
Abstract
Microbial L-asparaginase (ASNase) is an important anticancer agent that is used extensively worldwide. In this study, 40 bacterial isolates were obtained from the Red Sea of Saudi Arabia and screened for ASNase production using a qualitative rapid plate assay, 28 of which were producing large L-asparagine hydrolysis zones. The ASNase production of the immobilized bacterial cells was more favorable than that of freely suspended cells. A promising isolate, KKU-KH14, was identified by 16S rRNA gene sequencing as Bacillus licheniformis. Maximal ASNase production was achieved using an incubation period of 72 h, with an optimum of pH 6.5, an incubation temperature of 37 °C, an agitation rate 250 rpm, and with glucose and (NH4)2SO4 used as the carbon and nitrogen sources, respectively. The glutaminase activity was not detected in the ASNase preparations. The purified ASNase showed a final specific activity of 36.08 U/mg, and the molecular weight was found to be 37 kDa by SDS-PAGE analysis. The maximum activity and stability of the purified enzyme occurred at pH values of 7.5 and 8.5, respectively, with maximum activity at 37 °C and complete thermal stability at 70 °C for 1 h. The Km and Vmax values of the purified enzyme were 0.049995 M and of 45.45 μmol/ml/min, respectively. The anticancer activity of the purified ASNase showed significant toxic activity toward HepG-2 cells (IC50 11.66 µg/mL), which was greater than that observed against MCF-7 (IC50 14.55 µg/mL) and HCT-116 cells (IC50 17.02 µg/mL). The results demonstrated that the Red Sea is a promising biological reservoir, as shown by the isolation of B. licheniformis, which produces a glutaminase free ASNase and may be a potential candidate for further pharmaceutical use as an anticancer drug.
Similar content being viewed by others
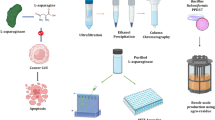


Introduction
ASNase (EC 3.5.1.1) is an important enzyme used in the pharmaceutical, biosensor and food industries1 and has anticarcinogenic potential for the treatment of acute lymphoblastic leukemia, lymphomas and other cancers2,3. ASNase selectively targets the metabolism of cancer cells by exploiting deficiencies in metabolic pathways and catalyzing the degradation of L-asparagine into L-aspartic acid and ammonia, causing nutrient starvation of cancer cells and bringing about their demise4. Thus, there is a need for novel and robust ASNases from new microorganisms that exhibit improved stability, lower glutaminase activity, high substrate affinity, and low Km values for use as therapeutics. Two types of commercial ASNase are currently in clinical use for chemotherapy, enzymes from Erwinia chrysanthemi and E. coli. However, these enzymes have drawbacks in that they exhibit low substrate specificity and high glutaminase activity5, the latter of which can cause liver dysfunction, pancreatitis, leucopenia, neurological seizures, and coagulation abnormalities that can lead to intracranial thrombosis or hemorrhages. Therefore, it is important to identify sources and methods of producing greater amounts of glutaminase free ASNases and exhibit high substrate affinity and therapeutic activity6,7. A wide range of microbes have been shown to be valuable sources of enzymes, such as Pichia pastoris, Saccharomyces cerevisiae, E. coli, Aerobacter, Pseudomonas, Bacillus, Xanthomonas, Serratia, and Streptomyces8. Recently, ASNases from E. coli and Erwinia carotovora produced by submerged fermentation were approved for use in medical applications by the United States Food and Drug Administration9,10. The Red Sea of the Saudi Arabia has yet to be thoroughly investigated with respect to its harboring microbes with biopharmaceutical and biotechnological potential11. Marine microbe-derived ASNases may be more effective and have fewer side effects as therapeutic agents for acute lymphoblastic leukemia treatment than traditional sources of these enzymes12. The enhanced production of novel glutaminase free ASNase from Pectobacterium carotovorum13 and Erwinia carotovora14 was achieved by submerged fermentation in batch and fed batch bioreactors. Cell immobilization is considered to be a promising approach for enhancing fermentation processes for ASNase production15. The benefits of using immobilized cells include the facilitation of continuous operations over an extended period of time, potential recycling of immobilized beads and a simpler means of harvesting these products, reactor productivity, higher catalysis efficiency and the development of economical methods, as well as lowering the cost of industrial processes7. The purification of ASNases is important to promote their characterization and therapeutic use with fewer adverse effects16. Thus, the goal of the present study was to isolate and screen marine bacteria for the production of glutaminase free ASNases from the Red Sea, Saudi Arabia. The optimization of enzyme production using free and immobilized cells and an evaluation of its properties and potential as an anticancer agent against different human cancer cell lines was also investigated.
Materials and Methods
Chemicals
The chemicals used for enzyme production and purification were purchased from Sigma-Aldrich Chemical Co., USA. Nessler’s reagent was purchased from Fluka (Buchs, Switzerland). The chemicals and markers used for the molecular identification of ASNases were obtained from Bio-Rad Laboratories (USA).
Sample collection
Between January and March 2017, water samples were collected from the Red Sea off the southwestern coast of Saudi Arabia from three stations: The Al-Shuqaiq coast (41° 59′22.5″E, 17° 42′35.0″N), the Al-Haridah coast (41° 55′17.8″E, 17° 45′58.5″N) and the Al-Birk coast (41° 31′40.5″E, 18° 13′49.4″N). The water samples were collected in 500-mL glass screw cap bottles and stored at 4 °C until processed.
Isolation of marine bacterial strains
Two methods were used to isolate marine bacteria: (1) the water samples (100 ml) were filtered through a Millipore membrane filter (0.22 µm), which was subsequently aseptically transferred onto agar-solidified medium; and (2) the water samples were serially diluted, and 100 µl of the 10−1,10−2, 10−3 and 10−4 dilutions were spread onto agar-solidified medium. All of the inoculated plates were incubated at 37 °C for 3 days. The composition of the asparagine agar17 was as follows (w/v): 0.6% beef extract, 1% peptone, 0.33% KH3PO4, 0.1% L-asparagine and 1.5% agar, pH 6.5.
Screening of bacterial isolates for ASNase production
The bacterial isolates were screened for ASNase production using the rapid plate assay method18, which was performed using asparagine agar medium containing 0.3 ml of 2.5% phenol red. After incubating at 37 °C for two to three days, the pink zone around the colonies was measured, and an enzyme index was calculated using the equation: Enzyme index = Pink zone (mm)/Colony diameter (mm).
Identification of promising isolates via 16S rRNA gene sequence analysis
Bacterial genomic DNA from the promising isolates was extracted from 5-ml cultures grown overnight in nutrient broth using a modified QIAamp DNA Mini kit (Qiagen Inc., Valencia, CA)19. The extracted DNA from each bacterial isolate was used as a template for amplification of the 16S rRNA gene using the universal primers 5′ CCA GCA GCC GCG GTA ATA CG 3′ and 5′ ATC GG(C/T) TAC CTT GTT ACG ACT TC 3′. The 50-µL PCR mixtures contained the following components: 10 mM Tris-HCl (pH 8.3), 50 mM KCl, 2.5 mM MgCl2, each dNTP at 0.2 mM, 1.25 IU of Taq polymerase, each primer at 0.2 µM, and 1 µL of DNA template, with the final volume brought up to 50 µL with water. PCR was performed using the following thermocycling program: 10 min of denaturation at 94 °C, followed by 35 cycles of 1 min denaturation at 94 °C, 1 min annealing at 55 °C, a 2 min extension at 72 °C, and a final extension for 10 min at 72 °C. Subsequently, 5 µL of the PCR products were analyzed on a 1.5% agarose gel made in 0.5× of Tris/Borate/EDTA (TBE) buffer and containing ethidium bromide. An electrophoresis unit was used to run the gel for 30 min at 150 V. The migrated bands were observed under UV light and photographed using a gel documentation system. To verify the presence of appropriately sized amplicons, the PCR products for each isolate were compared with a 1 kb DNA ladder. Products of the correct size were purified using a TaKaRa Agarose Gel DNA purification kit Ver. 2.0 and were sequenced in both directions using an ABI 3730 automated sequencer (Macrogen, Korea). The obtained sequences for the selected isolates were aligned and compared with the sequences deposited in GenBank (http://blast.ncbi.nlm.nih.gov/Blast.cgi). To determine the taxonomic position of the isolates, a phylogenetic tree was constructed with MEGA version 5.0 using a neighbor-joining algorithm. The Jukes-Cantor distance estimation method with bootstrap analyses for 1000 replicates was also performed. The nucleotide sequences of the amplified 16S rRNA genes of the strains reported in this study have been deposited in the GenBank nucleotide sequence database under the accession number MG580926.
Immobilization of B. licheniform is by entrapment in Ca-alginate
The freely suspended B. licheniformis cell inoculum was cultured in asparagine broth medium at 37 °C and 250 rpm for 24 h. To prepare the immobilized cell inoculum10, the wet cells were prepared after centrifugation (10,000 × g for 20 min) of the freely suspended cell inoculum and mixed with 50 ml of 3% (w/v) sodium alginate solution, after which the inoculum was stirred for 10 min to make the mixture uniform. The obtained mixture was extruded dropwise through a 5-ml pipette into a 1.5% (w/v) CaCl2 solution. The bacterial cells were entrapped when the alginate drops solidified and formed capsules upon coming into contact with the CaCl2 solution. Subsequently, the capsules were allowed to harden for 1.0 hour, after which they were washed with a sterile saline solution (0.09% NaCl) and preserved in CaCl2 solution in the refrigerator at 4 °C. All operations were performed aseptically under a laminar flow unit.
Scanning electron microscope (SEM)
Surface characterization and microscopic analysis of gold coated free bacterial cells and immobilized calcium alginate capsules were carried out using energy-dispersive analysis in a SEM (Jeol Jsm 6360 LA, Japan).
ASNase production
Erlenmeyer flasks (250 ml) containing 50 ml of asparagine broth medium were inoculated with 4 g of the alginate gel. For the freely suspended cell cultures, the asparagine agar medium was inoculated with bacterial cells equivalent to those used in the immobilized cultures. Fermentation was performed with the free and immobilized cells at 37 °C for three days at 250 rpm. Subsequently, the cultures were centrifuged (10,000 × g for 20 min) and the supernatants were used as the enzyme source. The cell growth in the freely suspended cultures and that of the cells immobilized in the alginate matrix was determined as cell dry weight. The total weight of the bacterial cell-containing alginate gel capsules was compared with the alginate capsules without cells to determine the cell biomass. Several factors affecting ASNase production were optimized by varying one factor at a time, namely, incubation period (12–96 h), pH (5.0–8.0), temperature (25–50 °C), agitation rate (0.0–300 rpm) carbon sources (glycerol, fructose, glucose, maltose, sucrose, dextrin and starch), and nitrogen sources (asparagine, yeast extract, urea, casein, ammonium sulfate, ammonium chloride, sodium nitrate and potassium nitrate).
Determination of ASNase and glutaminase activity
A reaction mixture containing 0.5 mL of enzyme, 0.5 mL of 0.05 M Tris-HCl buffer (pH 7.4), 0.5 mL of 0.04 M asparagine (L-glutamine was used as the substrate to assess glutaminase activity) and 0.5 mL of distilled water to bring the final volume to 2.0 mL was incubated for 30 min. The reaction was stopped by adding 0.5 mL of 1.5 M trichloroacetic acid, and the mixture was then centrifuged at 4025 × g for 10 min. The absorbance of a mixture containing 0.1 mL of the supernatant, 3.7 mL of distilled water and 0.2 mL of Nessler’s reagent was measured using a spectrophotometer at 500 nm20. One unit of ASNase activity was reported as the amount of enzyme that liberated 1 µmol of ammonia per ml per min.
Protein determination
The method of Lowry et al.21 was used to establish the enzyme concentration. A stock solution of standard protein, bovine serum albumin at a concentration of 1000 μg/mL, was prepared. Folin-Ciocalteu reagent was added to each sample, and after incubating for 30 min, the absorbance was measured at 660 nm.
Purification of ASNase
Ammonium sulfate was added to the cold crude enzyme to reach a saturation of 70%. The mixture was left overnight at 4 °C and then was centrifuged at 4025 × g for 20 min. The precipitate obtained was dissolved in the minimum volume of 50 mM Tris-HCl buffer (pH 7.4) and was dialyzed against the same buffer overnight at 4 °C to remove the salts. The dialyzed fraction was loaded onto a Sephadex G-100 column (45 × 1.5 cm) that was pre-equilibrated with 0.05 M Tris-HCl buffer (pH 8.6). The protein was eluted with 0.05 M Tris-HCl buffer (pH 7.4) containing 0.1 M KCl. The fractions were collected and assayed for protein and enzyme activity22. The fraction with the highest level of enzyme activity was lyophilized, and the resulting powder was stored at 4 °C.
Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE)
To generate whole cell lysates, cells were resuspended in 200 µL of lysis buffer (62.5 mM Tris/HCl, pH 6.8) (Mllinckrocit, USA), 2% sodium dodecyl sulfate (SDS), 15% glycerol, 5% 2-mercaptoethanol and 0.001% bromophenol blue (tracking dye). The whole cell lysates were boiled for 5 min at 100 °C and then were loaded onto a 15% SDS polyacrylamide gel and run at 100 V. The separated proteins were stained with Coomassie blue. Subsequently, the gel was transferred to de-staining solution to remove unbound dye from the gel and leave the stained proteins as visible blue bands, after which the gel was imaged using a Gel Documentation System.
Kinetic properties of the purified ASNase
The optimum pH for ASNase activity was determined over a pH range of 5.0 to 10.0. For pH stability studies, enzyme preparations at various pH values were incubated for 1 h, and then, the relative activity was determined under the standard assay conditions. The optimum temperature for enzyme activity was determined by assaying ASNase activity at different temperatures ranging from 30 to 100 °C. In addition, the thermal stability of the enzyme was determined by incubating the lyophilized enzyme at the same temperatures for 1 h and assaying ASNase activity3.The kinetic parameters for ASNase (Km and Vmax) were determined using L-asparagine as a substrate at concentrations ranging from 0.0 to 0.1 M. The Michaelis-Menten parameters were determined from Lineaweaver-Burk plots using the equation derived from linear-regression analysis of the curve3.
Anticancer assay
The cytotoxicity assay23 was performed using human cancer cell lines) MCF-7 breast, HCT-116 colon and HepG2 live) obtained from Vacsera (Egypt). The cells were maintained in Roswell Park Memorial Institute (RPMI) media supplemented with 100 µg/mL streptomycin, 100 U/mL penicillin and 10% heat-inactivated fetal bovine serum in a humidified atmosphere containing 5% CO2 (v/v) at 37 °C. In addition, the cells were sub cultured twice a week. The cytotoxicity of the enzyme was tested against selected cancer cell lines using the sulforhodamine B (SRB) assay. Exponentially growing cells were collected using 0.25% trypsin-EDTA and plated in 96-well plates at 2000 or 100 μL/well. The cells were exposed to different concentrations of ASNase (0.0–100 μg/mL) for 72 h and were subsequently fixed with 100 μL/well of trichloroacetic acid (10%) for 1 h at 4 °C. After extensive washing, the cells were exposed to a 0.4% SRB solution for 10 min in a dark place and then were washed with 1% glacial acetic acid to remove unbound dye. After drying overnight, the SRB dye was solubilized with 50 μL/well of 10 mM Tris-HCl (pH 7.4) for 5 min on a shaker at 1600 rpm. The optical density (OD) of each well was measured at 570 nm using a microplate reader. Control wells contained only cells and medium without treatment, and the positive control was doxorubicin. The IC50 values were calculated using sigmoidal concentration response curve fitting models that were generated with Sigma Plot.
Statistical analysis
Analysis of variance using one-way ANOVA was performed, and all significant differences at P ≤ 0.05 were determined using Minitab for Windows version 15. The error bars represent the standard error of the mean for n = 3. Sigma Plot was used to calculate the IC50 values.
Compliance with ethical standards
This article does not contain any studies with human participants or animals performed by any of the authors.
Results
Isolation and screening of marine bacteria producing ASNase from the Red Sea
Forty marine bacterial isolates were isolated from the three marine coastal sites via the Millipore membrane filter method while, serial dilution method was not feasible. The greatest number of bacteria were isolated from the Al-Haridah coast (15 isolates), followed by the Al-Birk coast (14 isolates) and the Al-Shuqaiq coast (11 isolates). All bacterial isolates were screened for their ability to produce ASNase using a qualitative rapid plate assay based on their ability to form a pink zone around colonies on asparagine agar plates containing a phenol red indicator. Twenty-eight isolates showed intense pink zones around the colonies. Four bacterial isolates exhibited a notable ability to produce ASNase as assessed by the diameters of the halos, including KKU-KH14 (1.75 mm) and KKU-KH17 (2.37 mm) from the Al-Haridah coast, KKU-KSh6 (2.0 mm) from the Al-Shuqaiq coast and KKU-KB 27 (1.7 mm) from the AL-Birk coast. By evaluating the morphological properties of the colonies displaying ASNase production potential through the plate assay, 10 isolates were selected for the subsequent in-depth studies.
ASNase production by free and immobilized cells of 10 promising bacterial isolates
The bacterial isolates with promising ASNase activity were subjected to further screening for the production of ASNase through submerged fermentation in a shaking incubator using two inoculum methods, freely suspended and alginate-immobilized bacterial cells. ASNase production was measured via spectrophotometry using Nessler’s reagent method. The results (Fig. 1) indicate that the immobilized cells exhibited greater enzyme production than the free cells. The amount of enzyme produced by free bacterial cells ranged from 1.56 to 8.1 U/mL, whereas that produced by immobilized cells ranged from 2.13 to 11.66 U/mL. The highest enzyme production was achieved by the bacterial isolate KKU-KH14 obtained from the Al-Haridah coast, producing 8.1 U/mL using the freely suspended cell inoculum and 11.66 U/mL using the immobilized cell technique. The morphological appearance of the free and immobilized bacterial cells in calcium alginate capsules were investigated using high power magnifications of a scanning electron microscope (SEM). The photographed free cells showed the bacilli appearance of the B. licheniformis strain (Fig. 2A). Additionally, no other bacterial morphologies were detected, which indicates the purity of the used bacterial sample. On the other hand, the spherical shape of the alginate capsules was a clear sign for the proper polymerization of the alginate polymer using calcium ions as a cross linker. Moreover, the proper gold coating of the capsules was under a condition of complete dryness of the scanned capsules, which in turn kept the bacterial cells inside the alginate capsules (Fig. 2B). The investigation of the bacterial cells inside the polymeric capsule was detected using a semi sphere of the prepared capsule (Fig. 2C). The bacterial cells were detected inside the polymeric semi sphere with slightly different shape than seen for free cells. The difference in the bacterial bacilli shape may be attributed to the complete coverage of the cells with the viscous polymeric material that transformed the bacterial shape into slight spherical shape (Fig. 2D). These results indicated the proper immobilization of the bacterial cells inside the polymeric capsules, in addition to the predicted prolonged shelf life of the dried immobilized capsules which have the ability to allow the bacterial cells to restore their activity after exposure to humidity conditions.

ASNase production by freely suspended and immobilized cells of promising bacterial isolates. 1: KKU-KH12, 2: KKU-KH14, 3: KKU-KH16, 4: KKU-KH17, 5: KKU-KH18, 6: KKU-KH20, 7: KKU-KH 38, 8: KKU-KB27, 9: KKU-KB36, 10: KKU-KSh6. The values in the same column followed by the letter (S) were not significantly different at P ≤ 0.05.

Scanning electron micrograph showing (A) free B. licheniformis cells (B) a capsule of immobilized bacterial cells in alginate polymeric material, (C) a semi-sphere capsule and (D) bacterial cells inside the cavities of the capsule.
Identification of strains via 16S rRNA gene sequencing and phylogenetic analysis
To identify and determine the correct phylogenetic position of the selected promising isolate, KKU-KH14, molecular genetic identification was performed. First, the genomic DNA was extracted from the isolated bacterial strain using a modified QIAamp DNA Mini kit. Amplification and sequencing of the 16S rRNA gene fragment was performed by Macrogen, Korea, using the universal primers 27F and 1492R. The obtained sequence data of the16S rRNA gene was compared with the sequences of 16S rRNA regions in GenBank using a BLAST search of the National Center for Biotechnology Information (NCBI) databases (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastn&PAGE_TYPE=BlastSearch&LINK-LOC=blasthome). An alignment of the 16S rRNA gene sequence of the isolate, along with sequences obtained from the BLAST search and MACROGEN results, revealed that the strain KKU-KH14 exhibited 99% similarity to B. licheniformis. Various sequences taken from the GenBank database were used to build the phylogenetic tree to determine the phylogenetic position of the strain (Fig. 3) and the accession number of bacterial strain sequence information is MG580926.

Phylogenetic relationship between the KKU-KH14 bacterial isolate and other 16S rRNA gene sequences of published strains.
Optimization of ASNase production
The optimization of ASNase production by immobilized cells of B. licheniformis was investigated using submerged fermentation in an incubator shaker. The cells were cultured in asparagine medium with an initial pH of 6.5 at 37 °C and 250 rpm.
Effect of time course
The results indicated that the growth of B. licheniformis cells entrapped in a calcium alginate matrix increased gradually over a period of up to 72 h of culturing at the stationary growth phase (Fig. 4). After 24 h of incubation, the production was only 19.10% of the maximal observed value, gradually increasing up to 36 h (30.84%), with exponential increase was observed at 72 h. Additional culturing after the optimum culture period did not increase enzyme production but rather strongly decrease in enzyme production and bacterial growth were observed.

Effect of time course. The values indicated with the same letter (s) on the same line were not significantly different at P ≤ 0.05.
Effect of initial pH
The enzyme production was altered according to the initial pH of the medium. The enzyme production increased up to pH 6.5, which was the most favorable value for ASNase production and bacterial growth (Fig. 5). However, beyond this pH value, the production markedly decreased. The ASNase production at pH 6.0 was comparable to that observed at pH 6.5, as the yield decreased by only 5.11%, while at pH 7.5 the yield decreased by 18.77%.

Effect of the initial pH. The values indicated with the same letter (s) on the same line were not significantly different at P ≤ 0.05.
Effect of incubation temperature
The results demonstrated a significant relationship between ASNase production and the incubation temperature up to 37 °C, where an ASNase yield of 11.74 U/ml was observed (Fig. 6). A significant reduction in enzyme production was observed compared to the optimum temperature value, decreasing by 5.19 and 32.19% at 42 and 47 °C, respectively.

Effect of the incubation temperature. The values indicated with the same letter (s) on the same line were not significantly different at P ≤ 0.05.
Effect of agitation rate
The maximal enzyme production was increased by agitation, with enzyme yield significantly increasing with shaking at 250 rpm (Fig. 7). Under static conditions, enzyme production decreased, reaching 11.93% of the maximal observed value. Furthermore, increasing the agitation rate above the optimum value was unfavorable for ASNase production.

Effect of the agitation rate. The values indicated with the same letter (s) on the same line were not significantly different at P ≤ 0.05.
Effect of carbon sources
Experiments were performed to assess the ability of the B. licheniformis isolate to utilize different carbon sources (glycerol, fructose, glucose, maltose, sucrose, dextrin and starch) for the production of ASNase. The carbon source used had a clear effect on ASNase production, with glucose being the most preferred carbon source for enzyme production, reaching 11.2% more than the maximal observed value (Fig. 8). Interestingly, the other tested carbon sources did not facilitate bacterial growth and enzyme production, with the lowest production of ASNase observed using glycerol and starch as carbon sources, reaching 40.36 and 53.31% less than the maximal observed value, respectively.

Effect of carbon sources. The values indicated with the same letter (s) in the same column were not significantly different at P ≤ 0.05.
Effect of nitrogen sources
The effect of different nitrogen sources (asparagine, yeast extract, urea, casein, ammonium sulfate, ammonium chloride, sodium nitrate and potassium nitrate) on ASNase production was investigated. The results showed that ammonium salts were the best source of nitrogen with respect to ASNase yield (Fig. 9) Ammonium sulfate was observed to be the optimum source of nitrogen for B. licheniformis, resulting in 35.56% increase in enzyme production. The yeast extract, urea and casein did not show any significant changes in enzyme production, whereas nitrate salts, sodium nitrate and potassium nitrate significantly reduced enzyme production between 71.01 and 72.86% of the maximal observed value.

Effect of nitrogen sources. The values indicated with the same letter (s) in the same column were not significantly different at P ≤ 0.05.
Purification of ASNase
ASNase extracted from B. licheniformis was purified by ammonium sulfate precipitation, dialysis and gel filtration chromatography using a Sephadex G-100 column (Table 1). In the twelve fractions obtained from the cell free extract, one fraction exhibited ASNase activity. The specific activity of the enzyme and the purity increased with every step of purification, whereas the total protein, total activity and yield decreased proportionally. For the crude enzyme, a yield of 114.4 mg of protein and 890 U of ASNase activity were obtained, with a specific enzyme activity approximately 7.78 U/mg. The purification of the crude enzyme yielded 11.5 mg of protein and 415 U of enzyme activity, with an increased specific activity of 36.08 U/mg. Furthermore, a 4.63-fold purification and a 46.62% yield of the enzyme were obtained.
The homogeneity and the molecular weight of the purified ASNase was determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The results demonstrated the purity of the ASNase preparation, as only a single distinctive protein band was observed with an apparent molecular weight of 37 kDa when compared with the standard molecular weight markers (Fig. 10). The SDS-PAGE and glutaminase activity assay results of the ASNase protein demonstrated the success of the enzyme purification and showed that the purified enzyme exhibited no glutaminase activity.

SDS-polyacrylamide gel electrophoresis of the purified ASNase from B. licheniformis. M: Protein marker; 1: Sephadex G-100-purified ASNase; 2: Ammonium sulfate-precipitated ASNase; 3: Cell free crude ASNase preparation.
Kinetic properties of the purified ASNase
The enzyme was active over broad pH range, with its activity gradually increasing up to pH 7.0 (Fig. 11a). The enzymatic activity decreased at other than optimal pH values, with up to 31.63 and 18.47% of the maximal activity retained at pH values of 9.0 and 5.0, respectively. The purified enzyme was stable at a pH range of 7.0 to 8.5, losing only 7.5 and 26.60% of its original activity at pH values of 9.0 and 10.0. The maximal activity of the purified ASNase was recorded at 37 °C, with the enzyme activity gradually decreasing at temperatures above this value (Fig. 11b). The lowest activity of the ASNase was observed at 80 °C, where an activity of 13.90 U/mL was measured. The ASNase was exhibiting thermostability over a temperature range of 30–70 °C. The parameters Km and Vmax were determined for the purified ASNase through a steady-state kinetic analysis (Fig. 11c). The plot of the reaction velocity versus substrate concentration exhibited a typical hyperbolic saturation curve that was fitted to the Michaelis-Menten equation, yielding the kinetics constant. The Lineweaver Burk plot showed that the Km and Vmax values of enzyme using L-asparagine as substrate were 0.049995 M and 45.45 μmol/ml/min, respectively.

Effect of pH (a) and temperature (b) on the activity and stability of ASNase. The values indicated with the same letter (s) were not significantly different at P ≤ 0.05. The plot of the reaction velocities (V) vs. substrate concentration fitted to the Michaelis-Menten equation and determination of the Km and Vmax values of purified ASNase by Lineweaver-Burk plot (c).
Anticancer activity of ASNase against cancer cell lines
The viability (%) of MCF-7, HepG-2 and HCT-116 cell lines was used as an indicator of cell toxicity after cells were treated with different concentrations of ASNase produced by B. licheniformis for 72 h. The resulting toxicity of the enzyme toward MCF-7, HepG-2 and HCT-116 cells was dose dependent since gradual increases in the dose of the ASNase enzyme resulted in a gradual inhibition of cell growth (Fig. 12). The purified ASNase enzyme was shown to selectively inhibited cancer cell reproduction without any cytotoxic effect toward the non-carcinogenic human cell line (normal cells) at the concentrations studied. Morphological changes in the cancer cell lines after treatment with bacterial ASNase were evaluated under an inverted microscope. The cells showed signs of detachment from the surface of the wells, which denoted cell death. The apoptotic cells also showed specific characteristics, such as cellular rounding, shrinkage, membrane blabbing and loss of cell adhesion. The cytotoxic effect of the B. licheniformis ASNase showed significant toxic activity toward HepG-2 cells (IC50 value of 11.66 µg/mL), which was more than the effect observed against MCF-7 and HCT-116 cells (IC50 values of 14.55 and 17.0226 µg/mL, respectively).

Dose response curves of B. licheniformis against MCF-7, HepG-2 and HCT-116 cancer cells.
Discussion
The current commercially available ASNases produced by bacteria can lead to hypersensitivity and toxicity during therapy, demonstrating the need to identify new sources of these enzymes24. In the present study, 40 marine bacterial isolates were obtained from the Red Sea coast in the southern region of Saudi Arabia using a Millipore filter method while the serial dilution method was unfavorable in view of the difficulty of isolating marine bacteria since most them uncultured. The marine environment of the Red Sea is a potent source of bacterial species that may be useful in the discovery of novel bioactive compounds7,25. In our study, 28 marine bacterial isolates showed the ability to produce ASNase to different extents. Marine bacteria are believed to be a promising source of anticancer ASNases through the use of submerged fermentation, which has unique characteristics26. Additional screening was conducted to assess ASNase production using freely suspended and immobilized cell techniques. The production of enzyme by immobilized bacterial cells was greater than that observed by the freely suspended cells, with an approximately 43.9%. Kattimani et al.27 reported that enzyme production of immobilized Streptomyces gulbargensis cells was enhanced by approximately 30% compared to conventional free cell fermentation. The grater ASNase production by immobilized cells could be due to the increased growth of cells in the alginate matrix, as well as the enzyme activities being maintained at higher levels than those of the free cells28. Cell immobilization is considered a promising approach for enhancing the production of ASNase, as this approach offers numerous benefits, including a sustained process over a long period of time, the re-use of immobilized beads, easy harvesting of products, reactor productivity, higher catalytic efficiency and lowered processing costs7.
The SEM photographed of the free and immobilized bacterial cells in calcium alginate capsules indicates the purity of the bacterial strain and the proper immobilization of the bacterial cells inside the polymeric capsules, in addition to the predicted prolonged shelf life of the dried immobilized capsules which have the ability to allow the bacterial cells to restore their activity after exposure to various conditions29.
Molecular identification of the bacterial 16S rRNA gene and phylogenetic analysis confirmed the classification of the selected isolate, KKU-KH14 as B. licheniformis, which belongs to the phylum Firmicutes. The predominant bacterial phyla in the Red Sea are Proteobacteria, Firmicutes, Fusobacteria, Bacteroidetes and Spirochetes30. ASNases have been identified in several marine bacteria while, the most abundant bacterial isolates were Bacillus sp. and Pseudomonas sp31. The abundance of Bacillus sp. throughout marine habitats is associated with its ability to produce spores, which exhibit high resilience to environmental stresses32.
The optimization conditions of ASNase production by the B. licheniformis identified in this study were in agreement with the results of a study by Kumar et al.33, who reported that ASNase production is strongly influenced by the composition of fermentation media and the culture conditions, including time, temperature, pH and agitation rate. The maximal production of the ASNase investigated in this study was observed at 72 hours, in the stationary phase of bacterial growth, indicating that enzyme production and bacterial growth were linked. The culturing period plays an essential role in ASNase productivity by bacterial strains, with optimal production observed for S. marcescens and B. methylotrophicus at 96 hours34, for Bacillus sp. R36 at 24 hours35, for Lactobacillus salivarius at 120 hours26 and for Bacillus subtilis at 36 hours36.
The pH of a medium significantly impacts numerous enzyme processes and the movement of different components across the microbial cell membrane37. The most suitable pH for ASNase production is dependent on the specific strain, differences in which could be due to fermentation conditions and the specific genetic characteristics of a microbial species38. In this study, the maximum ASNase production occurred at pH 6.5, a result that is similar to that observed by Pradhan et al.36, who reported that ASNase production by Bacillus subtilis occurred optimally at 6.5 pH. Moreover, Mahajan et al.39 reported that ASNase production by B. licheniformis occurs over a pH range of 5.5–7.5, with maximum enzyme production occurring at pH 6.0.
Maximum enzyme productivity of the ASNase evaluated in this study was obtained at 37 °C, similar to the ASNase from Bacillus subtilis36, whereas the optimal temperature for the production of enzyme by Streptomyces olivaceus was 35 °C38. In contrast, the optimal production of ASNases from Bacillus PG02 and Bacillus PG04 occurred at pH 6–7.5, which is close to physiological pH40. This feature is a major requirement for the antitumor activity of ASNases with optimal temperatures of 37 °C.
The agitation process is essential for optimal bacterial growth and ASNase production38. The optimal agitation rate for enzyme production was observed at 250 rpm. Because microorganisms growing in a submerged culture use oxygen dissolved in the medium, this is critical for the growth requirements of the organism and biosynthesis of specific end products. Maysa et al.35 revealed that oxidation-reduction mechanisms in the fermentation mixture may exert a chemical influence on the biosynthetic of ASNase productivity. The best agitation rate for ASNase production was determined to be 200 rpm for Streptomyces olivaceus38, 150 rpm for Bacillus subtilis36 and 250 rpm for Lactobacillus salivarius26.
The nutritional requirements for maximal ASNase synthesis varies among microorganisms, and the rate of synthesis varies in the same organism as a function of culture conditions6. Different carbon sources in media formulations were used to enhance growth and the synthesis of primary metabolites, such as enzymes. The ASNase production in cells of B. licheniformis was observed to depend on specific factors. Glucose was the best carbon source for the bacterial growth and ASNase production, similar to the results reported for Bacillus sp.41. In contrast, Wakil and Adelegan42 showed that the optimal carbon source was mannitol for ASNase production in Bacillus circulans, Streptococcus sp. and Bacillus polymyxa; maltose for Streptococcus sp. and Bacillus firmus; and sucrose for Paenibacillus validus, which also aided in stabilizing the enzyme.
Organic and inorganic nitrogen sources are microbial metabolized to produce amino acids, nucleic acids, proteins, and cell wall components to enhance the production of ASNase. The optimum nitrogen source for ASNase production in this study was (NH4)2SO4. Hosamani and Kaliwal43 reported that ammonium sulfate was the optimum nitrogen source for ASNase production in B. licheniformis, while Baskar and Renganathan44 reported that ammonium chloride was more favorable. In a previous study, the nitrogen source was the second most crucial factor affecting ASNase production and was also important for the growth of organisms42. The authors showed that the ideal nitrogen source for ASNase production by Bacillus circulans and Streptococcus sp. was casein, yeast extract for Bacillus polymyxa and Bacillus firmus, and NaNO3 for Streptococcus sp. and Paenibacillus validus. Yeast extract and casein were shown to promote cell growth and ASNase production, while increased concentrations of these components resulted in decreased ASNase synthesis, which may be due to the presence of a high substrate concentration and the induction of proteolytic enzyme42.
An ASNases was purified by ammonium sulfate precipitation, dialysis and gel filtration chromatography using Sephadex G-100 chromatography22. The precipitation ASNases via ammonium is the most widely used method to purify these enzymes, followed by dialysis to remove excess salt. Furthermore, complete purification of ASNases has been achieved using chromatographic techniques45. Specific activity (units/mg) is a measure of enzyme purity, the value of which increases as the purity of an enzyme increases since the amount of contaminating protein typically decreases. The results of this study agreed with the findings of Farag et al.7, where a Streptomyces fradiae NEAE-82 ASNase was purified and exhibited a final specific activity of 30.636 U/mg protein and was purified 3.33-fold29.
An SDS-PAGE analysis of the purified ASNase showed a single distinct protein band with a molecular weight of 37 kDa (Fig. 12). These results were in accordance with other reports46,47 for ASNases from B. licheniformis. The molecular weights of ASNases varies according to the microbial source of enzyme. The molecular weight of an ASNase from Streptomyces fradiae NEAE-82 was determined to be approximately 53 kDa29. In contrast, SDS-PAGE analysis of a purified ASNase from Pseudonocardia endophytic VUK-10 revealed a distinct single peptide chain with molecular weight of 120 kDa22. A purified Vibrio cholerae ASNase that was recombinant overexpressed in E. coli exhibited a molecular weight of approximately 36.6 kDa1. The results of SDS-PAGE and glutaminase activity assay for the purified ASNase in this study indicated that the enzyme was pure and free of glutaminase activity. These results were similar to those observed in numerous previous reports that described B. licheniformis as producing ASNases with low or no glutaminase activity39,46,47. The concern associated with the commercial use of ASNases is its high glutaminase activity, which can cause liver dysfunction, pancreatitis, leucopenia, neurological seizures, and coagulation abnormalities that lead to intracranial thrombosis13,14,46.
The optimal pH and temperature at which the optimal activity and stability of the ASNase evaluated in this study, showed that the enzyme could function well in the human body. Enzymes that are stable at physiological temperature and pH is a desirable characteristic for proteins that are used as therapeutics. Our results were similar to those of Ghasemi et al.,7 who observed that the optimum temperature for the activity of an ASNase from the marine bacterium Halomonas elongata was 37 °C, while the enzyme showed maximum activity over a wide pH range (6–9) that was similar to values observed in the human body. An ASNase from B. licheniformis was maximally active over a pH range of 6.0 to 10.0 at a temperature of 40 °C, with maximal enzyme stability observed at pH 9.046. In addition, El-Naggar et al.29 reported that ASNase are amidases that are generally active and stable at neutral and alkaline pH. They observed that the optimum temperature for the activity and stability of an ASNase from Streptomyces fradiae NEAE-82 occurred at 40 and 50 °C, respectively. In addition, the ideal temperature for the activity of a B. licheniformis ASNase occurred at 37 °C. The observed increase in the thermostability of this enzyme may be explained by changes in the charges of specific amino acids, which can destabilize the proteins47.
In this study, the kinetic constants Km and Vmax of the purified ASNase were determined by steady-state kinetic analysis fitted to the Michaelis-Menten equation. The Km reflects the affinity of the enzyme for its substrate while Vmax is a measure of substrate turnover and is expressed in units of product formed per unit of time22. The Lineweaver Burk plot results showed a high affinity of the enzyme toward the substrate, with a measured Km value of 0.049995 M and a Vmax value of 45.45 μmol/ml/min. Many factors affect the kinetic parameters of enzymes, such as the source, type, and form of an enzyme, as well as changes in enzyme conditions and assay procedures7. Sudhir et al.47 reported Km and Vmax values for an ASNase from B. licheniformis of 0.42 mM and 2778.9 μmol min, respectively, while Km and Vmax values of the enzyme purified from the same bacterium were measured as 1.4 × 105 M and 4.03 IU, respectively46. In addition, the Km and Vmax values for an ASNase from Streptomyces fradiae NEAE-82 were 0.01007 M and 95.08 Uml−1min−1, respectively29.
Cancer rates are rising worldwide, and breast cancer is considered to be the second leading cause tumor-related deaths in women in Saudi Arabia48, with colon cancer being the third leading cause of death, and a high mortality rate observed for breast cancer worldwide49. Liver malignancy is another common diagnosis in middle-eastern patients who suffer from chronic liver disease50. The toxicity of the ASNase produced by B. licheniformis in this study was evaluated for cancer cell lines, the results of which showed that the enzyme was highly toxic toward HepG-2 cells, more so than against MCF-7 and HCT-116 cells. These findings were comparable to those of other studies46,51, where ASNases from marine Bacillus sp. and B. licheniformis strains exhibited the highest cytotoxic effect against the cancer cell lines Jurkat clone E6-1, MCF-7 and K-562, with IC50 values of 0.22, 0.78 and 0.153 IU, respectively. However, the results of the current study were superior to those described previously7 for Halomonas elongate, where an ASNase inhibited the growth of human leukemia cell lines with IC50 values of 1–2 U/mL. Furthermore, an ASNase from Bacillus sp. R36 inhibited the growth of two human cell lines (HCT-116 and HepG-2), with IC50 values of 218.and 112.19 μg/mL, respectively35. In addition, Shafei et al.52 described an ASNase that inhibited the growth of three human cell lines, with values of IC50 values of 12.5, and 37 μg/mL against breast, hepatocellular and prostate carcinoma, respectively. ASNases have been shown to inhibit the glycosylation of several forms of newly synthesized proteins53. El-Naggar et al.29 suggested that ASNases can alter the interactions between the microvasculature of endothelial cells, colon cancer cells, and extracellular matrix components, resulting in colon cancer cell disruption. After being converted into oxaloacetic acid, asparagine is involved in the Krebs cycle, influencing cell metabolism54. In addition, intracellular asparagine contributes to the uptake of extracellular serine, which is utilized to synthesize nucleic acids55. Since the multiplication of the human leukemic cell lines and other tumor cells requires excessive amounts of these amino acids to produce sufficient energy and synthesize biomolecules, decreases in the supply of asparagine are likely to prevent the growth of these cells56. The microscopic observations made in this study further confirmed our results. Apoptotic cells exhibit condensed nuclear heterochromatin and partitioning of the cytoplasm and nucleus into membrane-bound vesicles25,57. In addition, Karpel-Massler et al.2 observed that cancer cell lines are susceptible to the repression of proliferation by ASNase, which stimulates apoptosis by decreasing mitochondrial membrane potential. Muñoz-Pinedo58 elucidated the action of anticancer enzymes that catalyze the conversion of specific amino acids into a form that is unavailable to cells, leading to starvation conditions. In addition, Ueno et al.59 showed that tumor cells do not have an L-asparagine synthetase gene and cannot produce L-asparagine such that they will starve and cause apoptosis to occur, and the absence of asparagine disrupts the cell cycle.
Conclusion
The results of this study demonstrate the potential of the marine environment of the Red Sea as a potent source of bacterial strains for the discovery of anticancer drugs. The identification of strains with improved ASNase yields has attracted a great deal of attention to improve the cost-effectiveness of these enzymes. Efficient production of ASNase was achieved through Ca-alginate immobilization of B. licheniformis cells. The rate of ASNase synthesis depends on complex interactions among pH, temperature, agitation and the presence of specific nutrients. The properties, kinetic parameters, and anticancer activity of the purified enzyme demonstrated its excellent potential for use as a potent anticancer agent. However, more detailed studies are needed to evaluate this possibility, including in vivo immunogenicity and half-life determination assays, pharmacokinetic and pharmacodynamic profiling in model animals, and human clinical trials.
Data Availability
The datasets generated during this study are available from the corresponding author upon reasonable request. The data that support the findings of this study are available from GenBank and the KKU microbial collection.
References
Radha, R., Arumugam, N. & Gummadi, S. N. Glutaminase free L-asparaginase from Vibrio cholerae: Heterologous expression, purification and biochemical characterization. Int. J. Bio. Mac. 111, 129–138 (2018).
Karpel-Massler, G. et al. Metabolic reprogramming of glioblastoma cells by L-asparaginase sensitizes for apoptosis in vitro and in vivo. Oncotarget 7, 33512–33528 (2016).
Ghasemi, A., Asad, S., Kabiri, M. & Dabirmanesh, B. Cloning and characterization of Halomonas elongata L-asparaginase, a promising chemotherapeutic agent. Appl. Microbiol. Biotech. 101, 7227–7238 (2017).
Martinez-Outschoorn, U. E., Peiris-Pagés, M., Pestell, R. G., Sotgia, F. & Lisanti, M. P. Cancer metabolism: a therapeutic perspective. Nat. Rev. Clin. Onco. 14, 11–31 (2017).
Duval, M. et al. Comparison of Escherichia coli–asparaginase with Erwinia-asparaginase in the treatment of childhood lymphoid malignancies: results of a randomized European Organisation for Research and Treatment of Cancer-Children’s Leukemia Group phase 3 trial. Blood 99, 2734–2739 (2002).
Sushma, C., Anand, A. P. & Veeranki, V. D. Enhanced production of glutaminase free L-asparaginase II by Bacillus subtilis WB800N through media optimization. Korean J. Chem. Eng. 34, 2901–2915 (2017).
Farag, A. M., Hassan, S. W., Beltagy, E. A. & El-Shenawy, M. A. Optimization of production of anti-tumor L-asparaginase by free and immobilized marine Aspergillus terreus. Egypt Aqu. Res. 41, 295–302 (2015).
Trilokchandran, B., Agrawal, P. & Krishna, V. Screening studies of microbial species for L-asparaginase production. J. Env. Res. Dev. 10, 712–716 (2016).
Badoei-Dalfard, A. L-asparaginase production in the pseudomonas pseudoalcaligenes strain JHS-71 isolated from Jooshan Hot-spring. Mol. Biol. Res. Commun. 5, 1–10 (2016).
Lopes, A. M. et al. Therapeutic L-asparaginase: upstream, downstream and beyond. Crit. Rev. Biotech. 37, 82–99 (2017).
Nielsen, J. et al. Building a bio-based industry in the Middle East through harnessing the potential of the Red Sea biodiversity. Appl. Microbiol. Biotech. 101, 4837–4851 (2017).
Lakshmi, T. V., Mallika, D. S., Amos, S. J. & Kasturi, K. Marine L-asparaginase: A novel microbial therapeutic approach for cancer. Int. J. Pharm. Sci. Rev. Res. 33, 228–234 (2015).
Kumar, S., Prabhu, A. A., Dasu, V. V. & Pakshirajan, K. Batch and fed-batch bioreactor studies for the enhanced production of glutaminase-free L-asparaginase from Pectobacterium carotovorum MTCC 1428. J. Prep. Biochem. Biotechnol. 47, 74–80 (2017).
Goswami, R., Veeranki, V. D. & Hegde, K. Enhanced production of novel glutaminase free recombinant L-asparaginase II of Erwinia Carotovora Subsp. Atroseptica Scri 1043 in Escherichia coli BL21 (DE3) in a batch and fed-batch culture. Austin. J. Biotechnol. Bioeng. 2, www.austinpublishinggroup.com (2015).
Vimal, A. & Kumar, A. Biotechnological production and practical application of L-asparaginase enzyme. Biotech. Gen. Eng. Rev. 33, 40–61 (2017).
El-Naggar, N. E., Deraz, S. F., El-Ewasy, S. M., Suddek, G. M. Purification, characterization and immunogenicity assessment of glutaminase free L-asparaginase from Streptomyces brollosae NEAE-115. BMC Pharmacol Toxicol. 19, https://doi.org/10.1186/s40360-018-0242-1 (2018).
Kamble, K. D. et al. Characterization of L-asparaginase producing bacteria from water, farm and saline soil. Biosc. Disc. 3, 116–119 (2012).
Gulati, R., Saxena, R. K. & Gupta, R. A rapid plate assay for screening L‐asparaginase producing microorganisms. Lett. in Appl. Microbiol. 24, 23–26 (1997).
Lu, J., Perng, C., Lee, S. & Wan, C. Use of PCR with universal primers and restriction endonuclease digestions for detection and identification of common bacterial pathogens in cerebrospinal fluid. J. Clin. Microbiol. 38, 2076–2080 (2000).
Imada, A., Igarasi, S., Nakahama, K. & Isono, M. Asparaginase and glutaminase activities of micro-organisms. Microbiol. 76, 85–99 (1973).
Lowry, O. H., Rosebrough, N. J., Farr, A. L. & Randall, R. J. Protein measurement with the Folin phenol reagent. J. Bio. Chem. 193, 265–275 (1951).
Mangamuri, U., Muvva, V. & Poda, S. Purification and characterization of L-asparaginase by Pseudonocardia endophytic VUK-10 isolated from Nizampatnam mangrove ecosystem. Int. J. Pharmacy. Pharm. Sci. 8, 281–285 (2016).
Skehan, P. et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Nat. Cancer Inst. 82, 1107–1112 (1990).
Kebeish, R., El-Sayed, A., Fahmy, H. & Abdel-Ghany, A. Molecular cloning, biochemical characterization, and antitumor properties of a novel L-asparaginase from Synechococcus elongatus PCC6803. Biochem. 81, 1173–1181 (2016).
Abdelfattah, M. S., Elmallah, M. I. Y., Hawas, U. W., El-Kassema, L. T. A. & Eid, M. A. G. Isolation and characterization of marine-derived actinomycetes with cytotoxic activity from the Red Sea coast. Asian Pac. J. Trop. Biomed. 6, 651–657 (2016).
Bhargavi, M. & Jayamadhuri, R. Isolation and screening of marine bacteria producing anti-cancer enzyme L-asparaginase. American J. Mar. Sci. 4, 1–3 (2016).
Kattimani, L., Amena, S., Nandareddy, V. & Mujugond, P. Immobilization of Streptomyces gulbargensis in Polyurethane Foam: A Promising technique for L-asparaginase production. Iran. J. Biotech. 7, 199–204 (2009).
Kar, S. & Ray, R. C. Statistical optimization of α-amylase production by Streptomyces erumpens MTCC 7317 cells in calcium alginate beads using response surface methodology. Polish. J. Microbiol. 57, 49–57 (2008).
El-Naggar, N. E. A., Deraz, S. F., Soliman, H. M., El-Deeb, N. M. & El-Ewasy, S. M. Purification, characterization, cytotoxicity and anticancer activities of L-asparaginase, anti-colon cancer protein, from the newly isolated alkaliphilic Streptomyces fradiae NEAE-82. Sci. Rept. 6, 27705431, https://doi.org/10.1038/srep32926 (2016).
Mustafa, G. A., Abd-Elgawad, A., Abdel Haleem, A. M. & Siam, R. Egypt’s red sea coast: phylogenetic analysis of cultured microbial consortia in industrialized sites. Front Microbiol. 5, 1–11 (2014).
Izadpanah, F., Homaei, A., Fernandes, P. & Javadpour, S. Marine microbial L-asparaginase: Biochemistry, molecular approaches and applications in tumor therapy and in food industry. Microbiol. Res. 208, 99–112 (2018).
Qeshmi, F. I., Javadpour, S., Malekzadeh, K., Rahimzadeh, M. & Jahromi, S. T. Isolation and molecular identification of L-asparaginase producing bacteria from persian gulf. Iran. J. Pub. Heal. 43, 133–139 (2014).
Kumar, S., Dasu, V. V. & Pakshirajan, K. Localization and production of novel L-asparaginase from Pectobacterium carotovorum MTCC 1428. Process Biochem. 45, 223–229 (2010).
Nongkhlaw, F. M. & Joshi, S. R. L-Asparaginase and antioxidant activity of endophytic bacteria associated with ethnomedicinal plants. Ind. J. Biotech. 14, 59–64 (2015).
Maysa, E. M., Amira, M., Gamal, E., Sanaa, T. & Sayed, E. I. Production, immobilization and anti-tumor activity of L-asparaginase of Bacillus sp. R36. J. Ameri. Sci. 6, 157–165 (2010).
Pradhan, B., Dash, S. K. & Sahoo, S. Screening and characterization of extracelluar L-asparaginase producing Bacillus subtilis strainhswx88, isolated from Taptapani hotspring of Odisha, India. Asian Pac. J. Trop. Biomed. 3, 936–941 (2013).
Sivasankar, P. et al. Efficient production of L-asparaginase by marine Streptomyces sp. isolated from Bay of Bengal, India. Afri. J. Microbiol. Res. 7, 4015–4021 (2013).
El-Naggar, N. E., Moawad, H., El-Shweihy, N. M. & El-Ewasy, S. M. Optimization of culture conditions for production of the anti-leukemic glutaminase by newly isolated Streptomyces olivaceus NEAE-119 using response surface methodology. Biomed Res. Int. 2015, 627031, https://doi.org/10.1155/2015/627031 (2015).
Mahajan, R. V., Saran, S., Kameswaran, K., Kumar, V. & Saxena, R. K. Efficient production of L-asparaginase from Bacillus licheniformis with low-glutaminase activity: optimization, scale up and acrylamide degradation studies. Biores. Tech. 125, 11–16 (2012).
Rahimzadeh, M., Poodat, M., Javadpour, S., Qeshmi, F. I. & Shamsipour, F. Purification, characterization and comparison between two new L-asparaginases from Bacillus PG03 and Bacillus PG04. Open Biochem. J. 10, 35–45 (2016).
Moorthy, V., Ramalingam, A., Sumantha, A. & Shankaranaya, R. T. Production, purification and characterisation of extracellular L-asparaginase from a soil isolate of Bacillus sp. Afr. J. Microbiol. Res. 4, 1862–1867 (2010).
Wakil, S. S. & Adelegan, A. A. Screening, production and optimization of L-asparaginase from soil bacteria isolated in Ibadan, South-western Nigeria. J. Basic. Appl. Sci. 11, 39–51 (2015).
Hosamani, R. & Kaliwal, B. B. L-asparaginase an anti-tumor agent production by Fusarium equiseti using solid state fermentation. Int. J. Drug Disc. 3, 88–99 (2011).
Baskar, G. & Renganathan, S. Application of latin square design for the evaluation and screening of supplementary nitrogen source for L-asparaginase production by Aspergillus terreus MTCC 1782. Ind. J. Sci. Tech. 2, 50–54 (2009).
Makky, E. A., Loh, Y. C. & Karim, M. R. Purification and partial characterization of a low molecular weight L-asparaginase produced from corn cob waste. Bio. Agric. Biotech. 3, 265–270 (2014).
Mahajan, R. V. et al. Purification and characterization of a novel and robust L-asparaginase having low-glutaminase activity from Bacillus licheniformis: in vitro evaluation of anti-cancerous properties. PlosOne 9, 99037, https://doi.org/10.1371/journal.pone.0099037 (2014).
Sudhir, A. P., Agarwaal, V. V., Dave, B. R., Patel, D. H. & Subramanian, R. B. Enhanced catalysis of L-asparaginase from Bacillus licheniformis by a rational redesign. Enzyme and Microbial Tech. 86, 1–6 (2016).
Malaikozhundan, B. et al. Biological therapeutics of Pongamia pinnata coated zinc oxide nanoparticles against clinically important pathogenic bacteria, fungi and MCF-7 breast cancer cells. Microb. Path. 104, 268–277 (2017).
Sabit, H., Samy, M. B., Said, O. A. & El-Zawahri, M. M. Procaine induces epigenetic changes in HCT116 colon cancer cells. Gen. Res. Int. 2016(8348450), 1155/2016/8348450 (2016).
Agha, A. et al. Hepatocellular carcinoma is the most frequent final diagnosis of focal liver lesions identified in a cross-sectional evaluation of patients with chronic liver disease in Saudi Arab. J. Cancer Res. 2015, 492782, https://doi.org/10.1155/2015/492782 (2015).
Arjun, J. K., Aneesh, B., Kavitha, T. & Krishnan, K. Therapeutic L-asparaginase activity of bacteria isolated from marine sediments. Int. J. Pharm. Sci. Drug Res. 8, 229–234 (2016).
Shafei, M. S. et al. Purification, characterization and kinetic properties of Penicillium cyclopium L-asparaginase: Impact of lasparaginase on acrylamide content in potato products and its cytotoxic activity. Cur. Tre. Biotech. Pharm. 9, 132–140 (2015).
Yu, M. et al. L‐asparaginase inhibits invasive anangiogenic activity and induces autophagy in ovarian cancer. J. Cell. Mol. Med. 16, 2369–2378 (2012).
Cooper, A. J. et al. ω-Amidase: An underappreciated, but important enzyme in L-glutamine and L-asparagine metabolism; relevance to sulfur and nitrogen metabolism, tumor biology and hyperammonemic diseases. Amino Acids 48, 1–20 (2016).
Krall, A. S., Xu, S., Graeber, T. G., Braas, D. & Christofk, H. R. Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nat. Commun. 7, 27126896, https://doi.org/10.1038/ncomms11457 (2016).
Hashimoto, K., Suzuki, F. & Sakagami, H. Declined asparagine synthetase mRNA expression and enhanced sensitivity to asparaginase in HL-60 cells committed to monocytic differentiation. Anticancer Res. 29, 1303–1308 (2009).
Elmallah, M. I. & Micheau, O. Marine drugs regulating apoptosis induced by tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). Mar. Drugs 13, 6884–6909 (2015).
Muñoz-Pinedo, C., El Mjiyad, N. & Ricci, J. Cancer metabolism: Current perspectives and future directions. Cell Death Dis. 3, https://doi.org/10.1038/cddis.2011.123 (2012).
Ueno, T. et al. Cell cycle arrest and apoptosis of leukemia cells induced by L-asparaginase. Leukemia 11, 1858–1861 (1997).
Acknowledgements
The authors are grateful to the King Abdul-Aziz City for Science and Technology (KACST) for funding this study (Grant number 1-17-01-010-0002).
Author information
Authors and Affiliations
Contributions
All of the authors contributed equally to this work. A.S. wrote the main text of the manuscript and prepared figures, M.Y. and K.A.A.-i. performed the enzyme production experiments, T.T. performed bacterial identification, and A.M. and S.E.E. performed the anticancer assays.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Alrumman, S.A., Mostafa, Y.S., Al-izran, K.A. et al. Production and Anticancer Activity of an L-Asparaginase from Bacillus licheniformis Isolated from the Red Sea, Saudi Arabia. Sci Rep 9, 3756 (2019). https://doi.org/10.1038/s41598-019-40512-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-40512-x
- Springer Nature Limited
This article is cited by
-
Production of highly cytotoxic and low immunogenic L-asparaginase from Stenotrophomonas maltophilia EMCC2297
AMB Express (2024)
-
Unlocking the Potential of Marine Asparaginase Sources
Thalassas: An International Journal of Marine Sciences (2024)
-
Application of sequential design for enhanced l-asparaginase synthesis from Ganoderma australe GPC191
World Journal of Microbiology and Biotechnology (2024)
-
Isolation, Identification, and Characterization of L-asparaginase-Producing Human Commensal Bacterial Strains: A Promising Next-Gen Probiotics
Applied Biochemistry and Biotechnology (2024)
-
Exploration of seaweed degradation potential of the prioritized microbes as a green saccharification technology
Biomass Conversion and Biorefinery (2024)