

Abstract>
Circulating tumour DNA (ctDNA) in blood plasma is an emerging tool for clinical cancer genotyping and longitudinal disease monitoring1. However, owing to past emphasis on targeted and low-resolution profiling approaches, our understanding of the distinct populations that comprise bulk ctDNA is incomplete2,3,4,5,6,7,8,9,10,11,12. Here we perform deep whole-genome sequencing of serial plasma and synchronous metastases in patients with aggressive prostate cancer. We comprehensively assess all classes of genomic alterations and show that ctDNA contains multiple dominant populations, the evolutionary histories of which frequently indicate whole-genome doubling and shifts in mutational processes. Although tissue and ctDNA showed concordant clonally expanded cancer driver alterations, most individual metastases contributed only a minor share of total ctDNA. By comparing serial ctDNA before and after clinical progression on potent inhibitors of the androgen receptor (AR) pathway, we reveal population restructuring converging solely on AR augmentation as the dominant genomic driver of acquired treatment resistance. Finally, we leverage nucleosome footprints in ctDNA to infer mRNA expression in synchronously biopsied metastases, including treatment-induced changes in AR transcription factor signalling activity. Our results provide insights into cancer biology and show that liquid biopsy can be used as a tool for comprehensive multi-omic discovery.
Similar content being viewed by others

Main
Cell-free DNA (cfDNA) is an emerging minimally invasive analyte across healthcare. In oncology, quantification and characterization of ctDNA in blood can inform early cancer diagnoses, select treatment, and monitor for relapse1. However the full potential for ctDNA to reveal tumour evolution and mechanisms of acquired resistance remains unknown. Prerequisite understanding of the somatic populations captured by ctDNA, their relationship to synchronous metastatic tissue, and their temporal dynamics during treatment has not been decisively established. Prior studies have focused on coding regions of known cancer genes, used shallow whole-genome sequencing (WGS), or only tracked the dynamics of individual mutations—all precluding high resolution of clonal architecture and unbiased analysis of putative resistance mechanisms2,3,4,5,6,7,8,9,10,11,12.
Nucleosome organization reflects transcription factor activity and gene expression. Intriguingly, nucleosome positioning can be inferred from cfDNA fragmentation patterns, which suggests that cfDNA may enable integrated genomic, transcriptomic and epigenomic profiling. However, the utility of cfDNA for phenotyping individual cancers is still unclear as previous studies lack deep clinical–genomic resolution and/or have not examined synchronous metastatic tissue, which is scarcely available outside of rapid autopsy programs13,14,15,16,17,18. Comprehensive dissection of these ctDNA features necessitates deep WGS of serial clinical samples from living patients with a high ctDNA fraction, which we have performed herein.
Deep clinical ctDNA whole genomes
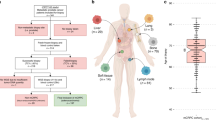
We performed deep WGS on 61 plasma cfDNA samples (median unique read depth: 187×) and 15 metastatic tissue biopsies (98×) from 33 patients with metastatic castration-resistant prostate cancer (mCRPC), as well as 9 additional cfDNA samples from 2 patients with metastatic neuroendocrine prostate cancer, 2 patients with metastatic bladder cancer and 5 control individuals with no detectable ctDNA (Fig. 1a,b and Supplementary Tables 1 and 2). All patients provided at least 1 cfDNA sample, and 21 patients provided multiple cfDNA samples that were collected serially at progression on different lines of therapy19,20. Thirteen metastatic tissue biopsies were collected on the same day as cfDNA20. The median ctDNA fraction based on genome-wide copy number states and heterozygous single-nucleotide polymorphism allele fractions (HSAF) was 47% (range: 17–82%) and correlated with prior deep targeted sequencing estimates6 (Pearson R = 0.82, P = 8.87 × 10−16) (Fig. 1c, Supplementary Methods and Supplementary Tables 1 and 3). In total, 687,293 somatic mutations and 16,200 structural rearrangements were identified across the 61 cfDNA and 15 tissue samples. The median ctDNA somatic mutation burden was 1.8 per Mb (range: 0.34–39.43) and was not correlated with ctDNA fraction (Pearson R = 0.008, P = 0.95), nor associated with collection time point (P = 0.18 by Mann–Whitney U test (MWU)), whole-genome duplication (WGD) (MWU P = 0.23) or polyclonality (MWU P = 0.07) (Fig. 1d and Extended Data Fig. 1a). Mitochondrial cfDNA was detected in all patients and had more variable fragment lengths compared to nuclear cfDNA (interquartile range: 108–248 bp versus 147–178 bp; MWU P < 0.001) and lacked mono- and di-nucleosomal peaks, consistent with an absence of nucleosome-mediated fragmentation bias16 (Extended Data Fig. 1b).

a, Study design. NEPC, neuroendocrine prostate cancer. b, Per-patient breakdown of plasma cfDNA and metastatic tissue samples. Rows indicate the ordinal timing of sequential collections. c, Plasma cancer fraction inferred by WGS correlates with previous estimates from deep targeted sequencing (73-gene panel). d, Per-sample overview of genomic information (main study cohort). The ‘non-integer copy number’ category only includes non-integer values of less than 4; signature weights are derived from bulk sequencing (signatures with weights less than 0.15 are grouped as ‘Other’). HRRd, homologous recombination repair defective; MMRd, mismatch repair defective; SNV, single-nucleotide variant. e, Comparison of evidence for subclonal copy number alterations detected in cfDNA (n = 58) versus tumour tissue (n = 15); samples that constituted admixtures of at least two major clonal populations with different WGD statuses were excluded. Representative examples of same-patient whole-genome tissue and cfDNA copy number profiles. LBD = ligand binding domain.
Molecular subtypes inferred from bulk ctDNA were representative of aggressive mCRPC20,21,22 (Fig. 1d and Extended Data Fig. 1a). Inactivating somatic alterations including structural rearrangements with an intronic or 5′-UTR breakpoint were common in tumour suppressors TP53 (73% of patients), PTEN (48%) and RB1 (18%) (Fig. 1d and Supplementary Table 4). ETS fusions were identified in 49% of patients (Extended Data Fig. 1c). Deleterious DNA repair defects were identified in nine patients and associated with characteristic phenotypes. BRCA2 defects (four patients) were linked to high chromosomal instability (including aneuploidy and an enrichment of structural variants) and signatures of homologous recombination repair deficiency (deletions with flanking microhomology and base-substitution signatures), whereas the tumour with mismatch repair defects exhibited somatic hypermutation and associated C>T base-substitution signatures23 (Fig. 1d and Extended Data Fig. 1d). CDK12 mutant tumours (four patients) were diploid, ETS fusion-negative and contained genome-wide tandem duplications24 (Supplementary Methods).
CtDNA copy number profiles frequently exhibited genomic regions whose copy number deviated from integer ploidies (accompanied by supportive shifts in HSAF), which suggests an admixture of multiple populations with distinct genomic copy number states. By contrast, metastatic tissue exhibited less subclonal copy number diversity, indicating greater population homogeneity (Fig. 1e).
Distinct populations resolved from ctDNA
Intratumoral heterogeneity can drive treatment failure25. Resolving subclonal heterogeneity and historical patterns of evolution may lead to improved understanding of disease and mechanisms of acquired treatment resistance26. Given evidence for intra-sample heterogeneity, we sought to identify dominant somatic populations in our samples. Multidimensional clustering of mutation cancer cell fractions (CCFs) across temporally or spatially distinct samples can reveal clonal composition and evolutionary history26,27 (Fig. 2a). We developed a custom subclonal reconstruction procedure optimized for our data and applied it to our patient cohort. Comprehensive step-by-step methodology, validation using orthogonal tools and in silico modelling (Supplementary Table 5), and raw data for all patients are provided in the Supplementary Methods. On average, 2.9 subclonal populations were detected per patient. Notably, the distinct genome-wide copy number landscapes of subclonal populations could be deduced in most patients (Supplementary Methods). Diversity in phylogenetic tree topology and molecular timing imply varied patterns of mCRPC evolution (Fig. 2b).

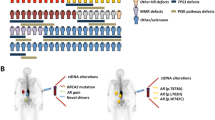
a, Overview of subclonal reconstruction methodology from bulk DNA. b, Per-patient illustration of inferred tumour populations. The top plots show cancer fraction and sample metadata. The third row illustrates the fractional representation of each subclone in a given patient sample (column). Phylogenetic tree branch length is proportional to the number of mutations assigned to a given population (truncal branch is scaled 0.25×). WGD events (annotated with stars) are positioned by their timing relative to mutations. Triplications (indicated by *) are not positioned by timing. c, Bar plot showing the proportion of the genome that is copy-altered (relative to expected ploidy accounting for WGD(s)), dichotomized by gains versus deletions in ctDNA and tissue. d, Bar plot showing the mean number of whole-chromosome copy number alterations (relative to expected ploidy) in diploid versus WGD samples; dots represent individual samples (dots are jittered along the x and y dimensions). e, Fraction of genomic regions that are copy-altered versus mean genome copy number. Samples that constituted admixtures of two or more major clonal populations with different WGD states were excluded. f,g, Box plots showing per-patient C>T ageing-associated (COSMIC 1) (f) and DNA-repair-defect-associated (COSMIC 3 or 6+15) (g) signature (n = 4) weights inferred from mutation subsets categorized by evolutionary timing. Connecting lines indicate same-patient clonal populations. MRCA, most recent common ancestor. h, Per-sample segmented copy number profiles of the AR gene and enhancer locus. LBD mutations and structural variants, and gene and enhancer copy number status, are annotated.
WGD can be associated with poor prognosis and distinct biological hallmarks, including potential therapeutic vulnerabilities28,29,30. Cancer cell population(s) with WGD were detected in 55% of patients and 55% of samples, including two patients with hexaploid and one patient with octaploid populations (Figs. 1d and 2b and Supplementary Methods). Chromosomal instability (primarily manifesting as deletions relative to expected ploidy after accounting for WGD) incrementally increased with the number of whole-genome doublings (median fraction of genome altered: 37% (diploid), 78% (tetraploid), 94% (hexaploid or octaploid); Kruskal–Wallis P = 4.6 × 10−11). Whole-chromosome copy alterations relative to expected ploidy were also more common in WGD samples (MWU P = 3.68 × 10−7) (Fig. 2c–e). WGD was predominantly truncal, present in the most recent common ancestor of all identified cancer cell populations in 72% of patients with WGD (Fig. 2b), and consistent with the similar frequency of WGD between cfDNA and tissue (56% versus 53%). By using the distinct mutant-allele copy numbers of pre- and post-WGD mutations, we estimated that on average 66% (range: 8–97%) of all truncal mutations preceded WGD. Of note, three patients had evidence for WGD occurring independently in two distinct subclonal lineages (Fig. 2b and Supplementary Information). Contemporary subclonal reconstruction methods assume that samples do not carry subclonal WGD status heterogeneity. Our data indicate that this assumption can be violated, particularly in ctDNA owing to increased subclonal heterogeneity26,28,31.
Subclones featured a lower predominance of the ageing-associated CG>TG mutation signature compared to the most recent common ancestor (median 8.4% versus 24.1%, MWU P = 1.0 × 10−5), and overall, the contribution of ageing-associated mutational processes monotonically decreased across phylogenetic epochs (ordered in time) (P = 0.003) (Fig. 2f). In patients with BRCA2 defects or mismatch repair deficiency (n = 4), the characteristic mutation signatures of these phenotypes were enriched in subclones compared to their truncal ancestor, which implies that subclone evolution continues to be shaped by truncal DNA repair dysfunctions23 (Fig. 2g and Supplementary Table 6).
AR alterations drive castration resistance, resulting in lethal disease32. Nearly all cfDNA samples in our cohort (95%) had some degree of AR alteration, including putative resistance mechanisms such as ligand-binding domain (LBD) missense mutations (18%), AR gene and/or upstream enhancer amplifications (48%) and LBD-truncating structural variants20,21,33,34,35,36 (7%) (Fig. 2h). The magnitude of AR gene and enhancer copy gain was highly variable between samples, ranging between 1–91 copies (gene body) and 1–118 copies (enhancer). AR alteration diversity within individual samples suggests the presence of multiple subclones with different AR genotypes.
ctDNA contribution of individual metastases
A hypothetical (but unconfirmed) advantage of ctDNA over tissue biopsy is the potential to inform on inter-lesional heterogeneity. We sought to define the relationship between the somatic populations captured by a single metastatic tissue biopsy and time-matched ctDNA. Twelve out of thirteen patients with same-day metastatic tissue and ctDNA samples contained mutation clusters (from subclonal reconstruction) that were truncal within the biopsied tissue but subclonal in ctDNA—the lowest CCF of these clusters in ctDNA corresponded to the contribution of the biopsied population to total ctDNA (Fig. 3a and Supplementary Information). In the remaining patient (DTB-205), all tissue-truncal mutations were also truncal in ctDNA, suggesting a contribution approaching 100%. The average ctDNA contribution of a biopsied metastatic tissue population was 19% (range 1–100%). This supports the hypothesis that ctDNA is derived from multiple metastases, each typically contributing a small proportion of total ctDNA.

a, Leveraging multi-sample subclonal reconstruction to estimate the proportion of ctDNA derived from somatic populations present in individual metastatic tissue biopsies. Three representative examples illustrating low, medium and high ctDNA contributions. b, Scatter plot illustrating high correlation between mCRPC driver gene copy number across matched cfDNA–tissue pairs; marker size reflects the proportion of ctDNA originating from the biopsied population. The Pearson correlation for each gene is shown. c, Mutation concordance (presence/absence) between cfDNA–tissue pairs. VAF, variant allele frequency. d, Per-patient somatic copy number differences between time-matched ctDNA and tissue. e, Average absolute ctDNA–tissue copy number differences across the genome (left) and the AR locus (right) on chromosome X. f, Scatter plot illustrating high correlation between AR gene and enhancer copy number between matched cfDNA–tissue pairs.
Recognizing that differences in clonal composition may affect genomic concordance between ctDNA and tissue, we compared mutation and copy number landscapes between the 13 same-patient sample pairs (Fig. 3b–f). Of the mutations independently detected in tissue (n = 89,554), 96.9% were redetected in cfDNA. Among mutations that were unique to either cfDNA (n = 1,420) or tissue (n = 2,748), but amenable to reciprocal redetection, only 7 were protein-truncating or established hotspots (Fig. 3c). This suggests that ctDNA can recapitulate the landscape of clonally expanded driver mutations, even when the fractional ctDNA contribution of individual lesions is small. Finally, 11 of 13 patients showed pervasive differences in copy number between time-matched cfDNA and tissue, consistent with evidence of differences in clonal composition. In patient DTB-119, the dominant cfDNA population was diploid, in contrast to a hexaploid tissue population (Fig. 3d). Despite broad differences in chromosomal copy number, mCRPC driver gene copy status was highly concordant between cfDNA and tissue, consistent with previous data and implying early truncal fixation37 (Fig. 3b).
AR genotypes were generally concordant between matched cfDNA and tissue but occasionally exhibited contrasting amplicon structures, implying the presence of different tumour populations with independently evolved AR amplifications (Figs. 3f and 2h and Extended Data Fig. 2a). Accordingly, the average absolute difference in copy number between cfDNA and tissue was greatest for chromosome X relative to other chromosomes, with the largest difference converging on the AR gene and enhancer (Fig. 3e). These data suggest that alterations in the AR locus are a critical substrate that drive clonal expansions in the context of AR-targeted therapy.
Serial ctDNA reveals treatment-driven selection for AR augmentation
Pre-treatment mCRPC contains a reservoir of heterogeneous AR alterations35,36. Narrow targeted sequencing of serial ctDNA suggests that activating AR genotypes undergo selection by AR signalling inhibitors and drive acquired resistance, but detailed genome-wide analyses to evaluate other putative resistance mechanisms have not been possible6. Therefore, we analysed 49 cfDNA samples that were collected at sequential clinical progressions from 21 patients with mCRPC who were undergoing treatment with AR signalling inhibitors (Fig. 1a,b). Across all 28 consecutive same-patient cfDNA pairs, the only highly recurrent changes in copy number, mutations or structural rearrangements were at the AR locus (Fig. 4a–c and Supplementary Table 7). This observation was not driven by any single patient; rather, 62% of patients showed a significant temporal increase in AR gene body and/or enhancer copy number, and/or LBD mutation switch (Fig. 4d and Supplementary Table 8). Whereas AR enhancer amplifications were primarily caused by tandem duplications, AR gene body amplification occurred through nested tandem duplications, inverted repeats, or a combination of both20,38 (Fig. 4c and Extended Data Fig. 2b). AR augmentation frequently occurred in the context of broad population restructuring, which was most pronounced in patients with deep treatment responses (Fig. 4e and Supplementary Information). Collectively, these results implicate the distinct AR genotypes of clones in driving their differential fitness under treatment and facilitating population shifts.

a, Genomic landscape across all 76 cfDNA and tissue samples (regardless of time point). Somatic copy number (top row), patients with genomic rearrangement breakpoints (1-Mb window; second row), patients with mutations (100-kb window; third row) and patients with protein-altering mutations (divided by the coding sequence (CDS) length in kb; bottom row). In the copy number panel (top row), red and blue colour indicate copy numbers that are larger and smaller, respectively, than the average base ploidy of all samples (accounting for WGDs). In the bottom row, only genes that are mutated in three or more patients are shown. b, Aggregate of acquired genomic changes between consecutive cfDNA samples after treatment with AR signalling inhibitors in 21 patients. Average copy number change after treatment (top row), patients with breakpoints acquired after treatment (1-Mb window; second row), patients with mutations acquired after treatment (100-kb window; third row) and patients with protein-altering mutations acquired after treatment (bottom row). In the bottom row, only genes with acquired mutations in two or more patients are shown. c, Average copy number (CN) change at the AR locus showing copy number increases that affect the enhancer and gene body. d, AR gene body and enhancer copy number in serial cfDNA samples from 21 patients. The estimated number of LBD mutant AR copies in each sample is indicated with coloured bar segments. Wild-type AR copies are shown in black. e, Clonal population dynamics (inferred from subclonal reconstruction) across treatment timelines. The x axis represents monotonically interpolated measurements of prostate-specific antigen (PSA) at four-week intervals, indicating changes in tumour burden. Subclone fractions are interpolated between cfDNA time points using exponential functions (see Supplementary Information, 'Visualization of tumour burden and subclone fractions over time'). Right, segmented AR locus copy number structures across consecutive cfDNA time points (gene body and enhancer are highlighted with colour).
cfDNA fragmentation recapitulates cell phenotypes
Nucleosomes protect DNA during apoptosis, which results in distinct cfDNA fragmentation patterns that indicate cell-of-origin nucleosome occupancy13,14,15,16,17,18 (Extended Data Fig. 1b). We used orthogonal methods (that is, windowed-protection score (WPS) and relative read-depth depletion) to generate per-sample nucleosome occupancy maps from cfDNA and correlated results with whole-transcriptome sequencing of same-day metastatic tissue biopsy.
For 13 patients with same-day metastatic tissue and cfDNA biopsy, we used tissue mRNA expression to rank genes into patient-specific expression deciles and examined the aggregated cfDNA fragmentation profiles of each decile (Fig. 5a). Highly expressed genes showed features consistent with active transcription—including nucleosome depletion at transcriptional start sites (TSSs), strong +1 occupancy and well-ordered nucleosomes extending into the first exon13 (Fig. 5b,c and Extended Data Fig. 3a–c). The magnitude of TSS nucleosome depletion was strongly correlated with patient-matched tissue mRNA expression (Spearman R = 0.99, P < 0.001) (Fig. 5d). Notably, we observed similarly high correlations in contrasting the fragmentation profiles for 61 cfDNA samples in our cohort against the median RNA expression in 101 mCRPC metastatic tissue samples, even after omitting housekeeping genes and genes highly expressed in haematopoietic cells20 (the main contributor to non-tumour cfDNA) (Spearman R = 0.97, P < 0.001) (Fig. 5e). Finally, we observed localized changes in cfDNA fragment-length distribution across the TSS of actively transcribed genes39 (Extended Data Fig. 4a–c). Collectively, this shows that cfDNA nucleosome occupancy profiles in samples with high ctDNA fraction mirror global transcriptomic patterns in metastatic lesions.

a, Evidence of nucleosome occupancy footprints in plasma cfDNA. Right, method for quantifying gene expression from TSS nucleosome depletion. b, The magnitude of cfDNA nucleosome depletion mirrors mRNA expression from patient-matched metastatic tissue whole-transcriptome sequencing at both the gene TSS (left) and the first exon–intron junction (right). Nucleosome depletion is shown using orthogonal methods (WPS and relative read-depth depletion). c, Per-sample cfDNA TSS nucleosome depletion stratified by patient-matched tissue mRNA expression. d, Spearman correlation of TSS and first exon–intron junction nucleosome depletion (in cfDNA) versus tissue gene expression percentile in 13 patients with synchronous cfDNA–tissue samples. Each line is an individual sample. e, Spearman correlation of TSS nucleosome depletion (in cfDNA) versus tissue gene expression percentile in 61 cfDNA samples including those without matched tissue (leveraging publicly available mCRPC tissue transcriptome data). Results for all genes after excluding housekeeping and haematopoietic lineage genes are shown in different colours. f, Differential first exon–intron junction read-depth depletion by first exon length (long exons represent top 25% of all RefSeq MANE first exons; short exons represent bottom 25%) across n = 61 cfDNA samples. g, Gene RNA expression correlates with gene body coverage depletion in cfDNA (n = 61) but not tissue (n = 16). h, Per-sample (row) cfDNA read-depth depletion at 3,224 ARBSs ordered by magnitude of nucleosome depletion. The AR copy number and presence of mutations are annotated on the left. Negative controls are shown at the bottom; ctDNA-positive mCRPC samples are shown at the top. i, Clonal population shifts in patient AE-180 across sequential progressions on enzalutamide and abiraterone. Population phylogeny shown on the right. j, Temporal decrease in AR signalling detected from average read-depth depletion around ARBS. The inset plot shows the temporal dynamics of lactate dehydrogenase (LDH; associated with liver metastases) and alkaline phosphatase (ALP) concentrations per upper-limit of normal (ULN), indicating changes in disease composition.
Metastatic tissue mRNA expression was also strongly correlated with cfDNA nucleosome depletion at the first exon–intron junction15 (Fig. 5d), although the magnitude of effect (relative to the TSS) was weaker and exhibited an association with first exon length (Fig. 5f). More broadly, highly expressed genes (including AR) showed systematically lower cfDNA sequencing coverage across the entire gene body relative to flanking regions (Fig. 5g and Extended Data Fig. 3d,e). This phenomenon was not observed in matched tissue and is consistent with cfDNA having decreased protection from degradation in regions of high RNA polymerase activity.
mCRPC can acquire resistance to AR inhibitors through a reduction in AR signalling dependency and the acquisition of a small-cell neuroendocrine phenotype40. This adaptive transformation cannot at present be inferred from DNA sequence profiling and is challenging to detect clinically, despite its implications for patient management. We measured cfDNA nucleosome occupancy at 3,224 prostate cancer AR-binding sites (ARBSs) to test the feasibility of using cfDNA to infer AR signalling activity in individual patients41,42. Most samples exhibited strong ARBS nucleosome depletion, which is suggestive of transcription factor binding and persistent AR signalling (Fig. 5h). ARBS nucleosome depletion correlated with average AR gene (Spearman R = 0.24, P = 0.05) and enhancer (Spearman R = 0.26, P = 0.04) copy number, but not with time-matched prostate-specific antigen (PSA) level (Spearman R = 0.10, P = 0.48), which is confounded by tumour burden and can disassociate from AR activity in advanced disease43,44 (Extended Data Fig. 3f–h). We performed additional 175× WGS on cfDNA from patients with metastatic bladder cancer and histologically confirmed small-cell neuroendocrine prostate cancer (typically AR-negative or AR-low) and observed weaker ARBS nucleosome depletion (Fig. 5h).
One patient with three cfDNA collections during sequential AR inhibition showed a temporal switch in clonal populations, marked by the emergence of visceral metastases and an RB1 splice-site mutation at clinical progression (potential indicators of neuroendocrine disease) (Fig. 5i,j). Notably, cfDNA nucleosome footprinting identified a commensurate loss of AR signalling (Fig. 5j). This is—to our knowledge—the first instance of leveraging cfDNA to observe dual genomic and transcriptomic evolution in a living patient who is developing resistance to treatment. Collectively, the signals we observed at TSS and ARBS suggest that cfDNA sequencing can help to identify clinically relevant differences in cancer phenotype.
Discussion
Here we have provided a deep WGS study of serial ctDNA and matched metastatic tissue. We show that samples with high ctDNA fractions permit dissection of metastatic cancer evolutionary histories, temporal subclonal dynamics, and the discovery of genomic and transcriptomic mechanisms of treatment resistance. This resolution was only previously achievable through invasive and repeated profiling of fresh tissue biopsies. Therefore, our work advances ctDNA profiling from an emerging tool for the detection of selected clinically actionable gene mutations, towards a modality for genome-scale discovery and deep clinical–biological insight in progressing metastatic cancers.
The populations that comprise ctDNA were typically complex and more heterogeneous than those found in bulk WGS of a synchronous metastasis (Figs. 1e and 2). We provide a comprehensive framework for robust subclonal reconstruction from serial ctDNA samples, leveraging allele-specific truncal copy number model fitting; mutation-based subclone quantification and phylogeny estimation (correcting for WGD and allelic status); and phylogeny-aware copy number deconvolution across samples. The accuracy of our clonal reconstruction framework is buttressed by multiple visualizations and in silico validations using simulated samples (Supplementary Information). In several instances, subclones within a patient contained distinct histories of WGD and aneuploidy—combinations that are challenging to resolve even with state-of-the-art tissue-profiling methodology26,28. Subclonal diversity may worsen disease prognosis by increasing opportunities for selection of resistant phenotypes25,45. Our results suggest that deep ctDNA WGS can be used in large cohorts of patients with high ctDNA fraction to determine the clinical relevance of metastatic tumour clonal composition and to understand how to exploit Darwinian evolutionary principles to improve cancer control. Notably, in several patients, a nondominant ancestral lineage that was present at baseline subsequently outcompeted daughter lineages at treatment progression through a selective sweep (Fig. 4e), which suggests that ancestral tumour lineages can retain clinical significance in late-stage disease when new treatments are initiated. We anticipate that the diminishing cost of WGS will enable higher sequencing depths (higher than 1,000×) in future studies, unlocking similar genome-wide clonal analysis in plasma samples with ctDNA fractions as low as 5%.
In almost all of the patients in our cohort, ctDNA was composed of multiple subclones with distinct genomic features, and each biopsied metastasis contributed only a small fraction of total ctDNA (Fig. 3a,d). Most genomic alterations of established functional importance in prostate cancer (for example, in TP53 and PTEN) are early truncal events, and were consequently shared between subclones and between ctDNA and tissue—this is corroborated by the relative homogeneity in driver gene alterations between metastases in published mCRPC autopsy studies37,46,47. Nevertheless, reliance on a single tissue biopsy may provide an incomplete view of polyclonal mechanisms of treatment resistance (for example, AR alterations or BRCA2 reversion mutations), misclassify alterations as truncal features (for example, WGD) or underestimate disease subclonal complexity48. Future work will be needed to understand how metastatic site (for example, bone versus soft tissue) and size relative to total tumour burden influence contribution to ctDNA, and whether emerging functional imaging techniques (for example, PSMA PET–CT (prostate-specific membrane antigen positron emission tomography combined with computed tomography) and [18F]FDG–PET (PET with 18F-fluorodeoxyglucose)) can help select lesions that are likely to reflect clinically dominant disease49. Nevertheless, serial ctDNA samples with a high cancer fraction may offer superior resolution of treatment-associated dynamics compared to serial metastatic tissue biopsies (which are pragmatically unfeasible), as the latter cannot distinguish temporal and spatial heterogeneity except in lesions that exhibit complete tumour eradication and subsequent repopulation.
AR signalling inhibitors are a mainstay of prostate cancer treatment but resistance is inevitable19,43. With the AR gene and enhancer as sole exceptions, we found no regions that contained recurrent mutations, structural rearrangements, or copy number changes that were acquired at treatment progression on AR-targeted therapy (in serial ctDNA), nor any regions of recurrent difference between patient-matched ctDNA and metastatic tissue (Figs. 4b and 3e). This strongly implies that mCRPC evolution is continually shaped by AR genotype, reiterating the sustained need for new therapies that target AR signalling, and proposing a minimally invasive practical tool for detecting emerging genomic mechanisms of resistance. Clinical suppression of AR signalling is essential for mCRPC management—all drugs are administered on a backbone of continuous androgen deprivation, and emerging standard-of-care and investigational agents (for example, 177Lu–PSMA-617, AR protein degraders and AR N-terminal domain inhibitors) offer renewed strategies for exploiting dependence on AR signalling. This constant selective pressure underscores the importance of understanding nuanced variation in AR genomic resistance mechanisms and underlying (sub)clonal diversity. More broadly, genomic mediators of resistance to clinically active targeted therapies are likely coupled to the specific pathways under selective pressure, which implies that ctDNA can nominate distinct mechanisms of acquired resistance in other cancers and therapeutic contexts (for example, PI3K-pathway or PARP inhibition in prostate and breast cancer)48,50,51.
cfDNA fragmentomics to profile transcription factor binding activity may help gauge reliance on transcription factor signalling for therapy selection (for example, broad chemotherapies versus agents that exploit transcription factor signalling addiction, such as AR pathway inhibitors or oestrogen receptor degraders in prostate and breast cancers), and could help to identify early emergence of non-genomic resistance mechanisms, including neuroendocrine-like phenotypes in lung and other cancers where lineage plasticity can drive acquired resistance52. cfDNA footprinting at gene TSSs can recapitulate broad patterns of mRNA expression in metastatic tissue, suggesting that cfDNA may also enable RNA subtyping in cancers where clinical relevance is established. Ultimately, we show that cfDNA sequencing enables parallel epigenomic and comprehensive genomic profiling, which could simplify workflows and conserve often-limited DNA yields. Targeted cfDNA assays that incorporate these biologically informative chromatin landmarks may circumvent the need for expensive deep WGS if profiling of TSSs or transcription factor binding sites is a primary goal, with the caveat that capacity for comprehensive dissection of other genomic information will be reduced15.
Highly expressed genes showed lower cfDNA sequencing coverage, possibly reflecting increased DNA vulnerability to apoptotic digestion owing to transcriptionally mediated nucleosome depletion (Fig. 5g and Extended Data Fig. 3d,e). Targeted panels for ctDNA genotyping—which are ubiquitous in research and rapidly entering clinical practice—may therefore underestimate true DNA copy number in highly expressed genes (including clinically relevant oncogenes like AR or ERBB2).
Our study has several limitations. First, samples were pre-selected for WGS on the basis of high ctDNA fraction from previous targeted sequencing. Mutation-based ctDNA fraction estimates from narrow targeted panels (in which WGD is often impossible to infer) may overestimate the ctDNA fraction of WGD tumours, potentially enriching our cohort for WGD53. The high frequency of WGD in our data is likely explained by both selection bias and genuine disease aggression, given the ubiquity of established clinico-genomic features that are independently linked to unfavourable outcomes (for example, TP53 defects and high ctDNA fraction)21,36. Second, metastatic tissue biopsy location was influenced by surgical accessibility and patient safety, and biopsies may therefore not necessarily represent bulk disease. Finally, patients in our cohort have multiple compounding poor prognosis characteristics that confound assessment of potential clinical outcomes differences between groups (for example, WGD status).
Our study reveals that plasma ctDNA-based subclonal reconstruction is associated with a unique set of challenges and opportunities. Heightened subclonal diversity invalidates many simplifying assumptions on which contemporary subclonal reconstruction methods rely, and regionally varying sequencing depth due to cfDNA nucleosome footprints may pose a challenge for ultra-sensitive copy number analysis. Conversely, serial ctDNA sampling also presents new opportunities, as the increased data dimensionality allows resolution of previously ambiguous scenarios, such as distinguishing between a WGD and a 50% CCF subclone27 (Supplementary Information). Notably, the fractal nature of clonal evolution means that the level of detail in reconstructions from bulk sequencing can vary according to sampling characteristics (for example, sequencing depth, breadth or number of samples)54.
We have generated a large repository of deep ctDNA WGS data, including data on patient-matched white blood cells. These data are available to researchers and will provide a resource for further discovery work using ctDNA.
Methods
Cohort
Patient samples were procured from two cohorts: (A) a recent prospective randomized phase II cross-over trial of abiraterone then enzalutamide (or reverse sequence) for first-line mCRPC (NCT02125357)19,36; or (B) the multi-centre Stand Up 2 Cancer–Prostate Cancer Foundation-funded West Coast Dream Team project (NCT02432001)20. All patients had histologically diagnosed prostate cancer (any histological subtype) or unambiguous clinical evidence of advanced prostate cancer (B only) and metastatic castration-resistant disease (Prostate Cancer Working Group 2 criteria; cohort A) at the time of first metastatic tissue or liquid biopsy19,55. For cohort A, serial plasma cfDNA samples were collected pre-treatment and at sequential clinical progressions (defined as first occurrence of confirmed PSA progression, radiographic progression as assessed by CT scan (RECIST 1.1 criteria) and/or bone scan (PCWG2 criteria), unequivocal clinical disease progression or unacceptable toxicity). For cohort B, same-patient metastatic tissue biopsy and peripheral blood draws were collected either at mCRPC diagnosis or at clinical progression on systemic therapy7,20. Tissue was sourced from metastatic bone, soft tissue or organ lesions by image-guided core needle biopsy as previously described20.
All samples (including plasma cfDNA, metastatic tumour tissue and leukocyte DNA) were subjected to initial targeted sequencing using an established panel of 73 prostate-cancer-relevant genes and/or whole-exome sequencing (WES)20,36. Initial sequencing enabled inference of prostate cancer driver gene genomics and ctDNA fraction (as a proportion of total cfDNA). Patients who provided at least two serial cfDNA samples (cohort A) or matched metastatic tissue and at least one cfDNA sample (cohort B) with at least one sample having cancer fraction of 30% or greater (cohorts A and B) were considered eligible for WGS (Fig. 1a). To maximize opportunity for subclonal reconstruction and deep genomic characterization, we prioritized patients with multiple high cancer fraction samples (from targeted sequencing). An additional five cohort A samples with less than 0.5% ctDNA from initial targeted sequencing were whole-genome-sequenced as negative controls. Four additional cfDNA samples from patients with metastatic urothelial carcinoma (n = 2) and metastatic histologically confirmed neuroendocrine prostate cancer (NEPC) (n = 2) were also whole-genome-sequenced as comparators; these supplementary samples were sourced from a provincial plasma cfDNA biobanking effort56. Samples that failed library generation or WGS were excluded. Sample inclusion and exclusion criteria were not pre-specified.
Approval for the collection and profiling of patient samples was granted by the University of British Columbia Research Ethics Board (cohort A) and the University of California San Francisco Institutional Review Board (cohort B). The study was conducted in accordance with the Declaration of Helsinki, and written informed consent was obtained from all patients before enrolment.
Blood and tissue collection, processing and DNA isolation
Whole blood was collected in 4 × 6-ml EDTA tubes or 2 × 9-ml Streck Cell-Free DNA BCT tubes. Samples collected in EDTA tubes were kept on ice or at 4 °C for a maximum of one hour, then centrifuged at 1,600 rpm for 10 min at 4 °C. Plasma was transferred to a new tube and spun for an additional 10 min at 1,600 rpm. Samples collected in Streck tubes were kept at room temperature and then centrifuged at 1,600g for 15 min. Buffy coat was aliquoted and plasma was transferred to a new tube and spun for an additional 10 min at 5,000g.
Plasma aliquots were stored at −80 °C before DNA extraction. cfDNA was extracted from up to 6 ml of plasma using the Qiagen Circulating Nucleic Acid kit. We included a one-hour lysis incubation at 60 °C and cfDNA was eluted in 60 μl water. After extraction, cfDNA was quantified with the Qubit 2.0 Fluorometer and Qubit dsDNA HS Assay Kit, or the Quantus Fluorometer and QuantiFluor ONE dsDNA system. Buffy coat aliquots were stored at −80 °C before DNA extraction. White blood cell (WBC) DNA was extracted from the buffy coat fraction using the Qiagen DNeasy Blood and Tissue kit or Promega Maxwell RSC Blood DNA kit and Maxwell RSC system, as per the manufacturer’s instructions.
Library preparation and sequencing
Sequencing libraries were prepared from 10–25 ng of plasma DNA input per sample. WBC DNA was sheared into approximately 180-bp fragments with a Covaris focused ultrasonicator or by enzymatic digestion prior to A-tailing, end repair, Illumina-compatible adapter ligation and PCR amplification. Library quantification was carried out with a NanoDrop spectrophotometer, and each library was run on an ethidium bromide gel to confirm success. Purified sample libraries were multiplexed into pools, diluted and sequenced in paired-end mode on Illumina X Ten instruments (V2.5 2×150 cycle kit).
Sequence alignment
Adapters in read 3′ ends were trimmed in paired-end mode using cutadapt v.1.11 with parameters -m 20 and -a/-A AGATCGGAAGAGC (ref. 57). Low-quality read tails (average base quality < 30) and low-quality bases (base quality < 20) were trimmed using the 'fasta trim by quality' and 'fasta mask by quality' subcommands of seqkit v.0.8. Reads were aligned against the hg38 reference genome using Bowtie v.2.3.0 with parameters --max-ins 1000 --score-min L,0,−0.6 (ref. 58). Duplicate fragments were marked using samblaster v.0.1.24 with default parameters and were omitted from further analyses59. Per-base read coverages in target regions were quantified using bedtools v.2.25.0 (ref. 60). cfDNA and WBC sample identities were verified on the basis of concordant single-nucleotide polymorphism (SNP) genotype profiles (see Supplementary Information, 'Validation of sample identities via SNP profile comparison').
Somatic and germline variant calling
We used Mutato v.0.7 (https://github.com/annalam/mutato) with arguments --alt-reads = 5 --alt-frac = 0.05 to generate a table of candidate single-nucleotide and indel variants, and their supporting read counts in all samples in the cohort. This table was post-processed using in-house scripts to identify somatic mutations and germline variants. Somatic mutations (base substitutions and indels) were called in cfDNA and tumour samples by searching for variants with eight or more supporting reads and a mutant allele fraction of 10% or higher. To remove germline SNPs, common sequencing or alignment errors and mutations of clonal haematopoietic origin, we required the mutant allele fraction to be at least 50× higher than the background error rate (that is, the average fraction of the mutant allele across all germline control samples, including 39 leukocyte samples and 5 non-cancerous cfDNA samples) and at least 10 times higher than the allele fraction in the patient’s matched WBC sample (with at least 10 reads covering the position). For base substitutions, the average mapping quality of mutation supporting reads was required to be at least 20, and the average distance of the mutant allele from the nearest read end was required to be at least 15 bases. Somatic mutation candidates with a population allele frequency of 0.5% or higher in the GNOMAD v.3.0 database were discarded. Protein-truncating mutations in clinically relevant prostate cancer genes were manually scrutinized for errors using Integrated Genomics Viewer (IGV) and validated against previous whole-exome and/or targeted sequencing variant calls20,36. Protein-level consequences of variants were predicted using ANNOVAR (v.20191024) using ANNOVAR RefGene annotations downloaded on 18th August 2020 (ref. 61). Mutational signatures were inferred using a Python implementation of DeconstructSigs v.1.47 and COSMIC mutational signatures (v.2). All single base-substitution signatures (SBSs) in the COSMIC v.2 catalogue were used for signature inference in all analyses (that is, no signatures were excluded upfront). We used default parameters for DeconstructSigs (which automatically disregards fitted signature weights below 6%)23,62. Microhomology counts were obtained using SigProfilerMatrixGenerator v.1.2.1 (ref. 63). Our mutation calling thresholds were calibrated to achieve a false discovery rate of 1.0% based on analysis of a cancer-negative control plasma cfDNA sample sequenced to 202× WGS depth. Further validation against GATK Mutect2 mutation calls is provided in the Supplementary Information.
For Fig. 4d and Supplementary Table 8, which show AR mutations, we required only 3 mutant reads with a variant allele frequency higher than 1% and at least 15 times the background error rate. These relaxed thresholds were only used for visualization, not in calculations of mutation frequencies and temporal changes within the cohort.
Rare and pathogenic germline variants were detected by searching for variants with an alternate allele fraction of at least 15%, and at least 8 supporting reads. Germline variants with a population allele frequency of 0.5% or higher in the GNOMAD v.3.0 database were discarded. DNA damage repair gene variant pathogenicity was assessed using ClinVar annotations (downloaded on 6th July 2020)64 (Supplementary Information).
Structural variant calling
Somatic structural rearrangements were identified in cfDNA and tumour samples using split-read methodology implemented in the Breakfast software v.0.5 (https://github.com/annalam/breakfast) with the --max-frag-len=1000 --anchor-len=30 options. A minimum of 5 unique junction-spanning reads were required. We discarded rearrangements with two or more supporting reads in the patient-matched leukocyte sample or in any samples from other patients (as rearrangement breakpoint positions are not recurrent between patients). Rearrangement breakpoints were annotated with adjacent Ensembl 84 genes. Rearrangements with breakpoints adjacent to a list of 177 known human cancer genes were collected into a spreadsheet and investigated by aligning the junction sequence using UCSC BLAT.
Tissue–cfDNA mutation concordance
To compare the presence and absence of mutations across same-day metastatic tissue and cfDNA samples, we defined mutations as concordant if they were independently detected in one sample and had at least one supporting mutant read in the other sample. Mutations were defined as discordant if they were independently detected in one sample and contained zero supporting reads in the other sample. However, variability in tumour content and sequencing depth can affect the sensitivity of mutation detection and result in false negative comparisons. To mitigate against potential false negatives, we only analysed variants amenable to reciprocal redetection. Specifically, mutations independently called in sample A were considered not amenable for detection in a paired sample B if the expected allele fraction MAFA × (FB/FA) was below 10%, where FB and FA were the tumour fractions of the B and A samples, respectively. In addition, if the probability of observing eight or more mutant reads in sample B (on the basis of a binomial distribution of observed read depth in B and expected allele fraction from sample A) was lower than 90%, the mutation was considered not amenable for detection6.
cfDNA nucleosome profiling
We applied a WPS to estimate nucleosome occupancy in cfDNA, modified from previously published methodologies13,14,15,16. In brief, the WPS is defined as the number of deduplicated cfDNA fragments that completely span a rolling 120-bp genomic window minus the number of cfDNA fragments partially overlapping the window (that is, containing an end-point within the interval). Only fragments of size 121–375 bp were queried, and improperly paired reads and/or reads with a mapping quality of lower than 30 were discarded. The raw WPS was zero-centred by subtracting its 1-kb rolling median and smoothed using a Savitzky-Golay filter13. The WPS corresponding to genomic regions with low sequencing coverage (median depth of lower than 50× in overlapping 1,000-bp bins) was masked from downstream analyses. Because the unadjusted WPS is theoretically proportional to sequencing read depth, we controlled for local coverage variation (thereby correcting for local copy number status) by dividing the WPS by the median depth of each initial rolling 120-bp interval. Given that the WPS is an approximately continuous periodic sinusoid, we reasoned that the Fourier components of the WPS with a period >300 bp or <50 bp were unlikely to reflect underlying chromatin biology. For additional noise suppression, we excluded the corresponding frequency ranges using a Butterworth bandpass filter.
To quantify nucleosome occupancy at precisely positionally defined biological features (that is, transcription start (TSS) and terminus sites (TTS), intron–exon junctions), we aggregated the normalized WPS from a set of genes and/or samples centred at individual features of interest (±1.5 kb upstream/downstream) and oriented 5′ to 3′ (for coding regions). We used scipy.signal find_peaks to identify positions with a high probability of aggregated nucleosome occupancy, and then calculated the area under the curve (AUC) of the ±2 peaks upstream or downstream of the feature of interest. The AUC score captures the degree of positional homogeneity of nucleosomes adjacent to a given chromatin landmark (among cfDNA-shedding cells) (Fig. 5a). To mitigate against the variable quality of the WPS signal—as well as to provide orthogonal validation of the WPS/AUC metric—we also leveraged a read-depth-based approach to measure nucleosome occupancy, similar to previous work14,16. For a set of regions and/or samples, we calculated relative read-depth depletion by normalizing to flanking regions (±1.5kb upstream or downstream of the chromatin feature of interest).
To measure gene-wide nucleosome depletion, we calculated relative coverage log ratios for gene bodies by normalizing to flanking regions (200-kb flanking regions with a 20-kb gap between the gene and the flanks). Coverage log ratios were calculated relative to a pooled white blood cell control for both cfDNA and tissue samples.
The positional coordinates of ARBS were less well-defined compared to gene structural features (e.g. TSS). To examine nucleosome occupancy at ARBS, we centred the WPS traces and read depth at the middle of each ARBS. Read-depth depletion was normalized using flanking tracts 1.5 kb upstream or downstream of the ARBS interval start or stop positions (rather than centre position). In our data, read-depth depletion appeared more informative for measuring ARBS nucleosome occupancy than WPS/AUC.
Evaluation of mitochondrial cfDNA
After initial alignment to the human reference genome using Bowtie v.2.3.0, we extracted reads mapping to the mitochondrial genome using SAMtools v.1.12 (htslib 1.12) (ref. 65). Nuclear DNA contains ancestral mitochondrial DNA (mtDNA) sequences (called nuclear mitochondrial DNA: NUMTs) that can result in read mismapping between the two genomic compartments. Therefore, to reduce the probability of NUMT contamination (that is, increase specificity for mtDNA), we separately realigned ChrM reads to both the nuclear and the mitochondrial reference genomes. We only preserved reads that realigned to the mitochondrial genome with a mapping quality of >35 and ≥10 higher than the mapping quality from nuclear DNA realignment. Picard 2.25.6 CollectInsertSizes was used to obtain cfDNA fragment sizes.
Statistical analyses and data visualization
Statistical tests and data analyses were conducted in Python v.3.7 (using pandas v.0.25.0, numpy v.1.16.4, scipy v.1.6.2 and statsmodels v.0.12.2), Julia v.1.5.1 (HypothesisTests 0.10.9 and Distributions 0.25.53), and R v.3.6.1 (dplyr 1.0.7). Descriptive statistics were used and sample size was not formally pre-specified. All hypothesis tests were two-tailed and required a 5% significance threshold; correction for multiple hypothesis testing was not performed. Visualizations were generated using matplotlib v.3.3.4 (Python) and ggplot 3.1.1 (R). All box plots are centred at the median and display interquartile ranges (IQRs) and minima and maxima extending to 1.5× IQR (as per convention); all raw data points are directly superposed where possible. The human figure in Fig. 1a was obtained and modified from Wikimedia Commons (original authors: P. J. Lynch, medical illustrator and C. C. Jaffe, cardiologist) available under a Creative Commons Attribution 2.5 generic license: https://en.wikipedia.org/wiki/File:Skeleton_whole_body_ant_lat_views.svg.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
The human reference genome hg38 was downloaded from UCSC. For cfDNA nucleosome depletion analyses, we used exon and TSS coordinates from the RefSeq Matched Annotation from NCBI and EMBL-EBI (MANE) database (genes not annotated in this database were omitted from analysis). Metastatic tissue RNA-sequencing data (cohort B) were obtained from previously published work (dbGaP study accession: phs001648.v2.p1)20. Gene sets representing housekeeping genes and highly expressed genes in haematopoietic lineages were derived from previously published work66 (Supplementary Tables 9 and 10). The 3,224 ARBSs were from previous chromatin immunoprecipitation followed by sequencing of 13 primary prostate cancer tissue samples41,42 (Supplementary Table 9). All de-identified WGS data have been deposited in the European Genome-Phenome Archive (EGA) under the accession code EGAS00001005783, and are available for download by contacting the corresponding authors. All other data supporting the findings of this study (source data) are available within the Article (including its Supplementary Information and Supplementary Tables).
Code availability
Custom computer code that was used for analysis is available on GitHub at https://github.com/annalam/cfdna-wgs-manuscript-code.
References
Ignatiadis, M., Sledge, G. W. & Jeffrey, S. S. Liquid biopsy enters the clinic—implementation issues and future challenges. Nat. Rev. Clin. Oncol. 18, 297–312 (2021).
Weber, Z. T. et al. Modeling clonal structure over narrow time frames via circulating tumor DNA in metastatic breast cancer. Genome Med. 13, 89 (2021).
Abbosh, C. et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 545, 446–451 (2017).
Parikh, A. R. et al. Liquid versus tissue biopsy for detecting acquired resistance and tumor heterogeneity in gastrointestinal cancers. Nat. Med. 25, 1415–1421 (2019).
Cresswell, G. D. et al. Mapping the breast cancer metastatic cascade onto ctDNA using genetic and epigenetic clonal tracking. Nat. Commun. 11, 1446 (2020).
Annala, M. et al. Evolution of castration-resistant prostate cancer in ctDNA during sequential androgen receptor pathway inhibition. Clin. Cancer Res. 27, 4610–4623 (2021).
Wyatt, A. W. et al. Concordance of circulating tumor DNA and matched metastatic tissue biopsy in prostate cancer. J. Natl Cancer Inst. 109, djx118 (2017).
Razavi, P. et al. Alterations in PTEN and ESR1 promote clinical resistance to alpelisib plus aromatase inhibitors. Nat. Cancer 1, 382–393 (2020).
Siravegna, G. et al. Radiologic and genomic evolution of individual metastases during HER2 blockade in colorectal cancer. Cancer Cell 34, 148–162 (2018).
O’Leary, B. et al. The genetic landscape and clonal evolution of breast cancer resistance to palbociclib plus fulvestrant in the PALOMA-3 trial. Cancer Discov. 8, 1390–1403 (2018).
Noorani, A. et al. Genomic evidence supports a clonal diaspora model for metastases of esophageal adenocarcinoma. Nat. Genet. 52, 74–83 (2020).
Woodcock, D. J. et al. Prostate cancer evolution from multilineage primary to single lineage metastases with implications for liquid biopsy. Nat. Commun. 11, 5070 (2020).
Snyder, M. W., Kircher, M., Hill, A. J., Daza, R. M. & Shendure, J. Cell-free DNA comprises an in vivo nucleosome footprint that informs its tissues-of-origin. Cell 164, 57–68 (2016).
Ulz, P. et al. Inference of transcription factor binding from cell-free DNA enables tumor subtype prediction and early detection. Nat. Commun. 10, 4666 (2019).
Zhu, G. et al. Tissue-specific cell-free DNA degradation quantifies circulating tumor DNA burden. Nat. Commun. 12, 1–11 (2021).
Ulz, P. et al. Inferring expressed genes by whole-genome sequencing of plasma DNA. Nat. Genet. 48, 1273–1278 (2016).
Peneder, P. et al. Multimodal analysis of cell-free DNA whole-genome sequencing for pediatric cancers with low mutational burden. Nat. Commun. 12, 3230 (2021).
Sun, K. et al. Orientation-aware plasma cell-free DNA fragmentation analysis in open chromatin regions informs tissue of origin. Genome Res. 29, 418–427 (2019).
Khalaf, D. J. et al. Optimal sequencing of enzalutamide and abiraterone acetate plus prednisone in metastatic castration-resistant prostate cancer: a multicentre, randomised, open-label, phase 2, crossover trial. Lancet Oncol. 20, 1730–1739 (2019).
Quigley, D. A. et al. Genomic hallmarks and structural variation in metastatic prostate cancer. Cell 174, 758–769 (2018).
Abida, W. et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl Acad. Sci. USA. 116, 11428–11436 (2019).
van Dessel, L. F. et al. The genomic landscape of metastatic castration-resistant prostate cancers reveals multiple distinct genotypes with potential clinical impact. Nat. Commun. 10, 5251 (2019).
Alexandrov, L. B. et al. The repertoire of mutational signatures in human cancer. Nature 578, 94–101 (2020).
Wu, Y.-M. et al. Inactivation of CDK12 delineates a distinct immunogenic class of advanced prostate cancer. Cell 173, 1770–1782 (2018).
Black, J. R. M. & McGranahan, N. Genetic and non-genetic clonal diversity in cancer evolution. Nat. Rev. Cancer 21, 379–392 (2021).
Dentro, S. C. et al. Characterizing genetic intra-tumor heterogeneity across 2,658 human cancer genomes. Cell 184, 2239–2254 (2021).
Tarabichi, M. et al. A practical guide to cancer subclonal reconstruction from DNA sequencing. Nat. Methods 18, 144–155 (2021).
Bielski, C. M. et al. Genome doubling shapes the evolution and prognosis of advanced cancers. Nat. Genet. 50, 1189–1195 (2018).
Quinton, R. J. et al. Whole-genome doubling confers unique genetic vulnerabilities on tumour cells. Nature 590, 492–497 (2021).
Cohen-Sharir, Y. et al. Aneuploidy renders cancer cells vulnerable to mitotic checkpoint inhibition. Nature 590, 486–491 (2021).
Zaccaria, S. & Raphael, B. J. Accurate quantification of copy-number aberrations and whole-genome duplications in multi-sample tumor sequencing data. Nat. Commun. 11, 4301 (2020).
Chen, C. D. et al. Molecular determinants of resistance to antiandrogen therapy. Nat. Med. 10, 33–39 (2004).
Henzler, C. et al. Truncation and constitutive activation of the androgen receptor by diverse genomic rearrangements in prostate cancer. Nat. Commun. 7, 13668 (2016).
Takeda, D. Y. et al. A somatically acquired enhancer of the androgen receptor is a noncoding driver in advanced prostate cancer. Cell 174, 422–432 (2018).
Li, Y. et al. Diverse AR gene rearrangements mediate resistance to androgen receptor inhibitors in metastatic prostate cancer. Clin. Cancer Res. 26, 1965–1976 (2020).
Annala, M. et al. Circulating tumor dna genomics correlate with resistance to abiraterone and enzalutamide in prostate cancer. Cancer Discov. 8, 444–457 (2018).
Baca, S. C. et al. Punctuated evolution of prostate cancer genomes. Cell 153, 666–677 (2013).
Viswanathan, S. R. et al. Structural alterations driving castration-resistant prostate cancer revealed by linked-read genome sequencing. Cell 174, 433–447 (2018).
Esfahani, M. S. et al. Inferring gene expression from cell-free DNA fragmentation profiles. Nat. Biotechnol. 40, 585–597 (2022).
Davies, A. H., Beltran, H. & Zoubeidi, A. Cellular plasticity and the neuroendocrine phenotype in prostate cancer. Nat. Rev. Urol. 15, 271–286 (2018).
Pomerantz, M. M. et al. The androgen receptor cistrome is extensively reprogrammed in human prostate tumorigenesis. Nat. Genet. 47, 1346–1351 (2015).
Morova, T. et al. Androgen receptor-binding sites are highly mutated in prostate cancer. Nat. Commun. 11, 832 (2020).
Rebello, R. J. et al. Prostate cancer. Nat Rev. Dis. Primers 7, 9 (2021).
Chi, K. N. et al. A prognostic index model for predicting overall survival in patients with metastatic castration-resistant prostate cancer treated with abiraterone acetate after docetaxel. Ann. Oncol. 27, 454–460 (2016).
Turajlic, S. et al. Deterministic evolutionary trajectories influence primary tumor growth: TRACERx Renal. Cell 173, 595–610 (2018).
Gundem, G. et al. The evolutionary history of lethal metastatic prostate cancer. Nature 520, 353–357 (2015).
Kumar, A. et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat. Med. 22, 369–378 (2016).
Quigley, D. et al. Analysis of circulating cell-free DNA identifies multiclonal heterogeneity of reversion mutations associated with resistance to PARP inhibitors. Cancer Discov. 7, 999–1005 (2017).
Unterrainer, M. et al. Recent advances of PET imaging in clinical radiation oncology. Radiat. Oncol. 15, 88 (2020).
Brandão, M., Caparica, R., Eiger, D. & de Azambuja, E. Biomarkers of response and resistance to PI3K inhibitors in estrogen receptor-positive breast cancer patients and combination therapies involving PI3K inhibitors. Ann. Oncol. 30, x27–x42 (2019).
Parikh, A. et al. Systematic liquid biopsy identifies novel and heterogeneous mechanisms of acquired resistance in gastrointestinal (GI) cancer patients. Ann. Oncol. 28, iii137 (2017).
Balanis, N. G. et al. Pan-cancer Convergence to a small-cell neuroendocrine phenotype that shares susceptibilities with hematological malignancies. Cancer Cell 36, 17–34 (2019).
Herberts, C. & Wyatt, A. W. Technical and biological constraints on ctDNA-based genotyping. Trends Cancer Res. 7, 995–1009 (2021).
Turajlic, S., Sottoriva, A., Graham, T. & Swanton, C. Resolving genetic heterogeneity in cancer. Nat. Rev. Genet. 20, 404–416 (2019).
Scher, H. I. et al. Trial design and objectives for castration-resistant prostate cancer: updated recommendations from the prostate cancer clinical trials working group 3. J. Clin. Oncol. 34, 1402–1418 (2016).
Warner, E. et al. BRCA2, ATM, and CDK12 defects differentially shape prostate tumor driver genomics and clinical aggression. Clin. Cancer Res. 27, 1650–1662 (2021).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10 (2011).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Faust, G. G. & Hall, I. M. SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics 30, 2503–2505 (2014).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010).
Rosenthal, R., McGranahan, N., Herrero, J., Taylor, B. S. & Swanton, C. DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 17, 31 (2016).
Bergstrom, E. N. et al. SigProfilerMatrixGenerator: a tool for visualizing and exploring patterns of small mutational events. BMC Genomics 20, 685 (2019).
Landrum, M. J. et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46, D1062–D1067 (2018).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021).
Eisenberg, E. & Levanon, E. Y. Human housekeeping genes, revisited. Trends Genet. 29, 569–574 (2013).
Acknowledgements
This work was funded by the Canadian Institutes of Health Research, the Canadian Cancer Society Research Institute, the Prostate Cancer Foundation, Prostate Cancer Canada, the Movember Foundation, the Jane and Aatos Erkko Foundation, the Academy of Finland Center of Excellence programme (project no. 312043), the Terry Fox New Frontiers Program and the BC Cancer Foundation. No funding sources were involved in the design or execution of the study. We thank S. Taavitsainen and R. Nätkin for their assistance, and are grateful to all participating patients and their families.
Author information
Authors and Affiliations
Consortia
Contributions
C.H.: conceptualization, data curation, software construction, formal analysis, visualization, methodology, writing (original draft), project administration, writing (review and editing). M.A.: conceptualization, data curation, software construction, formal analysis, visualization, methodology, writing (original draft), project administration, writing (review and editing). J.S.: conceptualization, data curation, software construction, formal analysis, visualization, methodology, writing (original draft), project administration, writing (review and editing). S.W.S.N.: data curation, software construction, formal analysis, visualization, methodology, writing (review and editing). X.E.C.: data curation, software construction, formal analysis, visualization, methodology, writing (review and editing). A.N.: data curation, formal analysis, software construction, writing (review and editing). O.V.K.: data curation, formal analysis, writing (review and editing). A.D.M.: software construction, formal analysis, visualization, methodology, writing (review and editing). K.B.: data curation, validation, methodology, writing (review and editing). E.S.: data curation, validation, methodology, writing (review and editing). C.Q.B.: data curation, validation, methodology, writing (review and editing). E.R.: software construction, methodology, writing (review and editing). J.V.W.B.: data curation, validation, writing (review and editing). N.A.L.: resources, writing (review and editing). M.N.: resources, writing (review and editing). R.A.: resources, writing (review and editing). E.J.S.: resources, writing (review and editing). M.E.G.: resources, writing (review and editing). D.A.Q.: resources, project administration, writing (review and editing). F.Y.F.: resources, project administration, supervision, writing (review and editing). K.N.C.: conceptualization, resources, project administration, supervision, funding acquisition, writing (review and editing). A.W.W.: conceptualization, resources, project administration, supervision, funding acquisition, writing (original draft), writing (review and editing). All authors reviewed and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
M.A. is a shareholder in Fluivia. R.A. has received honoraria from Clovis Oncology and has served in advisory or consulting roles for Dendreon, Advanced Accelerator Applications, Clovis Oncology, Axiom Biotechnologies, AstraZeneca, Pfizer, Merck, Amgen, Jubilant Pharmaceuticals and Alessa Therapeutics. R.A. also has research funding from Zenith Epigenetics, Novartis, Xynomic Pharma, Cancer Targeted Technology, Janssen, Merck, Abbvie, Amgen, AstraZeneca and BioXCel Therapeutics. E.J.S. has stock in Fortis, Harpoon and Teon, and has received honoraria for speaking engagements for Janssen and Johnson & Johnson. E.J.S. has also served in compensated advisory or consulting roles for Janssen, Fortis, Teon and Ultragenyx. M.E.G. is the founder of OncoGenex Technologies, Sustained Therapeutics and Sitka Pharmaceuticals, and has consulted for Astellas, AstraZeneca, Bayer, Janssen, Sanofi, Pfizer, Tersera, Roche and Genova Diagnostics. D.A.Q. has consulted for Varian and Circle Pharmaceuticals. F.Y.F. has served as a consultant or advisory board member for Janssen, Celgene, Blue Earth Diagnostics, Astellas, Myovant, Roivant, Bayer, Novartis and Foundation Medicine, has stock options in Artera and has stock options and is a scientific advisory board member for SerImmune and BlueStar Genomics. K.N.C. has research funding from Janssen, Astellas and Sanofi. K.N.C. also reports research funding and personal fees from Janssen, Astellas, AstraZeneca and Sanofi, as well as personal fees from Constellation Pharmaceuticals, Daiichi Sankyo, Merck, Novartis, Pfizer, Point Biopharma and Roche outside the submitted work. A.W.W. has served on advisory boards and/or received honoraria from AstraZeneca, Astellas, Janssen, and Merck. The laboratory of A.W.W. has a contract research agreement with ESSA Pharma. C.H., J.S., S.W.S.N., X.E.C., A.N., O.V.K., A.D.M., K.B., E.S., C.Q.B., E.R., J.V.W.B., N.A.L. and M.N. declare no competing interests.
Peer review
Peer review information
Nature thanks Martijn Lolkema, Adam Sowalsky and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Genomic features of ctDNA and metastatic tissue whole genomes.
(a) Per-sample overview of genomic information (grouped by same-patient sample pairs). Figure content is otherwise identical to Fig. 1d. (b) Fragment-length distribution of plasma cell-free DNA (cfDNA) of nuclear (left) and mitochondrial (right) origin. (c) Structures of E26 transformation-specific (ETS) family oncogenic fusions detected in the cohort. Solid colours denote intragenic regions of fusion partner genes (including untranslated regions) and lighter hues represent flanking intergenic 5’ upstream or 3’ downstream regions. The intergenic region between SLC45A3 and ELK4 is shown in grey. (d) Box plots illustrating higher microhomology-containing deletion counts (deletion length 3–5 bp with at least 2 bp of microhomology) (left) and structural variant counts (right) in samples with BRCA2 defects relative to wild-type (WT) samples.
Extended Data Fig. 2 AR gene and enhancer amplification mechanisms and amplicon structure in cfDNA and metastatic tissue.
(a) AR-neighbourhood copy number structure in same-day patient-matched cfDNA and metastatic tissue. Upper and lower plots display absolute copy number (i.e. corrected for tumour content) and uncorrected coverage log ratios, respectively. In this patient, the tissue sample shows a focal amplification of the AR enhancer region and a broad copy gain of the entire locus, whereas the cfDNA sample shows a more complex pattern of apparent nested copy gains. Despite these contrasting structures, the overall AR enhancer and gene body copy numbers are relatively similar (considering that the range of AR gene body absolute copy number across all samples was 1-91). (b) Absolute copy number across the AR gene and enhancer neighbourhood for three cfDNA samples and one tissue sample. Pileup of all per-sample structural arrangements shown below (indicating dominant mechanisms of copy amplification). Note evidence of local enhancer copy loss in patients, suggestive of enhanced cfDNA degradation owing to nucleosome depletion38. (c) Average copy number change at the AR locus across serial ctDNA collected during systemic treatment with AR signalling inhibitors. Structural variant breakpoints and tandem duplication events from all cfDNA and tissue samples (regardless of time point) are shown below, illustrating a convergence toward amplification of the AR gene and enhancer.
Extended Data Fig. 3 Expression-linked features and clinico-genomic correlates of nucleosome depletion.
(a–b) Correlation between transcription terminus site (TTS) nucleosome depletion and metastatic tissue mRNA expression decile for 61 cfDNA samples in our cohort, measured using relative read-depth depletion and WPS area under the curve. (c) Aggregate spatial read-depth profile of 61 cfDNA samples centred at the TTS, stratified into gene subsets by mRNA expression decile. (d) Aggregate spatial coverage log ratio profile of 61 cfDNA samples centred at gene bodies, with traces stratified into gene subsets representing median metastatic tissue mRNA expression deciles. Coverage log ratios are calculated relative to a pooled white blood cell normal. Traces are normalized to flanking regions (50 kb at ±2 Mb from the gene start/end) and are smoothed using locally weighted smoothing. (e) Examples of spatial gene body coverage depletion in cfDNA (left) but not patient-matched metastatic tumour tissue (right) in highly expressed genes (top). Gene bodies (TSS to TTS) are shaded grey. (f) In silico reverse dilution experiment illustrating the effect of ctDNA fraction on nucleosome depletion as measured by area under the WPS (±2 peaks surrounding the TSS). Vertical axis shows aggregated TSS nucleosome depletion (in 61 cfDNA samples) at 350 genes highly expressed in hematopoietic cells but lowly expressed in mCRPC tissue. Horizontal axis represents fraction of ctDNA sample sequencing coverage that has been randomly substituted for normal cfDNA not derived from prostate cancer (1 = complete substitution with healthy cfDNA). (g) Correlation between AR gene and enhancer copy number (horizontal axis) versus average read-depth depletion at ARBSs. (h) Correlation between time-matched PSA (ng ml−1) and WGS ctDNA fraction estimates versus average ARBS read-depth depletion.
Extended Data Fig. 4 Joint analysis of plasma cfDNA fragment locations and lengths around the TSSs of high- and low-expressed genes.
(a) cfDNA fragment density heat maps ('nucleograms') averaged across 63 mCRPC cfDNA samples in the cohort (left column), for four gene sets defined based on their expression quartile in mCRPC tissues. The middle column shows the same heat maps after eliminating interaction terms (i.e. outer product of the marginal distributions). In the right column heat maps, the interaction terms are isolated by subtracting the middle column heat maps from the left column heat maps. The analysis reveals enrichment of short cfDNA fragments at the TSS, and longer cfDNA fragments at immediate flanking regions. (b) Single gene nucleograms showing fragment localization and lengths near TSSs of four highly expressed genes (averaged across 63 mCRPC cfDNA samples). Nucleosome positions are well conserved and clearly visible, but the precise nucleosome localization pattern varies between genes. (c) cfDNA fragment length histograms within two 100 bp windows positioned relative to gene transcription start sites (TSS), grouped by gene expression level.
Supplementary information
Supplementary Information
This file contains a Supplementary Note (providing a list of Stand Up 2 Cancer (SU2C) / Prostate Cancer Foundation (PCF) West Coast Prostate Cancer Dream Team members and affiliations), as well as Supplementary Methods and associated Supplementary Figures 1–25. All technical validation experiments (and supporting data) are enclosed.
Supplementary Tables
Supplementary Tables 1–11, detailing (1) overview of clinical samples (2), time between serial cfDNA collections, (3) validation of truncal copy number models using Battenberg and ASCAT, (4) genomic rearrangements affecting known putative prostate cancer driver genes, (5) validation of subclonal reconstruction via blinded analysis of simulated whole genomes, (6) per-subclone fitted mutational signature weights from DeconstructSigs, (7) genes with a high frequency of protein-altering mutations (overall and acquired after AR targeted therapy), (8) pathogenic androgen-receptor point mutations, (9) 3804 housekeeping genes, (10) 350 genes highly expressed in haematopoietic lineages but minimally expressed in mCRPC tissue, (11) coordinates of 3224 AR-binding sites derived from ChIP–seq of primary prostate cancer tissue (hg38).
Rights and permissions
About this article

{kind=link}
Cite this article
Herberts, C., Annala, M., Sipola, J. et al. Deep whole-genome ctDNA chronology of treatment-resistant prostate cancer. Nature 608, 199–208 (2022). https://doi.org/10.1038/s41586-022-04975-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-022-04975-9
- Springer Nature Limited
This article is cited by
-
Patient-derived castration-resistant prostate cancer model revealed CTBP2 upregulation mediated by OCT1 and androgen receptor
BMC Cancer (2024)
-
Multimodal epigenetic sequencing analysis (MESA) of cell-free DNA for non-invasive colorectal cancer detection
Genome Medicine (2024)
-
Genomic profiling and clinical utility of circulating tumor DNA in metastatic prostate cancer: SCRUM-Japan MONSTAR SCREEN project
BJC Reports (2024)
-
Introduction and impact of routine whole genome sequencing in the diagnosis and management of sarcoma
British Journal of Cancer (2024)
-
The yin and yang of chromosomal instability in prostate cancer
Nature Reviews Urology (2024)