
Abstract
Loop-mediated isothermal amplification is a rather novel method of enzymatic deoxyribonucleic acid amplification which can be applied for the diagnosis of viruses, bacteria, and fungi. Although firmly established in viral and bacterial diagnosis, the technology has only recently been applied to a noteworthy number of species in the filamentous fungi and yeasts. The current review gives an overview of the literature so far published on the topic by discussing the different groups of fungal organisms to which the method has been applied. Moreover, the method is described in detail as well as the different possibilities available for signal detection and quantification and sample preparation. Future perspective of loop-mediated isothermal amplification-based assays is discussed in the light of applicability for fungal diagnostics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Information about the microbiological status of persons, materials, and the environment is a crucial factor in modern concepts such as human and animal healthcare, quarantine, food safety, environmental monitoring, or criminal forensics. All these areas have developed their arsenal of specialized methods for detection, enumeration, and identification of microorganisms, among which fungi may play a minor role when compared to the importance of bacteria. Nonetheless, since fungi do have an impact on human and animal health and nutrition as pathogens, toxicants, and spoilers, it would be a culpable mistake to ignore this group of organisms. The invention of deoxyribonucleic acid (DNA)-based technologies led to a general switch in diagnostic targets from multiplication of living cells to specific multiplication of nucleic acid. Hence, specificity of a diagnostic assay was no more depending on several different environmental factors but was reduced to only one factor, the nucleotide sequence of a DNA or ribonucleic acid (RNA) target. The first system for in vitro amplification of DNA was the polymerase chain reaction (PCR) in which heat denaturation of DNA double strands was used to permit exponential multiplication of the target sequence in discontinuous mode. Shortly after the invention of PCR, a multitude of alternative amplification techniques were published which used non-thermostable DNA or RNA polymerases under isothermal conditions for continuous amplification of a target nucleic acid (see reviews by Gill and Ghaemi 2008 and Yan et al. 2014). It can be assumed that much of the effort given to develop those systems was due to the fact that the PCR technology was rigidly covered by patents hampering general access to the fast growing market of nucleic acid-based diagnostics. Although meanwhile established as the Gold Standard in many areas of diagnosis and research, PCR turns out to be more suitable in situations in which accuracy is of importance rather than time to result. In many diagnostic situations, however, time to result is the most crucial factor since an information-based short-term decision has to be made, often with far-reaching consequences in terms of health, public safety, or economic considerations. Under such circumstances, some of the isothermal in vitro DNA amplification techniques may have an advantage over PCR since they can be operated at one continuous temperature obliviating thermal cycling equipment. Sample preparation, which is an important issue in PCR, can be kept to a minimum with some of the available isothermal methods and also signal detection is possible using simple equipment or the naked eye. Loop-mediated isothermal amplification (LAMP) of target DNA is an amplification technology with high diagnostic potential which is currently well on its way to replace PCR as a diagnostic tool in many fields such as clinical point of care diagnostics, phytoquarantine, and food safety. Although fungal organisms are rarely as acutely live threatening as some bacteria or viruses are, this group of microorganisms has a big impact on the health of humans, animals, and plants as well as on the safety of our food supply due to their production of mycotoxins. The following sections will, therefore, elucidate how LAMP can be used to improve fungal diagnosis in different fields of application.
The classical way of detecting and enumerating filamentous fungi and yeasts involves several steps of sample preparation, dilution, plating, incubation, enumeration, isolation, and identification. Various different media can be applied depending on the source to be analyzed and the group of fungi to which the analysis is aiming. Detailed descriptions of methods and media for the culture-based analysis of sources such like food (Pitt and Hocking 2009), soil (Alef and Nannipieri 1995), water (Mesaphy 2013), and air (O’Loughlin et al. 2013) have been published in the literature. Various different culture media are used to grow fungal isolates under different environmental conditions in order to identify them to species level based on culture characteristics, micro morphological features, and biochemical properties. Specificity of growth media can be improved by inhibition or even exclusion of unwanted groups of organisms or by selective enhancement of the organisms to be detected. Tsao (1970) published a comprehensive review about the general principles of developing selective fungal growth media. Development of automated detection and identification systems is strongly driven by clinical microbiologists due to their demand for rapid analysis and high throughput systems. To meet this demand, media were developed to selectively grow and identify fungal and yeast colonies of interest in one step by addition of chromogenic substrates (Odds and Bernaerts 1994; Baumgartner et al. 1996; Sendid et al. 2007; Ghelardi et al. 2008). Although more rapid culture-based identification systems exist for some groups of fungi, the routinely use of such methods for fungal diagnosis in clinical and industrial settings is generally hampered by their high requirement for time and specialization. In addition to culture based systems, immunochemical assays have been developed for identification of some filamentous fungi and yeasts as well as for diagnostic detection of specific antigens present in the blood stream of patients as an indicator for invasive fungal infections (Buckley et al. 1992). However, direct detection of filamentous fungi in formats such as enzyme-linked immunosorbent assay (ELISA) or lateral flow device (LFD) is limited by the fact that the mycelial surface does not carry antigenic structures which would enable sufficiently specific antigenic determination as present in other groups of organisms such as the Gram negative bacteria, in which the composition of pepticoglucan layers of the cell walls follows the taxonomical grouping of species (Schleifer and Kandler 1972). As for other groups of microorganisms, DNA sequence-based techniques have gained increasing importance in the diagnosis of fungal plant pathogens (McCartney et al. 2003; Atkins and Clark 2004), human pathogens (Yeo and Wong 2004; White et al. 2009), or fungi relevant to food safety and hygiene (Niessen 2008). Until recently, assays were mainly based on the concept of PCR. During recent years, amplification of DNA using isothermal conditions gained increasing attention due to the fact that they can be run with less effort and expense as compared to PCR. Loop-mediated isothermal amplification is a technique which has become especially important as an alternative technology for the detection of all groups of microorganisms, including viruses.
The LAMP reaction
LAMP is a novel approach to nucleic acid amplification. It relies on auto cycling strand displacement DNA synthesis performed by Bst DNA polymerase or similar thermophilic DNA polymerases under isothermal conditions with a set of four specifically designed primers hybridizing to six different parts of the target DNA sequence (Notomi et al. 2000). Figure 1 gives a schematic representation of the basic reaction steps involved. The method makes use of the large fragment of the Bst DNA polymerase from Geobacillus stearothermophilus. The large fragment of the enzyme contains the 5′–> 3′ polymerase activity but lacks 5′–> 3′ exonuclease activity. The enzyme is produced by an Escherichia coli strain containing a genetic fusion of the Bst DNA polymerase large fragment and the maltose binding protein (MBP) of E. coli. MBP is used for affinity purification and removed by cleavage of the fused proteins (Kong et al. New England Biolabs, unpublished data). Bst DNA polymerase large fragment displaces third strand DNA with high efficiency during primer-initiated polymerization of new DNA leaving a double stranded product and a single stranded DNA strand which can act as the matrix for further primer annealing and DNA polymerization. Since Bst DNA polymerase has a very high activity, vast amounts of high molecular weight DNA are produced within short time. The exceptionally high specificity of LAMP is due to the fact that a set of four primers with six binding sites must hybridize correctly to their target sequence before DNA biosynthesis occurs (see Fig. 1a). A third pair of primers (loop primers) can be added optionally to the reaction in order to further amplify the amount of DNA produced during LAMP (Nagamine et al. 2002). One of the primer pairs is constructed in such a way that the reverse complement of a binding site downstream of the F2c/B2c binding site (F1c/B1c) is attached to the 5′-end of a primer binding to that site. These composite primers are essential for the specificity of the amplification reaction and have, thus, to be chosen very carefully. Both parts of each FIP/BIP primer should be checked for cross reactivity by in silico analysis prior to application in LAMP reactions. A pair of outer primers (F3/B3) anneals upstream of the F2c/B2c binding site to displace the initial LAMP product strand from the DNA matrix. Specificity of outer primers can be regarded as being of lower importance since they are not involved in any of the following amplification reactions and a low number of base mismatches will not prevent amplification. The process is initiated by attachment of primers to the DNA target (see Fig. 1b). Primers are elongated and the second matrix strand is displaced from the target DNA. The newly synthesized product itself is displaced from the matrix strand by the F3/B3 product strand. Primers F3 and B3 have no further function once the amplification process has been initiated. As the final product of the amplification initiation step, a dumbbell structured single-stranded DNA is formed by hybridization of both ends of the molecule to complementary downstream sequences forming two loops. Starting from this structure, primers FIP and BIP continuously hybridize to newly formed binding sites and are elongated, displaced, and refolded while forming ever longer multimers of the basic dumbbell structure (see Fig. 1c). Loop primers are designed to hybridize to the single-stranded loop structures present in the dumbbell structures as well as in the multimeric DNA formed during autocycling DNA amplification. They prime the production of novel template DNA to which FIP/BIP primers can bind to initiate synthesis of even higher concentrations of DNA. Addition of loop primers, therefore, does not increase the speed of amplification but rather enables earlier detection of a LAMP signal as compared to a reaction run without loop primers.

Schematic representation of the LAMP reaction. a Primers, binding sites, and conditions. b Initiation of the LAMP reaction resulting in production of a double-loop stem structure (dumbbell structure). c Autocycling enzymatic DNA amplification during LAMP resulting in multimers of different size of the monomeric double-loop stem structure
LAMP-based assays for the detection of fungal organisms
The number of publications involving loop-mediated isothermal amplification as an alternative method in molecular diagnosis of microorganisms is steadily increasing (Niessen et al. 2013). However, publications dealing with LAMP-based detection and identification of fungal organisms (55 as of 2 September 2014) represent only a relatively small proportion in relation to the total number of publications involving LAMP (1,252 as of 2 September 2014, Science Citation Index). Table S1 (supplementary materials) gives an overview of published assays with species names, sequence source used to design primer sets, and several other details characterizing individual assays, including sample preparation. No reaction details such as incubation temperature were included in Table 1 since they are subject to variation and depend widely on the hardware used in individual labs. It will therefore be necessary to optimize incubation temperatures if assays are to be reproduced. Column 1 in Table 1 provides all species detected by a given primer set, including species which were found to cross react with specific assays. Table S1 (supplemental materials) provides sequences of all primers used in individual LAMP assays. All FIP/BIP primers were checked for correctness of sequences by comparing sequences with the corresponding gene as given in GenBank. The two parts of each primer are indicated by hyphenation in order to make in silico analysis of primer specificity easier for the reader.
LAMP assays for the detection of clinically relevant yeasts and filamentous fungi
Target organisms of LAMP assays can be grouped into three different categories according to their relevance, e.g., yeasts and fungi with clinical relevance in humans and animals, plant pathogenic fungi, and fungi relevant to the quality and safety of food and feed. As with other fields of applications of LAMP-based assays, also in the field of fungi the development started with the development of assays targeting clinically relevant filamentous fungi and yeasts. The first assay published on LAMP-based detection of a fungal organism was by Endo et al. (2004, see also Tatibana et al. 2009) who set up an assay for Paracoccidioides brasiliensis based on the sequence of the gp43 gene (GenBank accession no. HQ878437) which codes for a 43-kDa secreted glycoprotein involved in the attachment of the yeast form of the fungus to macrophages (Mendes-Giannini et al. 2000). A review of the pathology and ecology of the pathogen can be found in Restrepo (1985). A LAMP-based assay for diagnosis of Pneumocystis jirovecii was published by Uemura et al. (2008). The yeast-like fungus is an opportunistic pathogen in immunocompromised patients showing symptoms of Pneumocystis carinii pneumonia. LAMP analysis of sputum and bronchoalveolar lavage fluid samples showed that the sensitivity of the LAMP assay was considerably higher as compared to a nested-PCR assay for the fungus. Lucas et al. (2010) used primer sets based on the cap59 gene which harbors serotype associated alleles on which they based specific differentiation of several Cryptococcus species (Enache-Angoulvant et al. 2007). Recently, Ishikawa et al. (2014) described a LAMP assay for detection of Filobasidiella neoformans, the theleomorph of C. neoformans, which they applied to the detection of the mycelial form of the pathogen in a probiotic dairy product. Candida species, i.e., Candida albicans, Candida glabrata, Candida parapsilosis group, and Candida tropicalis are dimorphic yeasts known as causative agents of superficial or systemic candidal infections in humans (Fidel et al. 1999). Jensen Søe et al. (2013) developed isoPCR as a two-stage nested-like amplification technique that showed exceptionally high sensitivity and specificity combined with low operating time as compared to PCR-based assays of Candida yeasts. Talaromyces marneffei (syn. Penicillium marneffei) is the only (hypothermal) dimorphic species known among fungi with a Penicillium anamorphic state. The fungus causes systemic infections mostly in immunocompromised patients and has been found to be widely distributed as an AIDS related illness. Sun et al. (2010a) set up a LAMP assay which was based on the DNA sequence of the ITS region of the rRNA gene of T. marneffei and applied it to the analysis of skin, lung, and lymph node tissues of T. marneffei culture positive patients.
Beside yeast targeted assays, LAMP was also used for the diagnosis of filamentous fungi of clinical relevance. The dematiaceous mycelial fungus Ochroconis gallopava is an opportunistic zoonotic pathogen of concern mainly in immunocompromised or immunosuppressed patients with a mortality rate of 20 % after 6 month (Qureshi et al. 2012); hence, early diagnosis is of eminent importance. Ohori et al. (2006) developed a set of LAMP primers for the fungus which was based on the sequence of the D1/D2 domain within the 28S ribosomal RNA gene. The assay was applied successfully to the detection of O. gallopava in brain and spleen tissues of infected mice. Primers also picked up Ochroconis calidifuminalis, a sibling species of O. gallopava. Aspergillosis is a collective name for invasive human lung diseases caused by Aspergillus species (Patterson et al. 2000). Beside Aspergillus fumigates, the closely related Aspergillus nidulans and Aspergillus quadrilineata are mainly responsible for high mortality rates of the disease. Matsuzawa et al. (2010) developed sets of specific LAMP primers for either of the latter two species to enable correct diagnosis which is important for the proper treatment of patients since A. nidulans and A. quadrilineata display different susceptibility against antifungal drugs. Chromoblastomycosis is a collective designation for chronic, localized and slowly progressing subcutaneous fungal infections characterized by the development of brown-pigmented (demaciaceous) rounded sclerotic bodies in affected tissues (López Martínez and Mendéz Tovar 2007). Fonsecaea pedrosoi, Fonsecaea monomorpha, and Fonsecaea nubica (Najafzadeh et al. 2010) were found to be the most important ethiological agents worldwide. Therapy of the disease is strongly supported when taxonomical identification of the ethiological agent is available before treatment. Aimed at rapid identification of Fonsecaea agents of chromoblastomycosis, Sun et al. (2010b) set up a highly sensitive LAMP-based assay which gave a positive signal with all three species. Several species in the anamorphic genus Scedosporium and their teleomorphs in the genus Pseudoallesheria are known as causative agents of traumatically inoculated subcutaneous mycoses (Cortez et al. 2008; Pihet et al. 2009). Due to the fact that the relevant species differ widely in virulence and susceptibility to antifungal drugs, accurate identification and detection of the most relevant species are necessary. Lu et al. (2011) developed separate species specific LAMP assays for each of the six medically relevant pathogens within the Pseudoallesheria/Petriella complex of species.
LAMP-based assays for detection of plant pathogenic fungi
Botrytis cinerea (tel. Botryotinia fuckelina) is a necrotrophic fungus that affects the fruits of many plant species (Williamson et al. 2007), although wine grapes are supposed to be the most notable host leading to high economical losses (Elmer and Michailides 2007). Tomlinson et al. (2010a) developed a LAMP-based assay using primers designed from the nucleotide sequence of the ITS region of the rRNA gene of B. cinerea and applied it to the detection of the fungus in infected plants with high sensitivity. Several species within the genus Fusarium are well known plant pathogens. Fusarium oxysporum is one of the most abundant and widespread fungal species of the global soil mycobiota (Gordon and Martyn 1997). Genetically, the species represents a highly heterogeneous polytypic morphospecies (Waalwijk et al. 1996) containing formae speciales adapted to about 100 different host plants, among them ecomically important crops such as melon, tomato, cotton, and banana (Michielse and Rep 2009). LAMP assays have been set up for F. oxysporum f. sp. cubense as a wilt pathogen of banana plants (Panama disease). Using different target genes, Li et al. (2013a) and Zhang et al. (2013) independently published LAMP assays for the detection of tropical race 4 of the pathogen as the economically most important race and detected it in banana plantation soils with high sensitivity. LAMP-based assays for two other economically important formae speciales of F. oxysporum were published by Almasi et al. (2013) for f. sp. lycopersici (tomatoes) and by Peng et al. (2013) for f. sp. niveum (water melon). Fusarium mangiferae represents one of three lineages in the Gibberella fujikuroi complex of Fusarium species causing mango malformation (Lima et al. 2009). Pu et al. (2014) recently developed a LAMP-based assay for the sensitive detection of the pathogen in mango shoots. Guignardia citricarpa is pathogenic to a variety of Citrus species causing Citrus Black Spot on citreous fruits (EPPO 2009) and some other host. In order to replace time consuming microbiological analysis of the quarantine fungus, Tomlinson et al. (2013) set up a LAMP assay which they applied to the detection of the pathogen from citrus fruit lesions. Several closely related Glomus species are of pervasive importance to terrestrial ecosystems producing arbuscular mycorrhizas in plant roots (Rillig 2004). In order to detect four of the more important Glomus spp. together with Gigaspora rosea in one reaction, Gadkar and Rillig (2008) developed a group specific LAMP assay to detect presence of the fungi in carrot roots. Several species within the oomycetous genus Phytophthora are well known plant pathogens (Kroon et al. 2012). LAMP-based detection assays were developed for detection of quarantine species such as Phytophthora kernoviae and Phytophthora ramorum (Tomlinson et al. 2007, 2010b) in tissues of Rhododendron trees, Phytophthora melonis in cucumbers and potting soils (Chen et al. 2013) or Phytophthora sojae in tissues of soybean plants (Dai et al. 2012). Puccinia striiformis f. sp. tritici (PCT) is the causal agent of wheat stripe rust. The obligate biotrophic parasite is difficult to cultivate on artificial media. In order to facilitate management of the pest through early and sensitive detection in early growth stages, Huang et al. (2011) developed a LAMP-assay using primers specific for the ß-tubulin gene of the fungus and applied it to the detection of PCT in wheat leaves. Many species in the oomycetous genus, Pythium cause symptoms similar to those caused by Phytophthora spp., i.e., root rot and wilting. Fukuta et al. (2013, 2014) used LAMP-based assays for the detection of Phytophthora aphanidermatum and Phytophthora myriotylium in hydroponic fluids as well as in infected roots of tomato plants, respectively. Recently, Takahashi et al. (2014) published a set of LAMP primers which they applied in the detection of Phytophthora helicoides in roots of Poinsettia, an economical important ornamental plant. The ascomycete Sclerotinia sclerotiorum infects rapeseed and cotton plants. The LAMP-based assay published by Duan et al. (2014) was designed to analyse presence of the fungus in rapeseed plants and seeds with a detection limit in sub-femtogram range. Verticillium wilt is a disease known from more than 300 cultivated plant species. Verticillium dahliae and Verticillium albo-atrum are the most important pathogenic species within the genus. In order to test soil samples for the presence of V. dahliae, Moradi et al. (2014) recently set up a LAMP assay which detected both pathotypes of the fungus (D, ND) at the level of femtograms of DNA per reaction.
LAMP-based assays for detection of food relevant fungi and yeasts
The role of fungi and yeasts in the production of food and feed may be either positive, e.g., as starter cultures for food production or as biotechnological producers of food ingredients, or negative, e.g., as organisms inducing food poisoning or spoilage. The motivation behind all developments of LAMP-based assays published so far for the detection of filamentous fungi in food sources was the ability of the species to produce mycotoxins. The only exception so far is the assay developed by Denschlag et al. (2012, 2013) who detected the hyd5 gene in a group of closely related Fusarium species. The gene codes for a protein which is suspected to induce gushing (over foaming) of bottled beer (Sarlin et al. 2005). A variety of Aspergillus species are able to produce potent carcinogenic mycotoxins of the aflatoxin type (Bennett and Klich 2003). Luo et al. (2012) set up primer sets based on housekeeping genes to detect major aflatoxinogenic species (Aspergillus flavus, Aspergillus nomius, and Aspergillus parasiticus) on food commodities such as corn, groundnuts, Brazil nuts and green coffee beans. Specificity testing revealed cross reactions with closely related species especially with the A. flavus specific primer set. Comparison of LAMP results with aflatoxin contents showed that LAMP can be used to predict the presence or absence of aftlatoxins in Brazil nuts (Luo et al. 2014a). Storari et al. (2013) applied the nucleotide sequences of the pks genes within the ochtratoxin A biosynthetic pathways of Aspergillus carbonarius and Aspergillus niger to design species specific primer sets for both species and to analyze pure cultures obtained from vineyards for the presence of the gene. In order to detect and quantify producers of trichothecene mycotoxins among Fusarium spp. in cereal grain and malt, Denschlag et al. (2014) designed two sets of LAMP primers each of which was specific to a subset of species representing two chemotypes producing either type A or B trichothecenes. Multiplexing of both primer sets in one reaction enabled detection of the six most abundant species among trichothecene producers. Real-time LAMP results and concentrations of the mycotoxin deoxynivlenol (DON) found in corresponding samples were well correlated. Saccharomyces cerevisiae sensu stricto includes yeast strains used in a variety of artisan and industrial food and beverage fermentation processes. Aimed at discriminating the four economically important sibling species of S. cerevisiae, i.e., S. cerevisiae, Saccharomyces bayanus, Saccharomyces pastorianus, and Saccharomyces paradoxus (Guillamon et al. 1994) as well as Saccharomyces diastaticus, Hayashi et al. (2009) set up species specific primer sets for each of them an applied the LAMP assays for the discrimination of pure cultures of the yeasts and for the detection of S. bayanus as contaminant in wine and beer. Aimed at differentiating the most abundant species within Brettanomyces/Dekkera, Hayashi et al. (2007) designed primers based on sequence differences within the ITS region of the rRNA genes and set up LAMP-based assays to detect Dekkera bruxellensis in samples of beer and wine spiked with low numbers of yeast cells.
Selection of target sequences and LAMP primer design
The majority of LAMP-based assays for fungi and yeasts have been targeted to standard sequences which are readily available from public data bases. Housekeeping genes, such as acl1 (Niessen et al. 2012; Luo et al. 2014a), amy1 (Luo et al. 2012), tef1 (Ferdousi et al. 2014), and btub (Gadkar and Rillig 2008; Lu et al. 2011; Villari et al. 2013), or other genes of ubiquitous function in fungi, such as rodA (Matsuzawa et al. 2010) or ypt1 (Chen et al. 2013), are universal sources of sequence information available for a greater number of species and strains. Sequence information from genes coding for ribosomal RNA (rRBA) are even more universally available and have been used in nearly half of the total number of published LAMP assays. Different parts of the gene operon have been applied as sequence source. The ITS region as the universal fungal barcoding marker (Schoch et al. 2012) has been used particularly often (for example, Hayashi et al. 2007; Tomlinson et al. 2007; Sun et al. 2010b; Ishikawa et al. 2014; Kasahara et al. 2014). Other parts of the operon have been used less frequently such as the part coding for the 28S (26S) ribosomal subunit (Ohori et al. 2006; Inacio et al. 2008; Almasi et al. 2013; Jensen Søe et al. 2013) or the intergenic spacer region (Li and Cai 2011; Zhang et al. 2013; Kasahara et al. 2014). Beside their universal availability, rRNA sequences can be used to the advantage of higher assay sensitivity since the ribosomal genes are organized in multicopy clusters in eukaryotes ranging from a few to several hundred copies per genome (Prokopowich et al. 2003). Some of the LAMP assays developed so far have been targeted to certain properties of the detected fungi. In such cases, the selection of a sequence source is rather guided by considerations such like involvement of the target sequence in a particular biosynthetic pathway leading to the production of certain toxins (Storari et al. 2013; Denschlag et al. 2014) or other unwanted compounds (Denschlag et al. 2012). Lucas et al. (2010) selected the cap59 gene as LAMP target since it is involved in the extracellular trafficking of capsular glucuronoxylomannan in Cryptococcus spp. where its sequence variations correlate with the different serotypes of that medically important yeast. A similar approach was taken by Endo et al. (2004) who designed their LAMP primers from the sequence of gp43, a gene coding for a 43-kDa protein which is the main exocellular antigen recognized by sera from patients with paracoccidioidomycosis (Travassos et al. 1995). Niessen and Vogel (2010) used the gene coding for galactose oxidase (gaoA) as target for the detection of Fusarium graminearum since the enzyme is a typical feature characterizing this fungal species (Ögel et al. 1994). A few authors used genomic marker sequences of unknown function and localization as sequence source for primer design. Pu et al. (2014) and Moradi et al. (2014) used sequence characterized amplified regions (SCAR) which resulted from random amplification of genomic DNA of target organisms with random sequence PCR primers as the basis for primer design in LAMP assays detecting Fusarium mangiferae and Verticillium dahliae, respectively. A similar approach was taken by Li et al. (2013a) for LAMP-based detection of F. oxysporum f. sp. cubense TR4 in banana tissues and by Peng et al. (2013) for F. oxysporum f sp. niveum in cucumber plantation soil. Dai et al. (2012) applied sequences of the A3aPro deletion element of known localization but unknown function present in the genome of certain P. sojae races to detect the pathogen in soybean tissues in a LAMP assay and Duan et al. (2014) applied the sequence of a hypothetical protein of unknown function which was found by incident during annotation of the S. sclerotiorum genome.
Signal detection in fungal LAMP assays
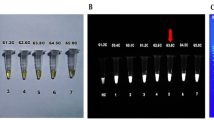
Detection of amplified product in LAMP has originally been accomplished by agarose gel electrophoresis which typically reveals a ladder like pattern of DNA fragments as shown in Fig. 2b. The smallest (monomer) fragment spans from the 5′ end of the Fc1 part of the forward inner primer (FIP) to the 5′end of the B1c part of the backward inner primer (BIP). Multimers and polymers of that monomeric structure are produced with sizes of a few hundred bp up to the formation of a smear of high molecular weight DNA of several kilobases in size. However, due to massive production of DNA during LAMP, the risk of cross contamination of samples by aerosolized product is very high and may compromise analytical results. This may be the reason why only few authors used this method to analyze the results of LAMP reactions (Ohori et al. 2006; Yarita et al. 2007; Lucas et al. 2010). A different approach to ex-tube detection of LAMP product was applied by Tomlinson et al. (2010b) and also by Vaagt et al. (2013). Both authors used LFD to detect LAMP product which was specifically double-labeled by using a pair of loop primers with either biotin or DIG/FITC in a commercially available standard format. However, also the use of LFD for detection necessitates opening of LAMP reaction vessels.

Different techniques frequently used for signal detection in LAMP. Detection of signals in LAMP reactions performed with a tenfold serial dilution of target fungal DNA. a Calcein fluorescence for end point determination of positive LAMP reactions, inspection under UV light. b Reactions shown in a after electrophoretic separation on agarose gel and staining with ethidium bromide. c Real-time monitoring of V13 fluorescence in the same serial DNA dilution, arbitrary fluorescence units plotted against time. Time to threshold (Tt) can be defined according to absolute signal values or derivative signal value, e.g., signal slope
In order to prevent cross contamination by aerosolized LAMP product, various different methods for in-tube detection of DNA amplification have been developed and were used also in LAMP assays targeted at fungal DNA. In-tube detection of amplified product does not involve opening of LAMP reaction vessels. Direct staining of double stranded DNA using fluorogenic intercalating dyes is one possible way to achieve this goal. Substances such as SYBR green 1 (Sun et al. 2010b; Huang et al. 2011; Lu et al. 2011; Jensen Søe et al. 2013; Vaagt et al. 2013), SYTO9 (Peng et al. 2013; Zhang et al. 2013; Pu et al. 2014), YOPRO1 (Uemura et al. 2008), PicoGreen (Tomlinson et al. 2007; Tomlinson et al. 2010b), or ethidium bromide (Almasi et al. 2013; Moradi et al. 2014) have been applied for the detection of LAMP product in fungal and yeast diagnostic assays with the first three fluorochromes applied mostly in real-time LAMP assays (see Fig. 2c). Since most of these substances substantially reduce productivity of the Bst DNA polymerase during LAMP reactions, they must be added after the reaction has run taking into account the risk of cross contamination due to opening of reaction vessels as previously described. In order to prevent inhibition of SYBR Green 1 in LAMP assays, Pu et al. (2014) placed the dye as a droplet in the lid of reaction vessels and mixed it by centrifugation after the reaction was finished. Intercalating fluorescent dyes which can be added before the reaction starts are the V13 01184 dye from Dyomics (Denschlag et al. 2012), the undisclosed fluorescent dyes present in ready-to-use master mixes provided with LAMP kits from OptiGene for applications running on real-time LAMP scanners of the Genie® series and in the Isothermal Master Mix from Nippon Gene (Tomlinson et al. 2010a; Tomlinson et al. 2013; Villari et al. 2013) or in LoopAmp kits from Eiken Chemicals (Tatibana et al. 2009). Also Gene Finder has been described as an intercalating fluorescent dye which has been used by Moradi et al. (2014) in an assay detecting Verticillium dahliae in soil samples. It is interesting to note, however, that the authors applied the fluorophore for visual detection of positive LAMP reactions since the compound results in a color change from orange to dark green when binding to double stranded DNA. Precipitation of the DNA produced during LAMP with a fluorescently labeled cationic polymer or the use of a specific fluorescently labeled probe have been shown to be an alternative for direct in-tube detection of the LAMP product (Mori et al. 2006). However, neither of the two methods has yet been used in LAMP assays involving fungal DNA.
Indirect in-tube detection of LAMP product is an alternative to the use of intercalating fluorescent dyes and labeled polymers or probes. It is based on the fact that pyrophosphate is a typical by-product of enzymatic DNA synthesis. The molecule forms an insoluble complex with bivalent cations such as Ca2+, Mg2+, or Mn2+. The turbidity which occurs upon precipitation of the complex was found to be proportional to the concentration of template DNA initially present in a LAMP reaction (Mori et al. 2001). As a consequence, monitoring of turbidity over time enables real-time monitoring of LAMP reactions (see Fig. 2c) and has been applied to the quantification of LAMP results (Mori et al. 2004). Turbidity assays have been used in several LAMP applications for the detection of fungi, either as end point analysis (Gadkar and Rillig 2008; Almasi et al. 2013; Fukuta et al. 2013; Moradi et al. 2014) or by real-time analysis using a dedicated turbidimeter (Hayashi et al. 2007; Denschlag et al. 2014; Ishikawa et al. 2014; Kasahara et al. 2014; Takahashi et al. 2014). In addition to haze formation, the ability of pyrophosphate to form a complex with bivalent cations can be applied during LAMP reactions to revert quenching of a complexometric fluorophore such as calcein (Diehl and Ellingboe 1956). Its fluorescence is initially quenched by the addition of manganese and subsequently restored by complexation of the metal ion to the pyrophosphate produced during DNA synthesis (Tomita et al. 2008). Fluorescence intensity was found to be proportional to the initial concentration of target DNA in LAMP reactions. Several assays involving calcein fluorescence to detect fungal DNA have been published (Niessen and Vogel 2010; Luo et al. 2012; Niessen et al. 2012; Luo et al. 2014a). Calcein fluorescence can also be used for real-time monitoring of LAMP reactions. However, the technology has not yet been applied for the analysis of fungal DNA. Apart from calcein, other complexometric dyes have been used which change color in the visible part of the spectrum. Goto et al. (2009) demonstrated the usefulness of hydroxynaphtol blue for indirect detection of DNA biosynthesis during LAMP with no need for UV light. The dye was applied for endpoint LAMP analysis of fungal DNA only in recent publications (Almasi et al. 2013; Dai et al. 2012; Storari et al. 2013; Duan et al. 2014; Moradi et al. 2014).
Quantification of fungal DNA using real-time LAMP
In contrast to PCR where DNA amplification is a function of the number of melting–amplification cycles (C t value), biosynthesis of DNA is a continuous process in LAMP reactions and is therefore a function of incubation time rather than cycle number. Because of its discrete mode of dependence, the concentration of target molecules used in a PCR can be calculated rather precisely from the concentration of amplification product observed after a known number of amplification cycles. The equation used to calculate copy numbers or DNA concentrations in qPCR is biased only by factors which influence amplification efficiency. Because of the dependence of DNA amplification on reaction time in LAMP, a theoretical calculation of target concentrations from a given concentration of amplification product as in qPCR is impossible. Quantitative estimation of target concentrations in a given LAMP reaction therefore has to be done by comparison of the time needed for the signal to reach a certain threshold (T t value) or, in analogy, to reach a positive signal (T p value). Few attempts were made so far to find quantitative correlations between LAMP signals and either cell counts (Hayashi et al. 2007), concentrations of purified template DNA (Denschlag et al. 2013; Peng et al. 2013; Vaagt et al. 2013), or concentrations of a plasmid standard containing the target DNA sequence (Zhang et al. 2013; Pu et al. 2014). Authors reported coefficients of correlation of <0.98 but standard deviations between repetitions were high in most cases. A reason for that fact could be that instruments used for real-time LAMP work with quite long data acquisition intervals (6 to 20 s) which might be generally inadequate in the case of a continuous process running at the speed of a usual LAMP reaction, where 2–5 min suffice from zero to maximum signal intensity.
Sample preparation
DNA polymerases used in any kind of amplification-based assay can interact with a variety of components introduced into the reaction together with sample of DNA to be analyzed. Inhibitors include constituents of body fluids such as hemoglobin, urea or heparin, food constituents such as phenolic compounds, glycogen, polysaccharides, enzymes, or fat as well as environmental materials such as humic acids, calcium, and heavy metals. Inhibitive interactions have been specifically well analyzed for taq DNA polymerase and other heat resistant DNA polymerases used in PCR (see reviews by Wilson 1997; Alaeddini 2012; Schrader et al. 2012). However, the mechanisms involved have been elucidated only for a limited number of compounds. In order to circumvent limitations imposed by inhibitive interactions, samples have to be highly processed in order to exclude inhibitors from the sample DNA used in PCR analysis (Schrader et al. 2012). Moreover, proper standards must be included in order to control the influence of a DNA sample on the outcome of the analysis since inhibitive potential of different compounds may widely vary between different PCR assays (Huggett et al. 2008). One of the features characterizing the LAMP reaction is its comparably low susceptibility to typical inhibitory compounds occurring in samples such as full bood, stool, soil or food materials (Boehme et al. 2006; Mori and Notomi 2008). Its robustness against variation of reaction conditions such as pH and temperature have been established (Francois et al. 2011).
Table 1 provides information about the DNA extraction procedures applied for LAMP analysis. With a few exceptions, authors apply highly purified genomic DNA (gDNA) for testing specificity and sensitivity of their LAMP reactions. For isolation and purification of gDNA from pure culture mycelia or yeast pure cultures, most authors basically use the method described by Raeder and Broda (1985) with several modifications. Genomic fungal DNA is precipitated by isopropanol or ethanol addition to a cell lysate commonly produced by mechanical maceration of mycelia or yeast cells to provide better accessibility of DNA. Maceration is mostly done by grinding of fresh, frozen or freeze dried mycelia or by application of sand, glass beads or steel ball bearings in a bead beating device. Alternatively, lysis can be accomplished by several cycles of freeze–thawing and/or boiling of cells (Lu et al. 2011). Extraction buffers are most commonly based on Tris–HCl as buffering agent with addition of NaCl to regulate ion strength and ethylenediaminetetraacetic acid as heavy metal chelator. Sodium dodecyl sulfate or benzyl chloride is added in order to facilitate cell wall and membrane disintegration (Fukuta et al. 2013). CTAB can be added to precipite sugars and polysaccharides from the extracts (Vaagt et al. 2013). Addition of polyvinylpolypyrrolidone (PVPP) is often necessary in cases where extracts contain polyphenols (Tomlinson et al. 2010a). Also, the combination of PVPP with Sephadex has been described for that purpose (Zhang et al. 2013). A lysis step at 55–65 °C is often integrated in order to intensify extraction procedures and improve DNA yield. To provide higher purity of precipitated DNA, organic compounds can be extracted from lysates with chloroform or mixtures of phenol and chloroform for additional protein precipitation (Sun et al. 2010b). Final DNA precipitation is by addition of half a volume of isopropanol or one volume of ethanol to the extract supernatant after maintaining high salt concentrations. Precipitated DNA is directly used in LAMP reactions or undergoes a further purification step using silica based spin column systems available from several commercial providers.
Preparation of highly purified DNA from pure cultures or from sample materials is a cumbersome business which takes a few hours if not days to be completed. Many authors have benefited from the fact that LAMP can be run with DNA of low purity with the effect that time needed for sample preparation can dramatically be reduced. To reach this goal, many authors use spin column based kit systems to extract, concentrate and purify LAMP ready target DNA from pure cultures or from contaminated sample materials (Niessen et al. 2012; Jensen Søe et al. 2013; Li et al. 2013a; Denschlag et al. 2014). An even simpler method of providing purified target DNA for LAMP reactions is the use of a FTA membrane (Whatman) to specifically bind gDNA and enable removal of non-DNA materials by a simple washing step. DNA bound to FTA membrane material can be added directly to a LAMP master mix to amplify target DNA (Lu et al. 2011). Similarly, Tomlinson et al. (2010b) applied a nitrocellulose membrane commonly used in LFD to bind and purify target DNA. LAMP was performed using a small segment of the membrane to provide the target for amplification. Other authors used raw cell or sample extracts after boiling in an extraction buffer or water with or without previous mechanical treatment from which an aliquot was directly added into the LAMP reaction (Hayashi et al. 2007; Lucas et al. 2010; Storari et al. 2013; Ishikawa et al. 2014). The highest degree of simplification in sample preparation is direct addition of materials into the LAMP master mix with no further handling necessary. This was demonstrated for pure culture materials (Luo et al. 2014a; Niessen and Vogel 2010; 2012; Denschlag et al. 2012) or for infected barley grains which were immersed directly into the master mix prior to the start of the LAMP reaction (Niessen et al. 2012).
Future perspectives of fungal LAMP
Review of the literature as given in the previous sections has demonstrated that the potential of LAMP is based on its sensitivity, rapidness and specificity, as well as convenience in practical implementations. Similar to other nucleic acid based methods, LAMP provides detection and identification in one reaction. Based on DNA amplification, the LAMP technique cannot distinguish whether or not fungi and yeasts detected are alive and multiplying. However, this fact may have serious implications in some areas such as medicine and food safety where treatment of patients or food and food raw materials is unnecessary if pathogens or spoiling organisms are not viable. However, no attempts have so far been made in that direction by application to fungal organisms. Only recently, Minenko et al. (2014) published the first paper describing the use of reverse transcription LAMP for the selective detection of a fungal mRNA template in a one-step reaction by directly adding fungal mycelia to the LAMP reaction or after mRNA isolation. Attempts to use sample treatment with ethidium monoazide or propidium monoazide prior to RNA isolation for the selection of live target cells as previously reported for living bacteria (Wang and Huo 2012; Wang et al. 2012) have not yet been used for fungi.
Within the near future LAMP-based diagnostics will neither replace sequencing-based identification nor the classical microbiological analysis of fungi and yeasts in the clinical, food, or environmental laboratory. However, it will provide a valuable tool in all such applications in which rapid and sensitive identification of organisms is a priority in order to facilitate decision making about actions to be taken, e.g., in medical point-of-care situations, sanitation and quarantine testing, and food quality control applications. Application of LAMP has been described now for major fungal pathogens in humans, animals, and plants. Only a few LAMP assays have yet been focused on organisms which compromise the quality of food and beverages and it can be anticipated that more such assays will be developed in the near future because the method is ideally suited for application in HACCP concepts in various industries. Ready to use kit systems have been patented mainly for bacteria and viruses but also systems for fungi or yeast will presumably become available in the future, especially for diagnosis of medically important organisms. Also the development and application for LAMP-based systems for diagnosis of quarantine organisms, especially plant pathogens, will become a growing field in the future since it will considerably speed up the release of plants and animals during import. Some such systems have already been developed (Tomlinson et al. 2010b; Li et al. 2013a; Tomlinson et al. 2013; Zhang et al. 2013; Moradi et al. 2014), but the list of organisms recommended for regulation as quarantine pests as approved by EPPO Council is long enough to keep scientists working late hours during the coming years.
As regards to speed of analysis, the recent release of the newly developed thermophilic Bst 2.0 DNA polymerases for LAMP applications has improved reaction speed a lot and similar types of enzymes have been released now from market competitors. Enzymes of that type can reduce reaction times by about 50 % according to the authors’ experience (Denschlag et al. 2013). In combination with modern portable devices for incubation and signal readout now available on the market, testing with a simple yes/no result will be possible within 10–15 min or even less with sensitivity similar to what has been shown in lab experiments. Other recently detected thermophilic DNA polymerases such as OmniAmp Pol expressed from the genome of PhyroPhage 3173 and modified for LAMP application may further improve the preparation of dried ready-to-use master mixes (Chander et al. 2014).
Another future trend in the application of LAMP for on-site testing is the use of micro fluidic instrumentation. Developments in that area are strongly driven by virological and bacteriological research. None of the developments has yet been applied or adapted in the field of mycological diagnostics. However, the field will certainly benefit a lot from the developments described below in applicactions such as medical diagnosis or diagnosis of quarantine plant panthogens. Recently, Ahmad and Hashsham (2012) reviewed the literature available on miniaturized nucleic acid amplification systems and their possible application for point-of-care analysis and described a variety of different systems already available or under development. At the current point of development, LAMP-based assays do not yet fulfill the WHO recommendation according to which point-of-care diagnostic devices developed for low-resource settings should follow the ASSURED criteria; affordable, sensitive, specific, user-friendly, rapid and robust, electricity free, and deliverable to end users (Peeling et al. 2006). Recent research towards the development of low equipment microLAMP shows progress towards this goal for future applications since electricity-free LAMP (tube based) was recently demonstrated by LaBarre et al. (2011), who used calcium oxide as heat source in combination with a phase change material in a NINA (non-instrumented nucleic acid amplification) process to create a constant temperature of 65 °C over 1 h without electricity and demonstrated the feasibility of the method to perform a LAMP reaction. Also such systems would be highly desirable for fungal diagnosis in clinical settings or in the field.
Besides making LAMP more easily operated, using the method for the analysis of multiple parameters in one run will be a future trend. Preliminary concepts have been demonstrated and prototype devices were published recently. It appears that two different approaches to multiplexing the LAMP reaction do currently develop in parallel. One approach is parallel multiplexing using microfluidic devices in which a sample is split into several subsamples which are than analyzed with multiple primer sets in separate LAMP reactions resulting in a positive/negative pattern readout (Fang et al. 2012; Zhu et al. 2012). Another approach is the use of simultaneous multiplexing using multiple primer sets in one reaction as demonstrated by Tanner et al. (2012) who applied the FRET-based assimilating probes technology previously described by Kubota et al. (2011). Separate detection of different amplification products obtained with different primer sets was achieved by integration of specific probes each of which was coupled to a different fluorescent marker corresponding to different channels of a fluorescence reader, e.g., real-time thermal cycler.
Some very recent developments show that LAMP has the potential to be a useful technology not only for the diagnosis of microorganisms. Combination of antibodies or aptameric probes for proteins or even low molecular weight compounds such as mycotoxins with a LAMP-based detection system can provide ultrasensitive systems for the analysis and quantification as an alternative to ELISA-based detection (immunoLAMP). Pourhassan-Moghaddam et al. (2013) combined antibody-based detection of proteins with LAMP-based amplification of a DNA marker sequence either attached to an antibody or specific aptamer or packed in attached liposomes which open upon binding and set the LAMP target free for subsequent amplification and detection. A similar system for specific quantification of ochratoxin A (OTA) was developed by Yuan et al. (2014) who used an OTA specific aptamer to react specifically with sensor bound toxin. The binding event was detected and quantified using a polynucleotide probe that was amplified in a subsequent LAMP reaction with Ru(phen)3 2+ based electrochemiluminiscent signal readout. The assay had a detection limit of 10 f. of OTA in a model solution. Xie et al. (2014) showed that the method is also applicable for the detection of the toxin in red wine samples. Other recently developed concepts have shown that LAMP can be combined with enzyme free DNA nanotechnologies such as catalytic hairpin assembly (Li et al. 2012) or enthropy driven DNA assembly such as strand displacement driven hybridization chain reaction (Li et al. 2013b). Implication of such newly developed technologies into the concept of LAMP-based detection systems as well as further completion of the portfolio of detectable species and groups will certainly cause a considerable improvement of the diagnostic tools for fungi and yeasts within the near future.
References
Abd-Elsalam KA, Bahkali AH, Moslem M, Amin OE, Niessen L (2011) An optimized protocol for DNA extraction from wheat seeds and loop-mediated isothermal amplification (LAMP) to detect Fusarium graminearum contamination of wheat grain. Int J Mol Sci 12:3459–3472
Ahmad F, Hashsham SA (2012) Miniaturized nucleic acid amplification systems for rapid and point-of-care diagnostics: a review. Anal Chim Acta 733:1–15
Alaeddini R (2012) Forensic implications of PCR inhibition—a review. Forensic Sci Int Gen 6:297–305
Alef K, Nannipieri P (1995) Methods in Applied Soil Microbiology and Biochemistry Academic press, London
Almasi, MA, Moradi, A, Ojaghkandi, MA, Aghaei, S (2013) Development and application of loop-mediated isothermal amplification assay for rapid detection of Fusarium oxysporum f sp lycopersici. J Plant Pathol Microbiol 4:177. doi:104172/2157-74711000177
Almoammar H, Bahkali AH, Abd-Elsalam KA (2013) One-hour loop-mediated isothermal amplification assay for the detection of quarantinable toxigenic Fusarium garmanirum. Afr J Microbiol Res 7:1179–1183
Atkins SD, Clark IM (2004) Fungal molecular diagnostics: a mini review. J Appl Genet 45:3–15
Baumgartner C, Freydière AM, Gille Y (1996) Direct identification and recognition of yeast species from clinical material by using albicans ID and CHROMagar Candida plates. J Clin Microbiol 32:454–456
Bennett JW, Klich M (2003) Mycotoxins. Clin Microbiol Rev 16:497–516
Boehme CC, Nabeta P, Henostroza G, Raqib R, Rahim Z, Gerhardt M, Sanga E, Hoelscher M, Notomi T, Hase T, Perkins MD (2006) Operational feasibility of using loop-mediated isothermal amplification for diagnosis of pulmonary tuberculosis in microscopy centers of developing countries. J Clin Microbiol 45:1936–1940
Buckley HR, Richardson MD, Evans EGV, Wheat LJ (1992) Immunodiagnosis of invasive fungal infection. J Med Vet Mycol 30(1):249–260
Chander Y, Koelbl J, PuckettJ, Moser MJ, Klingele AJ, Liles MR, Carrias A, Mead DA, Schoenfeld TW (2014) A novel thermostable polymerase for RNA and DNA loop-mediated isothermal amplification (LAMP). Front. Microbiol 5:p-p. doi: 103389/fmicb201400395
Chen Q, Li B, Liu P, Lan C, Zhan Z, Wenig Q (2013) Development and evaluation of specific PCR and LAMP assays for the rapid detection of Phytophthora melonis. Eur J Plant Pathol 137:597–607
Cortez KJ, Roilides E, Quiroz-Telles F, Meletiadis J, Antachopoulos C, Knudsen T, Buchanan W, Milanovich J, Sutton DA, Fothergill A, Rinaldi MG, Shea YR, Zaoutis T, Kottilil S, Walsh TJ (2008) Infections caused by Scedosporium spp. Clin Microbiol Rev 21:157–197
Dai TT, Lu CC, Lu J, Dong SM, Ye WW, Wang YC, Zheng XB (2012) Development of a loop-mediated isothermal amplification assay for detection of Phytophthora sojae. FEMS Microbiol Lett 334:27–34
Denschlag C, Vogel RF, Niessen L (2012) Hyd5 gene-based detection of the major gushing-inducing Fusarium spp in a loop-mediated isothermal amplification (LAMP) assay. Int J Food Microbiol 156:189–196
Denschlag C, Vogel RF, Niessen L (2013) Hyd5 gene based analysis of cereals and malt for gushing-inducing Fusarium spp by real-time LAMP using fluorescence and turbidity measurements. Int J Food Microbiol 162:245–251
Denschlag C, Rieder J, Vogel RF, Niessen L (2014) Real-time loop-mediated isothermal amplification (LAMP) assay for group specific detection of important trichothecene producing Fusarium species in wheat. Int J Food Microbiol 177:117–127
Diehl H, Ellingboe JL (1956) Indicator for titration of calcium in presence of magnesium using disodium dihydrogen ethylenediamine tetraacetate. Anal Chem 28:882–884
Duan Y, Ge C, Zhang X, Wang J, Zhou M (2014) A rapid detection method for the plant pathogen Sclerotinia sclerotiorum based on loop-mediated isothermal amplification (LAMP). Aust Plant Pathol 43:61–66
Elmer PAG, Michailides TJ (2007) Epidemiology of Botrytis cinerea in orchard and vine crops. In: Elad Y, Williamson B, Tudzynski P, Delen N (eds) Botrytis: biology, pathology and control. Springer, Dordrecht, pp 243–272
Enache-Angoulvant A, Chandenier J, Symoens F, Lacube P, Bolognini J, Douchet C, Poirot JL, Hennequin C (2007) Molecular identification of Cryptococcus neoformans serotypes. J Clin Microbiol 45:1261–1265
Endo S, Komori T, Ricci G, Sano A, Yokoyama K, Ohori A, Kamei K, Franco M, Miyaji M, Nishimura K (2004) Detection of gp43 of Paracoccidioides brasiliensis by the loop-mediated isothermal amplification (LAMP) method. FEMS Microbiol Lett 234:93–97
EPPO (European and Mediterranean Plant Protection Organization) (2009) Giugnardia citricarpa. EPPO Bull 39:318–327
Fang X, Chen H, Xu L, Jiang X, Wu W, Kong J (2012) A portable and integrated nucleic acid amplification microfluidic chip for identifying bacteria. Lab Chip 12:1495–1499
Ferdousi A, Shahhossein MH, Bayat M, Hashimi SJ, Gharhi M (2014) Comparison of polymerase chain reaction and loop-mediated isothermal amplification for diagnosis of Fusarium solani in human immunodeficiency virus positive patients. Afr J Biotechnol 13:1496–1502
Fidel PL Jr, Vazquez JA, Sobel JD (1999) Candida glabrata: review of epidemiology, pathogenesis, and clinical disease with comparison to C. albicans. Clin Microbiol Rev 12:80–96
Francois P, Tangomo M, Hibbs J, Bonetti EJ, Boehme CC, Notomi T, Perkins MD, Schrenzel J (2011) Robustness of a loop-mediated isothermal amplification reaction for diagnostic applications. FEMS Immunol Med Microbiol 62:41–48
Fukuta S, Takahashi R, Kuroyanagi S, Miyake N, Nagai H, Suzuki H, Hashizume F, Tsuji T, Taguchi H, Watanabe H, Kageyama K (2013) Detection of Pythium aphanidermatum in tomato using loop-mediated isothermal amplification (LAMP) with species-specific primers. Eur J Plant Pathol 136:689–701
Fukuta S, Takahashi R, Kuroyanagi S, Ishiguru Y, Miyake N, Nagai H, Suzuki H, Tsuji T, Hashizume F, Watanabe H, Kageyama K (2014) Development of loop-mediated isothermal amplification (LAMP) assay for the detection of Pythium myriotylum. Lett Appl Microbiol 59:49–57
Gadkar V, Rillig MC (2008) Evaluation of loop-mediated isothermal amplification (LAMP) to rapidly detect arbuscular mycorrhizal fungi. Soil Biol Biochem 40:540–543
Ghelardi E, Pichierri G, Castagna B, Barnini S, Tavanti A, Campa M (2008) Efficacy of chromogenic Candida agar for isolation and presumptive identification of pathogenic yeast species. Clin Microbiol Infect 14:141–147
Gill P, Ghaemi A (2008) Nucleic acid isothermal amplification technologies—a review. Nucleosides Nucleotides Nucleic Acids 27:224–243
Gordon TR, Martyn RD (1997) The evolutionary biology of Fusarium oxysporum. Ann Rev Plant Pathol 35:111–128
Goto M, Honda E, Ogura A, Nomoto A, Hanaki KI (2009) Colorimetric detection of loop-mediated isothermal amplification reaction by using hydroxy naphthol blue. Biotechniques 46:167–172
Guillamon JM, Barrio E, Huerta T, Querol A (1994) Rapid Characterization of four species of the Saccharomyces sensu stricto complex according to mitochondrial DNA patterns. Int J Syst Evol Microbol 44:708–714
Hayashi N, Arai R, Tada S, Taguchi H, Ogawa Y (2007) Detection and identification of Brettanomyces/Dekkera sp yeasts with a loop-mediated isothermal amplification method. Food Microbiol 24:778–785
Hayashi N, Minato T, Kanai K, Ikushima S, Yoshida S, Tada S, Taguchi H, Ogawa Y (2009) Differentiation of species belonging to Saccharomyces sensu stricto using a loop-mediated isothermal amplification method. J Am Soc Brew Chem 67:118–126
Huang C, Sun Z, Yan J, Luo Y, Wang H, Ma Z (2011) Rapid and precise detection of latent infections of wheat stripe rust in wheat leaves using loop-mediated isothermal amplification. J Phytopathol 159:582–584
Huggett JF, Novak T, Garson JA, Green C, Morris-Jones SD, Miller RF, Zumla A (2008) Differential susceptibility of PCR reactions to inhibitors: an important and unrecognised phenomenon. BMC Res Notes 1:70
Inacio J, Flores O, Spencer-Martins I (2008) Efficient identification of clinically relevant Candida yeast species by use of an assay combining panfungal loop-mediated isothermal DNA amplification with hybridization to species-specific olignonucleotide probes. J Clin Microbiol 46:713–720
Ishikawa H, Kasahara K, Sato S, Shimakawa Y, Watanabe K (2014) Simple and rapid method for the detection of Filobasidiella neoformans in a probiotic dairy product by using loop-mediated isothermal amplification. Int J Food Microbiol 178:107–112
Jensen Søe M, Rohde M, Mikkelsen J, Warthoe P (2013) IsoPCR: an analytically sensitive, nested, multiplex nucleic acid amplification method. Clin Chem 59:436–439
Kasahara K, Ishikawa H, Sato S, Shimakawa Y, Watanabe K (2014) Development of multiplex loop-mediated isothermal amplification assays to detect medically important yeasts in dairy products. FEMS Microbiol Lett 357:208–216
Kroon LPNM, Brouwer H, de Cock AWAM, Govers F (2012) The genus Phytophthora anno 2012. Phytopathology 102:348–364
Kubota R, Alvarez AM, Su WW, Jenkins DM (2011) FRET-based assimilating probe for sequence-specific real-time monitoring of loop-mediated isothermal amplification (LAMP). Biol Eng Trans 4:81–100
LaBarre P, Hawkins KR, Gerlach J, Wilmoth J, Beddoe A, Singleton J, Boyle D, Weigl B (2011) A Simple, inexpensive device for nucleic acid amplification without electricity—toward instrument-free molecular diagnostics in low-resource settings. PLoS One 6:e19738
Li Y, Cai SH (2011) Sensitive and rapid detection of the insect pathogenic fungus Metarhizium anisopliae var anisopliae by loop-mediated isothermal amplification. Curr Microbiol 62:1400–1404
Li B, Chen X, Ellington AD (2012) Adapting enzyme-free DNA circuits to the detection of loop-mediated isothermal amplification reactions. Anal Chem 84:8371–8377
Li B, Du J, Lan C, Liu P, Weng Q, Chen Q (2013a) Development of a loop-mediated isothermal amplification assay for rapid and sensitive detection of Fusarium oxysporum f. sp. cubense race 4. Eur J Plant Pathol 135:903–911
Li F, Zhang H, Wang Z, Li X, Li XF, Le XC (2013b) Dynamic DNA assemblies mediated by binding-induced DNA strand displacement. J Am Chem Soc 135:2443–2446
Lima CS, Pfenning LH, Costa SS, Campos MA, Leslie JF (2009) A new Fusarium lineage within the Gibberella fujikuroi species complex is the main causal agent of mango malformation disease in Brazil. Phytopathology 58:33–42
López Martínez R, Mendéz Tovar LJ (2007) Chromoblastomycosis. Clin Dermatol 25:188–194
Lu Q, Gerrits van den Ende AHG, Najafzadeh MJ, Melchers WJG, Bakkers JMJE, Li R, Sun J, de Hoog GS, Lackner M (2011) Identification of Pseudallescheria and Scedosporium species by three molecular methods. J Clin Microbiol 49:960–967
Lucas S, da Luz Martins M, Flores O, Meyer W, Spencer-Martins I, Ina J (2010) Differentiation of Cryptococcus neoformans varieties and Cryptococcus gattii using CAP59-based loop-mediated isothermal DNA amplification. Clin Microbiol Infect 16:711–714
Luo J, Vogel RF, Niessen L (2012) Development and application of a loop-mediated isothermal amlification assay for rapid identification of aflatoxinogenic molds and their detection in food samples. Int J Food Microbiol 159:214–224
Luo J, Taniwaki MH, Iamanaka BT, Vogel RF, Niessen L (2014a) Application of loop-mediated isothermal amplification assays for direct identification of pure cultures of Aspergillus flavus, A nomius, and A caelatus and for their rapid detection in shelled Brazil nuts. Int J Food Microbiol 172:5–12
Luo J, Niessen L, Vogel R (2014b) Rapid detection of aflatoxin producing fungi in food by real-time quantitative loop-mediated isothermal amplification. Food Microbiol 44:142–148
Matsuzawa T, Tanaka R, Horie Y, Gonoi T, Yaguchi T (2010) Development of rapid and specific molecular discrimination methods for pathogenic Emericella species. Jpn J Med Mycol 51:109–115
McCartney HA, Foster SJ, Fraaje BA, Ward E (2003) Molecular diagnostics for fungal plant pathogens. Pest Manag Sci 59:129–142
Mendes-Giannini MJS, Taylor ML, Bouchara JB, Burger E, Calich VLG, Escalante ED, Hanna SA, Lenzi HL, Machado MP, Miyaji M, Monteiro Da Silva JL, Mota EM, Restrepo A, Restrepo S, Tronchin G, Vincenzi LR, Xidieh CF, Zenteno E (2000) Pathogenesis II: fungal responses to host responses: interaction of host cells with fungi. Med Mycol 38(Supplement 1):113–123
Mesaphy S (2013) Development of media for growth and emumeration of fungi in water. In: Gupta VK, Tuohy MG (eds) Laboratory protocols in fungal biology—current methods in fungal biology. Springer Science + Business Media, New York, pp 201–210
Michielse CB, Rep M (2009) Pathogen profile update: Fusarium oxysporum. Mol Plant Pathol 10:311–324
Minenko E, Vogel RF, Niessen L (2014) Application of one-step reverse transcription loop mediated isothermal amplification (reverse transcription LAMP) for rapid detection of fungal gene expression in pure culture mycelia and in planta. Mycoscience 55:125–130
Moradi A, Almasi MA, Jafary H, Mercado-Blanco J (2014) A novel and rapid loop-mediated isothermal amplification assay for the specific detection of Verticillium dahliae. J Appl Microbiol 116:942–954
Mori Y, Notomi T (2008) Loop-mediated isothermal amplification (LAMP): a rapid, accurate, and cost-effective diagnostic method for infectious diseases. J Infect Chemother 15:62–69
Mori Y, Nagamine K, Tomita N, Notomi T (2001) Detection of loop-mediated isothermal amplification reaction by turbidity derived from magnesium pyrophosphate formation. Biochem Biophys Res Commun 289:150–154
Mori Y, Kitao M, Tomita N, Notomi T (2004) Real-time turbidimetry of LAMP reaction for quantifying template DNA. J Biochem Biophys Methods 59:145–157
Mori Y, Hirano T, Notomi T, Mori Y, Hirano T, Notomi T (2006) Sequence specific visual detection of LAMP reactions by addition of cationic polymers. BMC Biotechnol 6(3)
Nagamine K, Hase T, Notomi T (2002) Accelerated reaction by loopmediated isothermal amplification using loop primers. Mol Cell Probes 16:223–229
Najafzadeh MJ, Sun J, Vicente V, Xi L, Gerrits van den Ende AHG, de Hoog GS (2010) Fonsecaea nubica sp nov, a new agent of human chromoblastomycosis revealed using molecular data. Med Mycol 48:800–806
Niessen L (2008) PCR‐based diagnosis and quantification of mycotoxin‐producing fungi. Adv Food Nutr Res 54:81–138
Niessen L (2013) Loop-mediated isothermal amplification-based detection of Fusarium graminearum. In: O’Connor L, Glynn B (eds) Fungal diagnostics: methods and protocols. Methods in molecular biology, vol 968. Springer Science Business Media, New York, pp 177–193
Niessen L, Vogel RF (2010) Detection of Fusarium graminearum DNA using a loop-mediated isothermal amplification (LAMP) assay. Int J Food Microbiol 140:183–191
Niessen L, Gräfenhan T, Vogel RF (2012) ATP citrate lyase 1 (acl1) gene-based loop-mediated amplification assay for the detection of the Fusarium tricinctum species complex in pure cultures and in cereal samples. Int J Food Microbiol 158:171–185
Niessen L, Luo J, Denschlag C, Vogel RF (2013) The application of loop-mediated isothermal amplification (LAMP) in food testing for bacterial pathogens and fungal contaminants. Food Microbiol 36:191–206
Notomi T, Okayama H, Masubuchi H, Yonekawa T, Watanabe K, Amino N, Hase T (2000) Loop-mediated isothermal amplification of DNA. Nucleic Acids Res 28:e63. doi:10.1093/nar/28.12.e63
O’Loughlin MC, Turner KD, Turner KM (2013) Quantitative sampling methods for the analysis of fungi: air sampling. In: Gupta VK, Tuohy MG (eds) Laboratory protocols in fungal biology—current methods in fungal biology. Springer Science Business Media, New York, pp 337–342
Odds FC, Bernaerts R (1994) CHROMagar Candida, a new differential isolation medium for presumptive identification of clinically important Candida species. J Clin Microbiol 32:1923–1929
Ögel ZB, Brayford D, McPherson MJ (1994) Cellulose-triggered sporulation in the galactose oxidase-producing fungus Cladobotryum (Dactylium) dendroides NRRL 2903 and its re-identification as a species of Fusarium. Clin Microbiol Infect 16:711–714
Ohori A, Endo S, Sano A, Yokoyama K, Yarita K, Yamaguchi M, Kamei K, Miyaji M, Nishimura K (2006) Rapid identification of Ochroconis gallopava by a loop-mediated isothermal amplification (LAMP) method. Vet Microbiol 114:359–365
Patterson T, Kirkpatrick WR, White M, Hiemenz JW, Wingard JR, Dupont B, Rinaldi MG, Stevens DA, Graybill JR (2000) Invasive aspergillosis: disease spectrum, treatment practices, and outcomes. Medicine 79:250–260
Peeling RW, Holmes KK, Mabey D, Ronald A (2006) Rapid tests for sexually transmitted infections (STIs): the way forward. Sex Transm Infect 82(Suppl V):v1–v6
Peng J, Zhan Y, Zeng F, Long H, Pei Y, Guo J (2013) Development of a real-time fluorescence loop-mediated isothermal amplification assay for rapid and quantitative detection of Fusarium oxysporum f sp niveum in soil. FEMS Microbiol Lett 349:127–134
Pihet M, Carrere J, Cimon B, Chabasse D, Delhaes L, Symoens F, Bouchara JP (2009) Occurrence and relevance of filamentous fungi in respiratory secretions of patients with cystic fibrosis—a review. Med Mycol 47:387–397
Pitt JI, Hocking AD (eds) (2009) Fungi in food spoilage, 3rd edn. Springer Science Business Media, New York, p 519
Pourhassan-Moghaddam M, Rahmati-Yamchi M, Akbarzadeh A, Nejati-Koshki K, Hanifehpour Y, Joo SJ, Daraee H (2013) Protein detection through different platforms of immuno-loop-mediated isothermal amplification. Nanoscale Res Lett 8:485
Prokopowich CD, Gregory TR, Crease TJ (2003) The correlation between rDNA copy number and genome size in eukaryotes. Genome 46:48–50
Pu J, Xie Y, Zhang H, Zhang X, Qi Y, Peng J (2014) Development of a real-time fluorescence loop-mediated isothermal amplification assay for rapid and quantitative detection of Fusarium mangiferae associated with mango malformation. Phys Mol Plant Pathol 86:81–88
Qureshi ZA, Kwak EJ, Nguyen MH, Silveira FP (2012) Ochroconis gallopava: a dematiaceous mold causing infections in transplant recipients. Clin Transplant 26:E17–23
Raeder U, Broda P (1985) Rapid preparation of DNA from filamentous fungi. Lett Appl Microbiol 1:17–20
Restrepo AM (1985) The ecology of Paracoccidioides brasiliensis: a puzzle still unsolved. J Med Mycol 23:323–334
Rillig MC (2004) Arbuscular mycorrhizae and terrestrial ecosystem processes. Ecol Lett 7:740–754
Sarlin T, Nakari-Setälä T, Linder M, Penttilä M, Haikara A (2005) Fungal hydrophobins as predictors of the gushing activity of malt. J Inst Brew 111:105–111
Schleifer KH, Kandler O (1972) Peptidoglucan types of bacterial cell walls and their taxonomic implications. Bacteriol Rev 36:407–4077
Schoch CL, Seifert KA, Huhndorf S, Robert V, Spouge JL, Levesque CA, Chen W, Fungal Barcoding Consortium (2012) Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for fungi. Proc Natl Acad Sci U S A 109:6241–6246
Schrader C, Schielke A, Ellerbroek L, Johne R (2012) PCR inhibitors—occurrence, properties and removal. J Appl Microbiol 113:1014–1026
Sendid B, François N, Standaert A, Dehecq E, Zerimech F, Camus D, Poulain D (2007) Prospective evaluation of the new chromogenic medium CandiSelect 4 for differentiation and presumptive identification of the major pathogenic Candida species. J Med Microbiol 56:495–499
Storari M, von Rohr R, Pertot I, Gessler C, Broggini GAL (2013) Identification of ochratoxin A producing Aspergillus carbonarius and A. niger clade isolated from grapes using the loop-mediated isothermal amplification (LAMP) reaction. J Appl Microbiol 114:1193–1200
Sun J, Li X, Zeng H, Xie Z, Lu C, Xi L, de Hoog GS (2010a) Development and evaluation of loop-mediated isothermal amplification(LAMP) for the rapid diagnosis of Penicillium marneffei in archived tissue samples. FEMS Immun Med Microbiol 58:381–388
Sun J, Najafzadeh MJ, Vicente V, Xi L, de Hoog GS (2010b) Rapid detection of pathogenic fungi using loop-mediated isothermal amplification, exemplified by Fonsecaea agents of chromoblastomycosis. J Microbiol Meth 80:19–24
Takahashi R, Fukuta S, Kuroyanagi S, Miyake N, Nagai H, Kageyama K, Ishiguro Y (2014) Development and application of a loop-mediated isothermal amplification assay for rapid detection of Pythium helicoides. FEMS Microbiol Lett 355:28–35
Tanner NA, Zhang Y, Evans TC (2012) Simultaneous multiple target detection in real-time loop-mediated isothermal amplification. Biotechniques 53:81–89
Tatibana BT, Sano A, Uno J, Kamei K, Nishimura K, Igarashi T, Itano EN, Mikami Y, Miyaji M (2009) Detection of Paracoccidioides brasiliensis gp43 gene in sputa by loop-mediated isothermal amplification method. J Clin Lab Anal 23:139–143
Tomita N, Mori Y, Kanda H, Notomi T (2008) Loop-mediated isothermal amplification (LAMP) of gene sequences and simple visual detection of products. Nat Protoc 3:877–882
Tomlinson JA, Barker I, Boonham N (2007) Faster, simpler, more-specific methods for improved molecular detection of Phytophthora ramorum in the field. Appl Environ Microbiol 73:4040–4047
Tomlinson JA, Dickinson MJ, Boonham N (2010a) Detection of Botrytis cinerea by loop-mediated isothermal amplification. Lett Appl Microbiol 51:650–657
Tomlinson JA, Dickinson MJ, Boonham N (2010b) Rapid detection of Phytophthora ramorum and P. kernoviae by two-minute DNA extraction followed by isothermal amplification and amplicon detection by generic lateral flow device. Phytopathology 100:143–149
Tomlinson JA, Dickinson MJ, Boonham N, Ostoja-Starzewska S, Webb K, Cole J, Barnes A, Dickinson M, Boonham N (2013) A loop-midiated isothermal amplification-based method for confirmation of Guigniardia citricarpa in citrus black spot lesions. Eur J Plant Pathol 136:217–224
Travassos LR, Puccia R, Cisalpino P, Taborda C, Rodrigues EG, Rodrigues M, Silveira JF, Almeida IC (1995) Biochemistry and molecular biology of the main diagnostic antigen of Paracoccidioides brasiliensis. Arch Med Res 26:297–304
Tsao PH (1970) Selective media for the isolation of pathogenic fungi. Annu Rev Phytopathol 8:157–180
Uemura N, Makimura K, Onozaki M, Otsuka Y, Shibuya Y, Yazaki H, Kikuchi Y, Abe S, Kudoh S (2008) Development of a loop-mediated isothermal amplification method for diagnosing Pneumocystis pneumonia. J Med Microbiol 57:50–57
Vaagt F, Haase I, Fischer M (2013) Loop-mediated isothermal amplification (LAMP)-based method for rapid mushroom species identification. J Agric Food Chem 61:1833–1840
Villari C, Tomlinson JA, Battisti A, Boonham N, Capretti P, Faccoli M (2013) Use of loop-mediated isothermal amplification for detection of Ophiostoma clavatum, the primary blue stain fungus associated with Ips acuminatus. Appl Environ Microbiol 79:2527–2533
Waalwijk C, de Koning JRA, Baayen RP, Gams W (1996) Discordant groupings of Fusarium spp. from sections Elegans, Liseola and Dlaminia based on ribosomal ITS1 and ITS2 sequences. Mycologia 88:361–368
Wang D, Huo G (2012) Rapid detection of viable Escherichia coli O157 in raw milk using loop-mediated isothermal amplification with aid of ethidium monoazide. Adv Mater Res 343–344:1217–1221
Wang L, Zhong Q, Li Y (2012) Ethidium monoazide-loop mediated isothermal amplification for rapid detection of Vibrio parahaemolyticus in viable but non-culturable state. Energy Proc 17:1858–1863
White PL, Perry MD, Barnes RA (2009) An update on the molecular diagnosis of invasive fungal disease. FEMS Microbiol Lett 296:1–10
Williamson B, Tudzynski B, Tudzynski P, von Kan JAL (2007) Botrytis cinerea: the cause of grey mould disease. Mol Plant Pathol 8:561–580
Wilson IG (1997) Inhibition and facilitation of nucleic acid amplification. Appl Environ Microbiol 63:3741–3751
Xie X, Chai Y, Yuan Y, Bai L, Yuan R (2014) Development of an electrochemical method for ochratoxin A detection based on aptamer and loop-mediated isothermal amplification. Biosens Bioelectron 55:324–329
Yan L, Zhou J, Zheng Y, Gamson AS, Reombke BT, Nakayama S, Sintim HS (2014) Isothermal amplified detection of DNA and RNA. Mol BioSyst 10:970–1003
Yarita K, Sano A, Murata Y, Takayama A, Takahashi Y, Takahashi H, Yaguchi T, Ohori A, Kamei K, Miyaji M, Nishimura K (2007) Pathogenicity of Ochroconis gallopava isolated from hot springs in Japan and a review of published reports. Mycopathologia 164:135–147
Yarita K, Sano A, Samerpitak K, Kamei K, de Hoog GS, Nishimura K (2010) Ochroconis calidifluminalis, a sibling of the neurotropic pathogen O. gallopava, isolated from hot spring. Mycopathologia 170:21–30
Yeo SF, Wong B (2004) Current status of nonculture methods for diagnosis of invasive fungal infections. Clin Microbiol Rev 15:465–484
Yuan Y, Wei S, Liu G, Xie S, Chai Y, Yuan R (2014) Ultrasensitive electrochemiluminescent aptasensor for ochratoxin A detection with the loop-mediated isothermal amplification. Anal Chim Acta 811:70–75
Zhang X, Zhang H, Pu J, Qi Y, Yu Q, Xie Y, Peng J (2013) Development of a real-time fluorescence loop-mediated isothermal amplification assay for rapid and quantitative detection of Fusarium oxysporum f. sp. cubense tropical race 4 in soil. PLoS One 8:e82841
Zhu Q, Gao Y, Yu B, Ren H, Qiu L, Han S, Jin W, Jin Q, Mu Y (2012) Self-priming compartmentalization digital LAMP for point-of-care. Lab Chip 12:4755–4763
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 75 kb)
Rights and permissions
About this article

Cite this article
Niessen, L. Current state and future perspectives of loop-mediated isothermal amplification (LAMP)-based diagnosis of filamentous fungi and yeasts. Appl Microbiol Biotechnol 99, 553–574 (2015). https://doi.org/10.1007/s00253-014-6196-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-014-6196-3