
Abstract
L-asparaginase has been widely accepted as a standard anticancer drug for acute lymphoblastic leukaemia (ALL). Presently in L-asparaginase biotherapeutic applications, the main focus is developing new L-asparaginase with minimal or without any glutaminase activity to reduce the associated adverse drug reactions. In this study, Bacillus flexus strain SS (NCBI GenBank Accession Number MN420983) was identified as a promising producer of L-asparaginase. L-asparaginase production was optimized by response surface methodology statistical bioprocess modelling, and enzyme yield of 25.08 IU/mL was reached at the bioreactor scale. The purification of L-asparaginase included ammonium sulfate precipitation, ion-exchange chromatography and size exclusion chromatography yielding 5.27-fold purification. The SDS-PAGE and CE-SDS revealed the monomeric L-asparaginase with molecular weight of 33 kDa. The purified enzyme was highly specific to substrate L-Asn and free from glutaminase activity. The anticancer activity of purified L-asparaginase was found specific against tumor cell lines SKBR3 (IC50 = 0.8 µg/mL), WIL2-S (IC50 = 16.2 µg/mL), and TF-1 (IC50 = 47 µg/mL), but not against negative control HUVEC cells. Therefore, L-asparaginase from B. flexus SS with no glutaminase activity could be a potential new candidate for anticancer drug for ALL with reduced adverse effects.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
L-asparaginase is an amidohydrolase enzyme (E.C.3.5.1.1) and has been recognized worldwide for its use in the healthcare, biosensor, and food industries [1, 2]. As an anti-neoplastic drug, it is part of standard chemotherapy therapy for acute lymphoblastic leukaemia (ALL) in paediatric and adult age groups [3]. In addition to ALL, L-asparaginase is also used to treat other malignancies, including lymphosarcoma, chronic lymphocytic leukaemia, Hodgkin disease, acute myelomonocytic leukaemia, reticulosarcoma, and melanosarcoma [4].
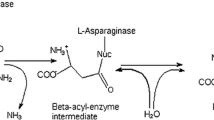
L-asparaginase catalyzes the conversion of L-Asn to Asp and ammonia, which prevents protein synthesis in tumor cells, making it an essential chemotherapeutic agent [5, 6]. The healthy cells can synthesize L-Asn with the help of enzyme asparagine synthetase from the central metabolic pathway making L-Asn a non-essential amino acid in them [3]. However, the tumor cells lack asparagine synthetase. They cannot synthesize L-Asn and depend on an extracellular plasma pool of L-Asn for their cell growth and development. The various approved L-asparaginase drugs are of bacterial origins, like colaspase obtained from Escherichia coli and crisantaspase produced by Erwinia chrysanthemi [7, 8].
The therapeutic uses of currently market approved L-asparaginase preparations reported adverse reactions or toxicity majorly attributable to glutaminase activity and bacterial endotoxin [9, 10]. One of the significant adverse effects in the majority of patients is hepatotoxicity [11]. Other side effects of L-asparaginase therapy include skin rashes, fever, pancreatitis, leucopoenia, neurological seizures, hepatic dysfunction, and abnormal coagulation tests, leading to haemorrhage or intracranial pressure thrombosis [4]. Therefore, a new L-asparaginase with improved immunological characteristics and fewer adverse reactions is highly needed for the current chemotherapeutics portfolio. In order to minimize the adverse reactions associated with L-asparaginase due to glutaminase activity and endotoxin content in the case of the enzyme derived from Gram-positive bacteria, this study aimed to screen bacteria for L-asparaginase production with minimal glutaminase activity. A statistical approach was used for optimization of L-asparaginase production. Subsequently, L-asparaginase was purified, characterized, and its anticancer activity was evaluated against various cancer cell lines (SKBR3 cells, WIL2-S, and TF-1).
MATERIALS AND METHODS
Reagents and chemicals. The chemicals used for enzyme production, i.e., L-Asn, phenol red, Nessler’s reagent, trichloroacetic acid, etc., were procured from Himedia, India. PCR reagents for molecular identification were obtained from New England Biolabs Inc., USA. Cell lines were purchased from ATCC.
Sample collections. Soil sampling was done from 5 different areas of the Himalayan region, including subtropical pine forest (Sarkaghat, Himachal Pradesh, India), rice field (Sadhot, Himachal Pradesh, India), Beas river sediment (Mandi, Himachal Pradesh, India), Ganga river sediment (Devprayag, Uttrakhand, India) and garden area (NOIDA, U.P., India).
Isolation of bacterial strains. The soil samples suspension were prepared by mixing 10 g of soil in 100 mL of sterile 50 mM K-phosphate buffer, pH 8.0. The flasks were shaken at 100 rpm for 10 min and kept undisturbed for 5 min. After centrifugation at 11 000 g for 10 min, the supernatant was serially diluted (from 10−1 to 10−5) and spread on nutrient agar plates having composition of 5 g/L peptone, 5 g/L NaCl, 1.5 g/L yeast extract, and 15 g/L agar powder. The plates were incubated for 24 h at 37°C. The purified isolates were preserved in 20% (vol/vol) sterile glycerol at -20°C.
Screening of bacterial isolates for L-asparaginase production. The qualitative screening of the bacterial isolates for L-asparaginase production was done in M‑9 production medium (g/L): Na2HPO4—6.0; KH2PO4—3.0; NaCl—0.5; L-Asn—10.0; glucose—2.0; MgSO4·7H20—0.5; CaCl2·2H2O—0.005. The medium was supplemented with 2% agar and 0.009% phenol red [12]. After the incubation of 24 h at 37°C, the zone of color change was measured.
L-asparaginase production by submerged fermentation (SmF). The isolates were seeded in 50 mL of production medium in 250 mL Erlenmeyer flasks. The flasks were incubated in a rotatory incubator shaker at 200 rpm and 37°C for 24 h. After incubation, the cells were centrifuged at 11 800 g and 4°C for 30 min and then lysed by sonication at amplitude of 35%, 20 s pulse-off and pulse-on for 15 min (Misonix Ultrasonic Sonicator S-400, Germany). The cell lysate was used for the estimation of L-asparaginase activity.
Enzyme activity assay. L-asparaginase enzyme activity was estimated using the Nesslerization method described previously by Imada et al. [13]. L-Gln was used as a substrate instead of L-Asn to assess the L‑glutaminase activity.
Identification of bacterial isolate. The promising isolate, MSD-11 showing high L-asparaginase and low glutaminase activity, was identified up to the genus level by various phenotypic characteristics, including colony morphology, Gram-staining, culture characteristics, and biochemical reactions as per Bergey’s manual of determinative bacteriology [14].
16S rRNA gene sequencing of MSD-11 and phylogenetic analysis. The extraction of genomic DNA, PCR, purification of the reaction mixture, and sequencing were done according to the method described earlier by Mihooliya et al. [12]. 16S rRNA gene sequencing was performed with an ABI Prism TM3130 XL genetic analyzer (Applied Biosystems, USA) using Big Dye Terminator v3.1 Cycle sequencing kit (Applied Biosystems, ThermoFisher Scientific, USA). The BLAST program analysed the 16S rRNA gene sequence of MSD-11 (https://blast.ncbi.nlm.nih.gov/Blast.cgi). The neighbor-joining method of MEGA 5.1 software was used to construct a phylogenetic tree of the 16S rRNA gene sequence of MSD-11 and other related microorganisms.
Optimization of culture conditions by one-variable-at-a-time (OVAT) analysis. The effect components of medium and bioprocess conditions on the production of L-asparaginase at the shake flask level were investigated initially by the OVAT method. The different physiological parameters were inoculum (1–10% of 1 × 108 CFU/mL), pH (4.0–10.0), temperature (25–45°C), agitation rate (100–350 rpm), 6 production media, and incubation period (6–48 h). The nutritional parameters studied included carbon sources (glucose, fructose, sucrose, maltose, and glycerol) and nitrogen sources (L-Asn, soybean meal, yeast extract, peptone, tryptone). The factors enhancing the L-asparaginase production were further optimized by Plackett-Burman design (PBD) and response surface methodology (RSM).
Selection of significant bioprocess variables using Plackett-Burman design (PBD). In PBD, the effect of 8 factors i.e. A: Na2HPO4 (g/L), B: KH2PO4 (g/L), C: NaCl (g/L), D: L-Asn (g/L), E: MgSO4 (g/L), F: CaCl2 (g/L), G: glucose (g/L), and H: glycerol (mL/L), on production of L-asparaginase were studied. Each variable was kept at low (–) and high (+) levels. PBD was established on the first-order model:
where, Y: response variable (L-asparaginase activity), β0: the model intercept, β0: the linear coefficient, and Xi: the level of the independent variables. All experiments were carried out in triplicates, and the mean activity of L-asparaginase was considered a response.
Bioprocess optimization by RSM containing central composite design (CCD). CCD of RSM was further used to optimize L-asparaginase activity considering 3 variables (A: Na2HPO4, B: KH2PO4, and C: L-Asn), which showed a significant positive effect on L-asparaginase activity in PBD. The variables A, B and C were set at 5 levels (− α, − 1, 0, + 1, + α), and a design of 15 experiments was generated. The variables central values considered in this design were: A (6 g/L); B (4.5 g/L); and C (30 g/L). The ANOVA and 3D response surface curves were used for studying the interactive effect of variables on L-asparaginase activity. All experiments were carried out in triplicates, and the mean activity of L-asparaginase was considered a response. The results of this design were evaluated via the second-order polynomial equation;
where, Y is the response predicted, Xi is the coded level of the independent variable, βij is the interaction coefficients, β0 is the regression coefficients, βii is the quadratic coefficients, and βi is the linear coefficient.
Statistical analysis. Design-Expert® 11.0 software (Stat-Ease Inc., USA) had been used to design experiments and statistical analysis.
Bioreactor scale optimization. The enzyme production was scaled up using optimized medium composition and process conditions in a parallel system bioreactor (2 L) (Dasgip GmbH, Germany) with 1 L working volume. The effect of rate of agitation and dissolved oxygen (DO) concentration on the production of asparaginase was investigated as the agitation effect on mass and heat transfer in a bioreactor could not be the same as compared to those in incubator shaker, and DO concentration was not measured at shake flask level. The bioreactor system contained 3 vessels of 2 L capacity, each operated at an agitation rate of 200, 350 and 500 rpm. Similarly, the DO concentrations of 10, 30, and 50% were maintained in different vessels. Polypropylene glycol as an antifoam agent was used for the control of foaming [15]. The airflow rate of 0.75 vvm was maintained in all bioreactors. The sampling was done at an interval of 3 h for analysis of various parameters.
Purification of L-asparaginase. The cells were harvested by centrifugation at 11 800 g for 30 min. The cell pellet was dissolved in 20 mM Tris-HCl buffer (pH 8.0). The lysis of cells was done using sonication (Misonix Ultrasonic Sonicator S-400, Germany) at 35% amplitude and 20-s pulse on and off. After centrifugation under the same conditions, the supernatant was used for further purification. The ammonium sulfate was added to its saturation level of 70%. The mixture was stored at 4°C overnight, followed by centrifugation at 2000 g for 20 min. The pellet was dissolved in 50 mM Tris-HCl buffer (pH 7.4) and dialyzed at 4°C overnight against the same buffer. The dialyzed protein was further purified by a DEAE-Sepharose resin packed column connected to a preparative chromatography system (Akta, GE Healthcare, USA). The same buffer was used for column pre-equilibration and 50 mM Tris-HCl buffer (pH 7.4) containing 0.1 M KCl was used for elution of the protein. The collected fractions were evaluated for protein concentration by the Lowry method and enzyme activity by the Nesslerization method [13]. The fraction having maximum L-asparaginase activity was lyophilized and stored at 4°C. The purified fraction of protein from ion-exchange chromatography was loaded on Superdex-200 Increase 10/300 column (GE Healthcare, USA) connected with the preparative chromatography system. The column was re-equilibrated with 50 mM Tris-HCl buffer (pH 8.6), and the system was run at 0.5 mL/min.
Characterization of purified L-asparaginase. SDS‑PAGE. The purified samples were suspended in 200 μL 62.5 mM Tris-HCl buffer, pH 6.8 (lysis buffer) containing 2% SDS, 15% glycerol, 5% 2-mercaptoethanol and 0.001% bromophenol blue. The lysates were heated at 100°C for 5 min and then loaded onto SDS polyacrylamide gel (15%) and run at 100 volts. The proteins bands separated were stained with silver staining [16].
Capillary electrophoresis-SDS (CE-SDS). Beckman Coulter PA 800 plus CE system (USA) with 32 Karat software was used for CE-SDS analysis. The bare fused silica capillary (SCIEX, USA) (50 μm ID), having a total length of 30.5 cm and an adequate length of 20 cm, was used to perform separation in CE-SDS. Before separating the sample and SDS-MW size standards (10 kDa to 225 kDa, SCIEX, USA), the capillary was rinsed with 0.1 M NaOH for 5.0 min, 0.1 M HCl for 1.0 min, and with deionized water for 1.0 min at 70 psi. The samples injection was done for 10 s at 10 kV. The proteins were detected at 220 nm by a UV detector with a data rate of 2 Hz [17].
Substrate specificity. The substrate specificity of purified L-asparaginase was investigated with various substrates (L-Asn, L-Gln, L-Asp, D-Gln, D-Asn, D-Asp) at a similar concentration of 25 mM.
Effect of pH and temperature. The purified enzyme activity was investigated at different pH (from 4.0 to 10.0) and at different temperatures (from 10 to 80°C) for obtaining the optima pH and temperature.
Estimation of anticancer activity of purified L-asparaginase. The cancer cell lines used to evaluate the anticancer activity of L-asparaginase were WIL2-S (Human spleen B lymphoblast, ATCC CRL-8885), TF-1 (Human bone marrow erythroblast, ATCC CRL-2003) and SKBR3 (Human Breast Cancer, ATCC HTB-30). HUVEC cells (Himedia, India, CL002) were used as control cells. WIL2-S and TF-1 cell lines were cultivated in RPMI-1640 medium (Sigma-Aldrich, USA) supplemented with 10% fetal bovine serum (FBS) (Sigma-Aldrich, USA). The growth medium for cultivation of SKBR3 cells was McCoy’s 5a medium (Sigma-Aldrich, USA) with 10% FBS and HUVEC cells were cultured in HiEndoXLTM Endothelial Cell Expansion Medium (Himedia, India) using 2% FBS. One hundred U/mL of penicillin and 100 μg/mL of streptomycin were added as antibiotics to the culture media, and cells were maintained at 37°C in a 5% CO2 incubator [18].
The anticancer effect was assessed using the Alamar blue (Sigma-Aldrich, USA) fluorescence-based inhibition of cell proliferation assay. The cell suspension (100 μL containing 2 × 104 cells) was seed per well in a 96-well plate. The cell lines were treated at various L-asparaginase concentrations (from 125 to 0.122 μg/mL) and incubated at 37°C and 5% CO2. WIL2-S and TF-1 cells were incubated for 48 h, whereas HUVEC and SKBR3 cells were incubated for 72 h and 96 h, respectively. After L-asparaginase treatment, 20 μL of the Alamar blue solution were added, and cells were incubated for 4 h at 37°C. The assay was read in a plate reader (Spectramax Gemini, Molecular Devices, USA) at 530 (excitation) and 590 nm (emission). The GraphPad Prism 9 software was used for nonlinear 4PL curve fit analysis and a one-way ANOVA-Dunnet test [18].
RESULTS
Screening and isolation of bacterial strain for L‑asparaginase production. The screening of soil samples from 5 different locations resulted in a total of 113 L-asparaginase-producing bacterial isolates (coded as MSD-1 to MSD-113). The maximum number of bacteria was isolated from the soil sample taken from Beas river sediment at Mandi (30 isolates), followed by those from subtropical pine forest, Sarkaghat (27 isolates), Ganga Beas river sediment at Devprayag (25 isolates), garden area Amity University NOIDA (22 isolates) and rice field of Sadhot (9 isolates). Based on the diameter of the pink zone on phenol red agar plates, 7 bacterial isolates exhibiting a high ability to produce L-asparaginase were selected (Table 1).
Upon quantitative screening, isolate MSD-11 showed the highest L-asparaginase activity of 3.48 IU/mL and the lowest glutaminase activity of 0.03 IU/mL, therefore, it was selected for further characterization studies.
Characteristics of bacterial isolate MSD-11. The colony morphology of isolate MSD-11 on the soybean casein digest agar (SCDA) plate was flat and smooth with undulate margins (Fig. S1). The morphological, physiological, and biochemical characteristics of MSD-11 are listed in Table S1. As per Bergey’s manual of determinative bacteriology, the findings of morphological, physiological, and biochemical tests concluded the MSD-11 as Bacillus species [19].
16S rRNA gene sequencing and phylogenetic analysis. The phylogenetic tree of the 16S rRNA gene sequence of MSD-11 and related microorganisms (Fig. 1) showed that MSD-11 was highly similar to Bacillus flexus strains like B. flexus strain ysm25 (Accession no. KF800780), B. flexus strain SS. SNC 05 (Accession no. MF347994), and B. flexus strain STM18 (Accession no. KY393011) [19].

Phylogenetic tree of 16S rRNA gene sequence of bacterial isolate MSD-11 and related bacterial species constructed by neighbor-joining algorithm using MEGA (version 5.1) software. The 16S rRNA gene sequence (1010 bp) of MSD-11 was submitted at the NCBI GenBank database under the accession number MN420983.
Therefore, as per the morphological, biochemical and 16S rRNA gene sequencing data, MSD-11 isolated from the subtropical pine forest region of Sarkaghat, H.P., India, was identified as B flexus strain SS where SS was used to signify the place of isolation of the strain. The 16S rRNA gene sequence (1010 bp) was submitted at the NCBI GenBank database under the accession number MN420983.
OVAT. In OVAT studies, the effect of bioprocess parameters on production of L-asparaginase at shake flask level revealed the optimum temperature of 37°C, pH of 7.0, inoculum concentration of 4% (vol/vol), an agitation rate of 300 rpm, and incubation time of 24 h in production medium containing (g/L): Na2HPO4—6.0, KH2PO4—3.0, NaCl—0.5, MgSO4·7H2O—0.50, CaCl2·2H2O—0.015, L-Asn—30.0, and glucose—2.0. After OVAT, the enzyme production was 10.47 IU/mL, which increased by 3-fold over that under unoptimized conditions (3.48 IU/mL).
PB experimental design. In PBD, the significance of 8 factors (A – H) was studied by building a 12 runs design (Table S2). In PBD, L-asparaginase activity showed variations ranging from 6.9 to 14.62 IU/mL. Among 8 factors tested, 3 factors, A, B and D, were found to be highly significant upon Pareto chart analysis (Fig. 2). The model was found fit (R2 = 0.9963) and significant (adjusted R2 = 0.9627) (Table S3). The regression equation of PBD factors is as follows:
where Y is L-asparaginase production, and the independent variables are A, B, C and D.

L-asparaginase bioprocess optimization. (a) Plot of predicted versus actual values of L-asparaginase production Plackett-Burman design. (b) The Pareto chart showing the order of significance of the variables in Plackett-Burman design affecting L-asparaginase production. (c) Normal probability plot of internally studentized residuals. (d) Plot of predicted versus actual values of L-asparaginase production in RSM-CCD. The plot of predicted versus the actual values of L-asparaginase production, displaying the data points close to the fitted line supported a significant correlation between the predicted and actual L-asparaginase production results.
RSM experimental design. The CCD of RSM contained 15 experiments for 3 significant variables qualified based on PBD results with low, high, and coded levels (Table S4). The maximum L-asparaginase production (19.85 IU/mL) was obtained in the cultivation medium supplemented with (g/L): Na2HPO4—9.0, KH2PO4—4.5, and L-Asn—30.0. The ANOVA (Table S5) was used for the analysis of the results, and the regression equation for this design was as follows:
where Y is the production of L-asparaginase, and the independent variables are A, B, and C.
The ANOVA results showed a model F-value of 169.62, and probability F-value (p > F) less than 0.0001 indicated that this model was a significant and proven fit as represented by R2 value (0.9967) and p-value (0.2373). The normal probability plot for studentized residuals and predicted value of L-asparaginase production versus actual values are shown in Fig. 2. The data points depicted in the normal probability plot of the residues were normally distributed, representing a good model fit. In 3D response surface plots, the highest and lowest levels of Na2HPO4, KH2PO4, and L-Asn showed low L-asparaginase production (Fig. S2). The highest L-asparaginase production was found at the mid moderate levels.
Bioreactor scale optimization. The three bioreactor vessels (2 L) were run under optimized conditions (i.e., medium composition and culture conditions) except for the agitation rate and DO concentrations varied across vessels. The effects of agitation rate on L-asparaginase showed the highest enzyme production of 20.27 IU/mL at 24 h at an agitation rate of 350 rpm. In contrast, L-asparaginase production of 15.46 IU/mL and 16.48 IU/mL were obtained at agitation rates of 200 rpm and 500 rpm, respectively (Fig. 3). Studies on the effects of DO concentration on enzyme production showed the maximum activity of 25.08 IU/mL when DO concentration was maintained at 30% (Fig. 3). L-asparaginase activities of 21.39 IU/mL and 22.84 IU/mL were obtained when DO concentrations were maintained at 10 and 50%, respectively.

L-asparaginase production (IU/mL) from MSD-11(B. flexus SS) in bioreactor. Bioreactor profile at agitation rate of 200 (a), 350 (b) and 500 (c) rpm. Similarly, bioreactor profile at DO concentration of 10 (d), 30 (e) and 50% (f). 1—Enzyme activity; 2—OD600; 3—pH.
The overall increase in L-asparaginase production from B. flexus strain SS after optimization at bioreactor scale was 7.2-fold (25.08 IU/mL) over that obtained under the initial unoptimized production conditions (3.48 IU/mL).
Purification of L-asparaginase. The cell lysate containing 20076 IU and 1219.0 mg of protein (specific activity of 16.46 IU/mg) was used for purification. Ammonium sulfate precipitation followed by dialysis resulted in 625 mg of partially purified L-asparaginase protein with a specific activity of 28.05 IU/mg and a recovery yield of 87.3% with 1.70-fold purification (Table 2). This was followed by purification using two chromatography steps, ion-exchange chromatography (IEX) by DEAE-Sepharose and size exclusion chromatography (SEC) by Superdex-200 Increase 10/300 column (Fig. 4). The purified fraction from SEC contained 89.3 mg protein with overall enzyme activity of 7749 IU (specific activity of 86.77 IU/mg), indicating 5.27-fold purification (Table 2).

Purification and characterization of L-asparaginase from B. flexus SS. (a) DEAE-Sepharose chromatography of ammonium sulfate precipitated fraction. (b) Superdex-200 profile of L-asparaginase with standard proteins (insets). (c) SDS-PAGE of L-asparaginase produced at different purification stage (lane 1—molecular weight markers, lane 2—crude extract, lane 3—ammonium sulfate fraction, lane 4—purified enzyme). (d) CE-SDS analysis of purified L-asparaginase. (e) Purified L-asparaginase activity in the presence of different substrates. (f) Effect of pH on purified enzyme activity. (g) Effect of temperature on purified L-asparaginase activity.
Characterization of L-asparaginase. SDS-PAGE. The purified monomeric L-asparaginase from B. flexus SS showed a single protein band at 33 kDa, indicating homogeneity of the purified protein (Fig. 4c). L-asparaginase molecular weight is reported to vary depending on the microbial source. L-asparaginase from E. coli [20], Erwinia carotovora [21], Bacillus lichenifromis [3], Bacillus coagulans [22], Pseudomonas aeruginosa 50071 [23], Bacillus strain DKMBT10 [24], Streptomyces fradiae NEAE-82 [25], and Streptomyces gulbargensis [26], exhibited molecular weight of 37, 33.5, 37, 84, 160, 45, 53, and 85 kDa, respectively.
CE-SDS. CE-SDS analysis showed a single peak in electropherogram of purified L-asparaginase from B. flexus SS detected at migration time of 13.09 min, which corresponded to the molecular weight of 33 kDa (Fig. 4d). The results for molecular weight of purified L-asparaginase by CE-SDS conformed to the estimation done by SDS-PAGE analysis.
Substrate specificity. Among different substrates investigated, the purified enzyme showed the maximum (100% relative) activity when L-Asn was used as a substrate. The purified L-asparaginase did not show enzyme activity in the presence of substrate L-Asp, D‑Gln, and D-Asp (Fig. 4e). The lack of activity in the presence of L-Gln signifies that L-asparaginase in the current study is glutaminase-free.
Effect of pH and temperature. The purified enzyme showed the maximum enzyme activity at a pH of 8.0 (100% relative activity), and the lowest activity was obtained at a pH of 4.0 (4.1% relative activity) (Fig. 4f). On the other hand, in the study of the effect of temperature, the purified enzyme showed a maximum activity of 97.6% at 37°C (Fig. 4g).
Anticancer activity. The purified L-asparaginase showed in vitro anticancer activity against human tumor cell lines used in this study, and the IC50 value ranged from 0.8 to 47 µg/mL (Fig. 5). The strongest cytotoxic activity was found against SKBR3 cell lines (IC50 of 0.8 µg/mL), followed by WIL2-S and TF-1 cells (IC50 of 16.2 and 47 µg/mL, respectively). There was no significant inhibition against normal HUVEC cells as the percentage cell viability at maximum (125 µg/mL) working concentration of L-asparaginase was 95%. This was almost the same as that of untreated cells, and therefore, the IC50 value was not calculated in the 4PL curve to fit the analysis. The p‑value = 0.0010 in ANOVA (Dunnett test) indicates a significant difference in multiple comparisons of differences in the group, means % cells viability of tumor cells to HUVEC cells (Fig. S3).

Purified L-asparaginase anticancer activity estimated by Alamar blue (resazurin) fluorescence-based inhibition of proliferation assay against WIL2-S (a), TF-1 (b), SKBR3 (c), HUVEC (d) cell lines. Cytotoxic effect found against cancer cells (WIL2-S, TF-1, SKBR3 cells) was accompanied by its absence in control non-cancerous cells (HUVEC) cells as shown in their respective 4 parameter logistic (4PL) curve fitting-regression results plotted between log of L-asparaginase concentration (Log conc.) (µg/mL) versus relative fluorescent units (RFU). The difference in mean % viability the control cells versus % viability of cancer cells (WIL2-S, TF-1, SKBR3 cells) was found significant by ANOVA (Dunnett test) at 95% conference interval (p value= 0.0010).
DISCUSSION
L-asparaginase production from B. flexus SS was strongly influenced by the fermentation media composition and culture conditions, pH, temperature, agitation rate, and DO concentration. The optimal production of L-asparaginase at the bioreactor level was found at an agitation rate of 350 rpm. Maysa et al. [27] showed that L-asparaginase productivity might be affected by the chemical influence exerted by the oxidation-reduction process in the fermentation. The optimal DO concentration for the enzyme production in this study was found at 30%. The obtained results were in line with one of the previously reported studies by Mihooliya et al. [15]. L-Asn acts as a nitrogen source and an inducer making it a key component in L-asparaginase production. The decrease in the L-asn optimal concentration in the production medium could affect L-asparaginase production. Similarly, increasing in this concentration beyond the optimum level could have led to too much formation of ammonia, which might have an adverse effect on L-asparaginase production [15].
L-asparaginase was purified using ammonium sulfate precipitation, dialysis, subsequently by two-step chromatography, DEAE-Sepharose, and Superdex-200. The final purified enzyme contained 89.3 mg protein with specific L-asparaginase activity of 86.77 IU/mg and overall, 5.27-fold purification. In this study, the purification was higher in comparison to the findings by Alrumman et al. [3], where the enzyme from B. licheniformis was purified by similar procedure to 4.63-fold purification.
The anticancer activity of L-asparaginase from B. flexus SS against cancer cell lines showed that it was highly cytotoxic against SKBR3 compared to WIL2-S and TF-1. These findings were in line with those described previously by Mostafa et al. [28], where L‑asparaginase from marine Bacillus velezensis exhibited the anticancer activity against breast adenocarcinoma cell lines MDA-MB-231 (IC50 of 12.6 μg/mL) and MCF-7 cells (IC50 of 17.3 μg/mL). Furthermore, Shafei et al. [29] described L-asparaginase as inhibiting the growth of 3 human cell lines, with IC50 values of 12.5, 14 and 37 μg/mL against breast, hepatocellular and prostate carcinomas, respectively. The findings of this study were superior to those reported in one of the previous studies where L-asparaginase from Bacillus sp. R36 showed anticancer effect against human cell lines HCT-116 (IC50 of 218 μg/mL) and HepG-2 (IC50 of 112.19 μg/mL) [30]. The possible mechanism of anticancer activity of L-asparaginase is still not very clear. Ueno et al. [31] reported that tumor cells lack the L-Asn synthetase gene disabling them to produce L-Asn, making them starve and causing apoptosis. One study reported that L-asparaginase inhibits glycosylation of several new synthesized proteins [30]. El-Naggar et al. [25] suggested that L-asparaginase can disrupt colon cancer cells by altering the interactions among the microvasculature of colon cancer cells, endothelial cells, and extracellular matrix components. Hashimoto et al. [32] advocated that the proliferation of the human leukemic needs high amounts of Asn to produce enough energy and for biomolecules synthesis; therefore, reduction in Asn supply would prevent the human leukemic cells growth. In addition, Karpel-Massler et al. [33] reported the susceptibility of cancer cells to the suppression of proliferation by L‑asparaginase, which results in cell apoptosis by reducing mitochondrial membrane potential.
The current marketed L-asparaginase formulations for ALL therapy are majorly derived from Gram-negative bacteria, like E. coli (colaspase) and E. chrysanthemi (crisantaspase) [7, 8, 34]. These bacteria are strongly immunogenic, thereby neutralizing the therapeutic effects. Further, adverse symptoms are reported in greater than 50% of cases. Another problem associated with the marketed L-asparaginase preparations is their high L-glutaminase activity causing liver dysfunction, pancreatitis, leukopenia, neurological seizures, and coagulation abnormalities leading to intracranial thrombosis or haemorrhage [4, 7, 9–11, 35]. Therefore, the newer L-asparaginase with improved characteristics and minimal L-glutaminase activity is desired to overcome the current crisis in L-asparaginase therapy for ALL. The monomeric molecular weight of purified L-asparaginase from B. flexus SS was 33 kDa which was found to be close to that of the subunit molecular weight of E. coli enzyme (32 kDa) and Erwinia preparations (40 kDa) [34, 35]. The L-asparaginases from E. coli and Erwinia were reported to have L-glutaminase activity up to 10% [35]. Therefore, one of the major advantages of purified L‑asparaginase from B. flexus SS was no glutaminase activity. Another benefit of this enzyme is that it is produced from Gram-positive bacteria, thereby eliminating the possibilities of side-effects from Gram-negative endotoxins. Therefore, L-asparaginase from Gram-positive B. flexus SS reported in this study could be a potential candidate for ALL therapy owing to its improved characteristics to that current commercial L-asparaginase.
* *
In conclusion, to the best of our knowledge, this is the first report wherein L-asparaginase derived from B. flexus SS isolated from the Himalayan subtropical region of H.P. (India) was studied with improved characteristics for anticancer applications. The maximum L-asparaginase production was optimized at the bioreactor (2 L) scale and found to be 25.08 IU/mL with a 7.2-fold increase in enzyme activity over unoptimized conditions. The purification of L-asparaginase was achieved to 5.27-fold purification with a specific activity of 86.77 IU/mg and glutaminase free. The L-asparaginase demonstrated the cytotoxic effect against the cancer cell lines, i.e., SKBR3, WIL2-S, and TF-1 with IC50 values of 0.8, 16.2), and 47 µg/mL, respectively.
REFERENCES
Mahajan, R.V., Saran, S., Kameswaran, K., Kumar, V., and Saxena, R.K., Bioresour. Technol., 2012, vol. 125, pp. 11–16.
Nunes, J.C.F., Cristóvão, R.O., Ebinuma, V.C.S., Faria, J.L., Silva C.G., Neves, M.C., et al., Encyclopedia, 2021, vol. 1, no. 3, pp. 848–858.
Alrumman, S.A., Mostafa, Y.S., Al-Izran, K.A., Alfaifi, M.Y., Taha, T.H., and Elbehairi, S.E., Sci. Rep., 2019, vol. 9, no. 1, p. 3756.
Chand, S., Mahajan, R.V., Prasad, J.P., Sahoo, D.K., Mihooliya, K.N., Dhar, M.S., and Sharma, G., Biotechnol. Appl. Biochem., 2020, vol. 67, no. 4, pp. 619–647.
Mahajan, R.V., Kumar, V., Rajendran, V., Saran, S., Ghosh, P.C., and Saxena, R.K., PLoS One, 2014, vol. 9, no. 6, article ID e99037.
Lubkowski, J. and Wlodawer, A., FEBS J., 2021, vol. 288, pp. 4183–4209.
Vimal, A. and Kumar, A., Biotechnol. Genet. Eng., 2017, vol. 33, no. 1, pp. 40–61.
Stefano, V.D., Za, T., Ciminello, A., Betti, S., and Rossi, E., Thromb. Haemost. 2015, vol. 113, no. 2, pp. 247–261.
Brumano, L.P., Silva, F.V.S., Costa-Silva, T.A., Apolinário, A.C., Santos, J.H.M.P., Kleingesinds, E.K., et al., Front. Bioeng. Biotechnol., 2019, vol. 6, no. 212, pp. 1–22.
Dai, Z.J., Huang, Y.Q., and Lu, Y., Transl. Pediatr., 2021, vol. 10, pp. 244–255.
Kamal, N., Koh, C., Samala, N., Fontana, R.J., Stolz, A., Durazo, F., et al., Hepatol. Int., 2019, vol. 13, no. 5, pp. 641–648.
Mihooliya, K.N., Nandal, J., Swami, L., Verma, H., Chopra, L., and Sahoo, D.K., Enzyme Microb. Technol., 2017, vol. 107, pp. 72–81.
Imada, A., Igarasi, S., Nakahama, K., and Isono, M., J. Gen. Microbiol., 1973, vol. 76, pp. 85–99.
Bergey, D.H. and Holt, J.G., Bergey’s Mannual of Determinative Bacteriology, Baltimore: Lippincott Williams and Wilkins, 1994, 9th ed.
Mihooliya, K.N., Nandal, J., Kumari, A., Nanda, S., Verma, H., and Sahoo, D.K., 3 Biotech, 2020, vol. 10, no. 4, p. 148.
Mattos, A., Jager-Krikken, A., Haan. M., Beljaars, L., and Poelstra, L., J. Controlled Release, 2012, vol. 162, pp. 84–91.
Salas-Solano, O., Tomlinson, B., Du, S., Parker, M., Strahan, A., and Ma, S., Anal. Chem., 2006, vol. 78, no. 18, pp. 6583–6594.
Chand, S., Kumar, B., Prathap, V.M., Singh, S., and Mahajan, R.V., Curr. Sci., 2018, vol. 114, no. 12, pp. 2513–2518.
Priest, F.G., Goodfellow, M., and Todd, C., J. Gen. Microbiol., 1988, vol. 134, no. 7, pp. 1847–1882.
Khushoo, A., Pal, Y., Singh, B.N., and Mukherjee, K.J., Protein Expr. Purif., 2004, vol. 38, no. 1, pp. 29–36.
Kamble, V.P., Rao, R.S., Borkar, P.S., Khobragade, C.N., and Dawane, B.S., Ind. J. Biochem. Biophys., 2006, vol. 43, no. 6, pp. 391–394.
Law, A.S. and Wriston, J.C., Arch. Biochem. Biophys., 1971, vol. 147, pp. 744–752.
El-Bessoumy, A.A., Sarhan, M., and Mansour, J., J. Biochem. Mol. Biol., 2004, vol. 37, pp. 387–393.
Moorthy, V., Ramalingam, A., Sumantha, A., and Shankaranaya, R.T., Afr. J. Microbiol. Res., 2010, vol. 4, pp. 1862–1867.
El-Naggar, N.E., Deraz, S.F., Soliman, H.M., El-Deeb, N.M., and El-Ewasy, S.M., Sci. Rep., 2016, vol. 6, p. 32926.
Amena, S., Vishalakshi, N., Prabhakar, M., Dayanand, A., and Lingappa, K., Braz. J. Microbiol., 2010, vol. 41, pp. 173–178.
Maysa, E.M., Amira, M., Gamal, E., Sanaa, T., and Sayed, E.I., J. Am. Sci., 2010, vol. 6, no. 8, pp. 157–165.
Mostafa, Y., Alrumman, S., Alamri, S., Hashema, M., Al-izran, K., Alfaifi, M., et al., Electron. J. Biotechnol., 2019, vol. 42, pp. 6–15.
Shafei, M.S., Heba, A., El-Refai, H.A., Mostafa, H., El-Refai, H.A., El-Beih, F.M., et al., Curr. Trends Biotechnol. Pharm., 2015, vol. 9, pp. 132–140.
Yu, M., Henning, R., Walker, A., Kim, G., Perroy, A., Alessandro, R., et al., J. Cell. Mol. Med., 2012, vol. 16, pp. 2369–2378.
Ueno, T., Ohtawa, K., Mitsui, K., Kodera, Y., Hiroto, M., Matsushima, A., et al., Leukemia, 1997, vol. 11, pp. 1858–1861.
Hashimoto, K., Suzuki, F., and Sakagami, H., Anticancer Res., 2009, vol. 29, pp. 1303–1308.
Karpel-Massler, G., Ramani, D., Shu, C., Halatsch, M., Westhoff, M., Bruce, J.N., et al., Oncotarget, 2016, vol. 7, pp. 33512–33528.
Koerholz., D., Brueck., M., Nuernberger. W., Juergens, H., Goebel, U., and Wahn, V., Eur. J. Hematol., 1989, vol. 42, pp. 417–424.
Narta, U.K., Kanwar, S.S., and Azmi, W., Crit. Rev. Oncol. Hematol., 2007, vol. 61, pp. 208–221.
ACKNOWLEDGMENTS
The authors acknowledge the National Institute of Biologicals (Ministry of Health and Family Welfare, NOIDA, Uttar Pradesh, India) and Amity University (NOIDA, Uttar Pradesh, India) for providing facilities, space and resources for this work.
Funding
This research received no external funding.
Author information
Authors and Affiliations
Contributions
Conceptualization of the research—G.S., J.P.P and D.K.S.; Methodology—S.C., K.N.M., G.S; Software work—S.C., K.N.M., D.K.S.; Validation work—S.C., K.N.M, D.K.S., G.S.; Formal analysis—S.C., K.N.M, D.K.S., J.P.P., G.S.; Investigation—S.C., K.N.M.; Resources—G.S. J.P.P., Data curation—G.S., J.P.P., D.K.S.; Writing-draft preparation- S.C., K.N.M..; Writing—review and editing—G.S., D.K.S., J.P.P.; Visualization—G.S., D.K.S., J.P.P.; Supervision—G.S., J.P.P.; Project administration—G.S., J.P.P. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
The authors declare no conflicts of interest. All authors read and approved the manuscript for publication. This article does not contain any studies involving animals or human participants performed by any of the authors.
Availability of data and material. The datasets generated during this article are included in the manuscript and supplementary information. Any further enquiries can be directed to the corresponding author.
Supplementary Information
Rights and permissions
About this article

Cite this article
Chand, S., Mihooliya, K.N., Sahoo, D.K. et al. L-asparaginase from Bacillus flexus strain SS: Isolation, Screening, Production Process Optimization, Purification, and Anticancer Activity. Appl Biochem Microbiol 58, 416–427 (2022). https://doi.org/10.1134/S0003683822040032
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0003683822040032