mixed with 250 μl DNAzol, (Molecular Research Center, Cincinnati, OH, USA) and 250 mg glass beads (Sigma-Aldrich, St. Louis, MO, USA), and homogenized using Precellys 24 (Bertin, Montigny-le-Bretonneux, France) at 5500 rpm for 20 s. The homogenate was transferred to a new tube and 250 μl DNAzol and 1 μl Proteinase K (20 mg/ml) (Merck Bioscience, Darmstadt, Germany) were added and mixed by inversion (25 times). After incubation for 1 h at 50 °C, the mix was centrifuged at 15,400 × g for 10 min at 4 °C, and 300 μl of the resulting supernatant was transferred to a new tube. After adding 150 μl of 99% ethanol, and mixing by inversion (50 times), a new centrifugation was performed (20,400 × g for 10 min at 4 °C). Then discarding the resulting supernatant, the pellet was washed twice with 70% ethanol, dissolved with 20 μl of sterilized distilled water (SDW), and incubated at 60 °C for 15 min. The DNA concentration was then quantified using NanoVue Plus (GE, Boston, MA, USA).
Preparation of artificially-infected midgut homogenates
Ten-fold serial dilutions of the spore suspension were prepared and 1 μl of each diluted solution was added to 9 μl of midgut homogenate of Bombus ardens ardens workers, which were confirmed to be free of N. bombi by PCR. The concentration of these solutions, designated “artificially-infected midgut homogenates” ranged from 4.57 × 104 to 4.57 spores/μl.
Amplification and sequencing
The PCR assay for detecting N. bombi and N. ceranae was performed using 1.0 μl of 10× Ex Taq Buffer (TaKaRa, Shiga, Japan), 0.8 μl of 2.5 mM each dNTP mixture (TaKaRa), 0.2 μl of 10 μM each forward and reverse primers for Small subunit (SSU) rRNA38,53 (Table 1), 0.05 μl of 5 U/μl Ex Taq HS (TaKaRa), 1 μl of template DNA, and 6.75 μl of SDW. The PCR profile for detection of N. bombi comprised initial denaturation at 94 °C for 3 min, followed by 40 cycles at 94 °C for 30 s, 50 °C for 30 s, and 72 °C for 30 s, and a final extension at 72 °C for 5 min. For detection of N. ceranae, the same PCR profile described by Martín-Hernández et al.53 was used. After electrophoresis and observation under UV light, the PCR products were purified using a QIAquick PCR Purification Kit (Qiagen, Hilden, Germany). The purified DNA was used for direct sequencing reactions with BigDye Terminator Kit ver. 3.1 (Applied Biosystems, Foster City, CA, USA). Sequencing was performed in the 3700 DNA analyzer (Applied Biosystems). The obtained sequences were confirmed as the N. bombi and N. ceranae SSU rRNA.
LAMP
All LAMP experiments were conducted in a clean bench with autoclaved equipment and filter chips. The primer set used in the present study was designed using Primer Explorer 5 (https://primerexplorer.jp/) (Fig. 1 and Table 1). The LAMP reaction was performed using 12.5 μl of 2 × Premix [0.5 μl Tris-HCl (pH 8.8), 0.25 μl 1 M KCl, 0.25 μl 1 M (NH4)2SO4, 0.2 μl 1 M MgSO4, 0.25 μl 10% Tween20 (Wako, Osaka, Japan), 0.35 μl of each 100 mM dATP, 100 mM dTTP, 100 mM dCTP, and 100 mM dGTP (Promega), 4 μl of 5 M Betaine (Sigma-Aldrich), and 5.65 μl SDW], 5.0 μl of 5 × Primer mix (F3: B3: FIP: BIP: LF: LB: DW = 1: 1: 8: 8: 4: 4: 74; the concentration of each primer was 100 μM), 1.0 μl of Fluorescent Detection Reagent (Eiken-Chemical, Tokyo, Japan), 1.0 μl of 8 U/μl Bst DNA polymerase (NipponGene, Tokyo, Japan), 1.0 μl of template DNA, and 4.5 μl of SDW. When the midgut homogenate was used as the template, it was added to the LAMP reaction solution using a toothpick to reduce contamination. The reaction was performed using the Genie®ІІ (OptiGene, West Sussex, NJ, USA) at 63 °C for 40 min and terminated by heating at 98 °C and then cooling to 80 °C at 0.05 °C/s.
Collection of wild individuals and infection diagnosis by PCR and LAMP
Workers of B. terrestris and B. hypocrita hypocrita (eight of each species) were collected at Biei-Cho, Hokkaido, Japan in August 2018. Direct LAMP was conducted using their midgut homogenates, which was added to the reaction reagent using a toothpick, and PCR and LAMP assays were conducted using 1 μl of extracted DNAs (c. 5–10 ng/μl) from them. In this experiment, LAMP was performed using both the Genie®II and a block incubator (BI-535, ASTEC, Fukuoka, Japan). When the block incubator was used, the fluorescence of LAMP products were checked under UV light.
The additional direct LAMP assays using 73 B. hypocrita hypocrita collected in Koshu City, Yamanashi, Japan, were conducted using their midgut homogenates added to the reaction reagent using a toothpick. Furthermore, PCR using 1 μl of extracted DNA and microscopy using 1 μl of midgut homogenates were also conducted. There were no replicates for this additional experiment.
References
Klein, A. M. et al. Importance of pollinators in changing landscapes for world crops. P. R. Soc. B 274, 303–313 (2007).
Potts, S. G. et al. Global pollinator declines: trends, impacts and drivers. Trends Ecol. Evol. 25, 345–353 (2010).
Delaplane, K. S. & Mayer, D. F. Crop Pollination by Bees (2000).
Williams, P. H. The distribution and decline of British bumble bees (Bombus Latr.). J. Apicult. Res. 21, 236–245 (1982).
Biesmeijer, J. C. et al. Parallel declines in pollinators and insect pollinated plants in Britain and the Netherlands. Science 313, 351–354 (2006).
Fitzpatrick, Ú. et al. Rarity and decline in bumblebees-a test of causes and correlates in the Irish fauna. Biol. Conserv. 136, 185–194 (2007).
Colla, S. H. & Packer, L. Evidence for decline in eastern North American bumblebees (Hymenoptera: Apidae), with special focus on Bombus affinis Cresson. Biodivers. Conserv. 17, 1379–1391 (2008).
Goulson, D., Lye, G. C. & Darvill, B. Decline and conservation of bumble bees. Annu. Rev. Entomol. 53, 191–208 (2008).
Cameron, S. A. et al. Patterns of widespread decline in North American bumble bees. PNAS 108, 662–667 (2011).
Kosior, A. The decline of the bumble bees and cuckoo bees (Hymenoptera: Apidae: Bombini) of Western and Central. Europe. Oryx 41, 79–88 (2007).
Cameron, S. A., Lim, H. C., Lozier, J. D., Duennes, M. A. & Thorp, R. Test of the invasive pathogen hypothesis of bumble bee decline in North America. PNAS 113, 4386–4391 (2016).
James, T. Y. et al. Reconstructing the early evolution of fungi using a six-gene phylogeny. Nature 443, 818–822 (2006).
Cali, A. & Takvorian, P. M. Developmental morphology and life cycles of the microsporidia. Microsporidia: Pathogens of Opportunity (eds. Weiss, L. M. and Becnel J. J.), 71–133 (John Wiley & Sons, Inc., 2014).
Fujiwara, T. Infectivity and pathogenicity of Nosema bombycis to larvae of the 313 silkworm. J. Sericult. Sci. Japan 48, 376–380 (1979).
Vilcinskas, A., Stoecker, K., Schmidtberg, H., Röhrich, C. R. & Vogel, H. Invasive harlequin ladybird carries biological weapons against native competitors. Science 340, 862–863 (2013).
Zander, E. Tierische parasite als Krankheitserrenger bei der biene. Leipziger Bienezeitung 24, 164–166 (1909).
Fries, I., Feng, F., Silva, A., Slemenda, S. B. & Pieniazek, N. J. Nosema ceranae n. sp. (Microspora, Nosematidae), morphological and molecular characterization of a microsporidian parasite of the Asian honey bee Apis cerana (Hymenoptera, Apidae). Eur. J. Protistol. 32, 356–365 (1996).
Higes, M., Martína, R. & Meana, A. Nosema ceranae, a new microsporidian parasite in honeybees in Europe. J. Invertebr. Pathol. 92, 93–95 (2006).
Higes, M., García-Palencia, P., Martín-Hernández, R. & Meana, A. Experimental infection of Apis mellifera honeybees with Nosema ceranae (Microsporidia). J. Invertebr. Pathol. 94, 211–217 (2007).
Fantham, H. B. & Porter, A. The morphology, biology and economic importance of Nosema bombi, N. sp., parasitic in various humble bees (Bombus spp.). Ann. Trop. Med. Parasit 8, 623–638 (1914).
Durrer, S. & Schmid-Hempel, P. Shared use of flowers leads to horizontal pathogen transmission. P. R. Soc. B 258, 299–302 (1994).
Fries, I. et al. Molecular characterization of Nosema bombi (Microsporidia: Nosematidae) and a note on its sites of infection in Bombus terrestris (Hymenoptera: Apoidea). J. Apicult. Res. 40, 91–96 (2001).
Otti, O. & Schmid-Hempel, P. Nosema bombi: A pollinator parasite with detrimental fitness effects. J. Invertebr. Pathol. 96, 118–124 (2007).
Rutrecht, S. T. & Brown, M. J. F. Differential virulence in a multiple-host parasite of bumble bees: resolving the paradox of parasite survival? Oikos 118, 941–949 (2009).
Cordes, N. et al. Interspecific geographic distribution and variation of the pathogens Nosema bombi and Crithidia species in United States bumble bee populations. J. Invertebr. Pathol. 109, 209–216 (2012).
Jabal-Uriel, C. et al. First data on the prevalence and distribution of pathogens in bumblebees (Bombus terrestris and Bombus pascuorum) from Spain. Span. J. Agric. Res. 15, e05SC01 (2017).
Takahashi, J., Takeuchi, M., Matsumoto, K. & Nomura, T. Prevalence of Microsporidia isolated from honeybee and bumblebees in Japan. The bulletin of the Research Institute of Advanced Technology Kyoto Sangyo University 12, 59–68 (2013).
Murray, T. E., Coffey, M. F., Kehoe, E. & Horgan, F. G. Pathogen prevalence in commercially reared bumble bees and evidence of spillover in conspecific populations. Biol. conserv. 159, 269–276 (2013).
Collado, J. G. S., Higes, M., Barrio, L. & Martín-Hernández, R. Flow cytometry analysis of Nosema species to assess spore viability and longevity. Parasitol. Res. 113, 1695–1701 (2014).
Notomi, T. et al. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 28, e63 (2000).
Nagamine, K., Hase, T. & Notomi, T. Accelerated reaction by loop-mediated isothermal amplification using loop primers. Mol. Cell. Probe. 16, 223–229 (2002).
Tomita, N., Mori, Y., Kanda, H. & Notomi, T. Loop-mediated isothermal amplification (LAMP) of gene sequences and simple visual detection of products. Nat. Protoc. 3, 877–882 (2008).
Li, Q. et al. Loop-mediated isothermal amplification (LAMP) method for rapid detection of cry1Ab gene in transgenic rice (Oryza sativa L.). Eur. Food Res. Technol. 236, 589–598 (2013).
Watts, M. R. et al. A Loop-Mediated Isothermal Amplification (LAMP) assay for Strongyloides stercoralis in stool that uses a visual detection method with SYTO-82 fluorescent dye. Am. J. Trop. Med. Hyg 90, 306–311 (2014).
Ohtsuka, K., Yanagawa, K., Takatori, K. & Hara-Kudo, Y. Detection of Salmonella enterica in naturally contaminated liquid eggs by Loop-Mediated Isothermal Amplification, and characterization of Salmonella Isolates. Appl. Environ. Microb 71, 6730–6735 (2005).
Poon, L. L. M. et al. Sensitive and inexpensive molecular test for falciparum malaria: Detecting Plasmodium falciparum DNA directly from heat-treated blood by Loop-Mediated Isothermal Amplification. Clin. Chem 52, 303–306 (2006).
Komatsu, K. et al. Detection of Magnaporthe oryzae chrysovirus 1 in Japan and establishment of a rapid, sensitive and direct diagnostic method based on reverse transcription loop-mediated isothermal amplification. Arch. Virol. 161, 317–326 (2016).
Klee, J., Tay, W. T. & Paxton, R. J. Specific and sensitive detection of Nosema bombi (Microsporidia: Nosematidae) in bumble bees (Bombus spp.; Hymenoptera: Apidae) by PCR of partial rRNA gene sequences. J. Invertebr. Pathol. 91, 98–104 (2006).
Arbulo, N. et al. High prevalence and infection levels of Nosema ceranae in bumblebees Bombus atratus and Bombus bellicosus from Uruguay. J. Invertebr. Pathol. 130, 165–168 (2015).
Plischuk, S. et al. South American native bumblebee (Hymenoptera: Apidae) infected by Nosema ceranae (Microsporidia), an emerging pathogen of honeybees (Apis mellifera). Env. Microbiol Rep 1, 131–135 (2009).
Ptaszyńska, A. A., Borsuk, G., Woźniakowski, G., Gnat, S. & Małek, W. Loop-mediated isothermal amplification (LAMP) assays for rapid detection and differentiation of Nosema apis and N. ceranae in honeybees. FEMS Microbiol. Lett. 357, 40–4839 (2014).
Reabel, S. Molecular diagnostic methods for detection of Encephalitozoon cuniculi in pet rabbits; atrium.lib.uoguelph.ca/xmlui/handle/10214/5275 (2012).
Maddox, J. V. & Solter, L. F. Long-term storage of infective microsporidian spores in liquid nitrogen. J. Eukaryot. Microbiol. 43, 221–225 (1996).
Malonea, L. A., Gatehouse, H. S. & Tregidga, E. L. Effects of time, temperature, and honey on Nosema apis (Microsporidia: Nosematidae), a parasite of the honeybee, Apis mellifera (Hymenoptera: Apidae). J. Invertebr. Pathol. 77, 258–268 (2001).
Li, X., Palmer, R., Trout, J. M. & Fayer, R. Infectivity of microsporidia spores stored in water at environmental temperatures. J. Parasitol. 89, 185–188 (2003).
Graystock, P. et al. The Trojan hives: pollinator pathogens, imported and distributed in bumblebee colonyies. J. Appl. Ecol. 50, 1207–1215 (2013).
Blaker, E. A., Strange, J. P., James, R. R., Monroy, F. P. & Cobb, N. S. PCR reveals high prevalence of non/low sporulation Nosema bombi (microsporidia) infections in bumble bees (Bombus) in Northern Arizona. J. Invertebr. Pathol. 123, 25–33 (2014).
Cha, J. O., Talha, A. F. S. M., Lim, C. W. & Kim, B. Effects of glass bead size, vortexing speed and duration on Eimeria acervulina oocyst excystation. Exp. Parasitol. 138, 18–24 (2014).
Thekisoe, O. M. M. Stability of Loop-Mediated Isothermal Amplification (LAMP) reagents and its amplification efficiency on crude Trypanosome DNA templates. J. Vet. Med. Sci. 71, 471–475 (2009).
Niessen, L. Current state and future perspectives of loop-mediated isothermal amplification (LAMP)-based diagnosis of filamentous fungi and yeasts. Appl. Microbiol. Biot 99, 553–574 (2015).
Niessen, L. The application of loop-mediated isothermal amplification (LAMP) assays for the rapid diagnosis of food-borne mycotoxigenic fungi. Curr. Opin. Food Sci 23, 11–22 (2018).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol 30, 2725–2729 (2013).
Martín-Hernández et al. Outcome of colonization of Apis mellifera by Nosema ceranae. Appl. Environ. Microb 73, 6331–6338 (2007).
Acknowledgements
We thank Hosaka Y. and Tanaka S. for their invaluable help in the field. This study was supported by a research grant provided by The Oshita Foundation.
Author information
Authors and Affiliations
Contributions
Y.K. and T.Y. designed experiments with the help of M.N., K.K. and M.N.I. Y.K. performed experiments, analyzed data. Y.K. and M.N.I. wrote the paper. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kato, Y., Yanagisawa, T., Nakai, M. et al. Direct and sensitive detection of a microsporidian parasite of bumblebees using loop-mediated isothermal amplification (LAMP). Sci Rep 10, 1118 (2020). https://doi.org/10.1038/s41598-020-57909-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-57909-8
- Springer Nature Limited