

Abstract
Pathogenic fungi reside in the intestinal microbiota but rarely cause disease. Little is known about the interactions between fungi and the immune system that promote commensalism. Here we investigate the role of adaptive immunity in promoting mutual interactions between fungi and host. We find that potentially pathogenic Candida species induce and are targeted by intestinal immunoglobulin A (IgA) responses. Focused studies on Candida albicans reveal that the pathogenic hyphal morphotype, which is specialized for adhesion and invasion, is preferentially targeted and suppressed by intestinal IgA responses. IgA from mice and humans directly targets hyphal-enriched cell-surface adhesins. Although typically required for pathogenesis, C. albicans hyphae are less fit for gut colonization1,2 and we show that immune selection against hyphae improves the competitive fitness of C. albicans. C. albicans exacerbates intestinal colitis3 and we demonstrate that hyphae and an IgA-targeted adhesin exacerbate intestinal damage. Finally, using a clinically relevant vaccine to induce an adhesin-specific immune response protects mice from C. albicans-associated damage during colitis. Together, our findings show that adaptive immunity suppresses harmful fungal effectors, with benefits to both C. albicans and its host. Thus, IgA uniquely uncouples colonization from pathogenesis in commensal fungi to promote homeostasis.
Similar content being viewed by others

Main
The intestinal microbiota contains fungal commensals that are potentially pathogenic. Prominent members of this community include opportunistic pathogens such as Candida species, which are capable of causing deadly disseminated infections4. Certain intestinal fungi exacerbate diseases such as inflammatory bowel disease (IBD)5,6. However, commensal fungi are benign in most healthy individuals. The forces that maintain homeostatic interactions between fungi and host immunity are not well defined. IgA is one of the effector molecules produced by the intestinal immune system and multiple studies have demonstrated the importance of IgA in the maintenance of homeostasis with bacteria7. Although systemic anti-fungal antibody responses have been documented in detail8,9, and certain fungi promote mucosal antibody responses10,11, little is known about how antibodies directly regulate fungi within their commensal niche. To address this knowledge gap, we studied mouse and human IgA responses against common fungal species in the gut and used the powerful genetic tools that are available for studying fungi to identify IgA-targeted effectors that influence intestinal homeostasis.
Intestinal IgA targets Candida species
The reactivity of intestinal IgA to four commensal fungi—Candida glabrata, Candida albicans, Saccharomyces cerevisiae and Candida tropicalis—was tested using human faecal samples. Most samples contained fungal-reactive antibodies and IgA dominated the response (Extended Data Fig. 1a). One form of IBD, Crohn’s disease, is associated with serum antibodies called ASCAs (anti-Saccharomyces cerevisiae antibodies), which target cell-wall components in Saccharomyces and Candida species12, and therefore we compared ASCAs in our samples. IBD status did not affect the levels of fungal-reactive IgA or IgA ASCAs in the faeces (Extended Data Fig. 1c-f), which is in contrast to the elevated levels of ASCAs observed in the serum and an increased reactivity to S. cerevisiae in patients with Crohn’s disease (Extended Data Fig. 1c–f). Although not altered by IBD, IgA was significantly less reactive towards S. cerevisiae and most reactive towards C. albicans (Fig. 1a, Extended Data Fig. 1b). Together, these results suggest that homeostatic intestinal IgA targets specific members of the fungal community, and that serum and mucosal Ig responses are distinct.

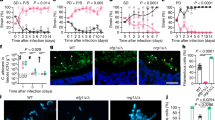
a, Human faecal IgA binding to cultured fungi quantified by flow cytometry (n = 30 healthy, n = 23 Crohn’s disease (CD) and n = 17 ulcerative colitis (UC)). AU, arbitrary units. b, IgA binding to faecal fungi (GFP–yeast, iRFP−, CFW+) (n = 4 S. cerevisiae (Sc)-colonized and n = 3 C. albicans (Ca)-colonized mice per group). c, IgA per mg of intestinal contents, assessed by enzyme-linked immunosorbent assay (ELISA) in small intestine (SI), caecum and colon from GF or monocolonized mice four weeks after inoculation. d, e, Colon lamina propria IgA+ plasma cells (PCs) (IgA+CD138+CD45+CD3−CD19− live cells) (d) and Peyer’s patch GC B cells (GL-7+FAS+IgD−CD19+ live cells) (e) from monocolonized mice four weeks after inoculation (n = 4 mice per group). f, Intestinal IgA reactivity to cultured C. albicans or S. cerevisiae quantified by flow cytometry (n = 4 GF, n =4 C. albicans-colonized and n = 4 S. cerevisiae-colonized mice per group). Data in b–f are representative of two experiments. g, Volcano plot of the ratio of caecal C. albicans transcripts in monocolonized wild-type (WT) and Rag1−/− mice four weeks after inoculation. SAP4 (Padj = 6.43 × 10−73) excluded from plot. h, GSEA of genes upregulated in hyphae (from a previous study1). i, Anti-C. albicans antibody (green) staining of C. albicans in antibiotic-treated wild-type and Rag1−/− mice four weeks after inoculation. Images from caecum (n = 5 mice per group; one experiment). j, Faecal C. albicans in antibiotic-treated wild-type and μMT−/− mice three weeks after inoculation (n = 5 mice per group; one experiment). Scale bars, 50 μm (i, j). k, Imaging flow cytometry images of IgA+ C. albicans from monocolonized mice. IgA+ and IgA− populations assessed via object circularity score. Bright-field (BF), calcofluor white (CFW), IgA, and CFW and IgA composite. Data are pooled from three B6 mice monocolonized with C. albicans three weeks after inoculation and are representative of two experiments. P values calculated using two-way ANOVA with Tukey’s test (b) or Sidak’s test (c, f, i), one-way ANOVA with Tukey’s test (d, e), Friedman test with paired Dunn’s multiple comparisons (a), two-sided unpaired t-test (j), Wald’s test with multiple test correction (g) or the ‘fgsea’ R package that corrects for multiple tests (h). Data are mean ± s.d. (c–f, i, j).
Mono-association of germ-free (GF) mice with C. albicans induced a specific IgA response in both C57BL/6 and Swiss Webster (SW) mice, which was characterized by the binding of IgA to C. albicans in the faeces, the induction of total and C. albicans-specific IgA, increased numbers of IgA plasma cells in the colon and increased numbers of Peyer’s patch lymphocytes, which include germinal centre B cells (GC B cells). By contrast, IgA and immune cell populations in mice colonized with S. cerevisiae were similar to those in GF mice (Fig. 1b–d, Extended Data Fig. 2a–f). Colonization with C. albicans, but not S. cerevisiae, induced a serum IgA and/or IgG1 response9 (Extended Data Fig. 2g, h), despite similar colonization levels (Extended Data Fig. 2i). A collection of 124 strains of S. cerevisiae13 that was screened for the induction of IgA through pooled colonization of GF mice still failed to induce IgA (Extended Data Fig. 3a–c). C. albicans is not unique in its ability to induce intestinal IgA responses, as C. glabrata also induced total and C. glabrata-specific IgA in addition to Peyer’s patch GC B cells and T follicular helper (TFH) cells (Extended Data Fig. 2j–m). C. albicans- and C. glabrata-induced IgA was species-specific and did not cross-react with other species in vitro (Fig. 1f, Extended Data Fig. 2n). In addition, the binding of IgA to C. albicans was significantly reduced in T-cell-deficient mice (Tcrb−/− mice) relative to wild-type mice (Extended Data Fig. 2o), suggesting that the C. albicans-induced IgA is T-cell-dependent. Together, these data demonstrate that several different fungi induce intestinal IgA responses in humans and mice, but that the response is species-dependent.
IgA targets adhesive fungal effectors
To study how T-cell-dependent IgA responses influence C. albicans, RNA was isolated from monocolonized wild-type or T- and B-cell-deficient Rag1−/− mice for RNA sequencing (RNA-seq). Despite similar colonization between the genotypes (Extended Data Fig. 4a), 25% of C. albicans genes were differentially regulated (q ≤ 0.05) between wild-type and Rag1−/− mice (Fig. 1g, Supplementary Table 1), and were enriched for genes that regulate pathogenesis, symbiosis and adhesion (Fig. 1g, Extended Data Fig. 4b, c). Virulence factors, such as the ALS adhesins, candidalysin (ECE1) and the SAP proteases, which promote tissue invasion and damage in disseminated infection models, were upregulated in Rag1−/− mice14. Carbohydrate and amino acid transporters were significantly downregulated in Rag1−/− mice (Extended Data Fig. 4c, d), suggesting that nutrient acquisition may be altered by adaptive immune responses. Notably, many of the upregulated genes in Rag1−/− mice are known to be specifically expressed during the C. albicans yeast-to-hyphal morphological transition, in which C. albicans forms elongated hyphae that are specialized for adhesion and tissue invasion15. Indeed, gene set enrichment analysis (GSEA) revealed a significant enrichment of hyphal-associated genes in the Rag1−/− mice (Fig. 1h; normalized enrichment score (NES) = 2.94 and Padj = 3.6 × 10−4), together suggesting that adaptive immune responses suppress the expression of genes associated with C. albicans hyphae. Imaging of C. albicans morphology revealed an increase in the proportion of C. albicans hyphal cells in Rag1−/− mice (Fig. 1i, Extended Data Fig. 4e). Control of C. albicans morphology was dependent on B cells, as B-cell-deficient μMT −/− (μMT is also known as Ighm) mice (Extended Data Fig. 4f) are also unable to suppress C. albicans hyphae (Fig. 1j). Imaging flow cytometry (ImageStream) revealed that elongated hyphae dominated the IgA-bound population in the faeces. Furthermore, IgA+ fungal cells were quantifiably less circular, or yeast-shaped, than the IgA− population (Fig. 1k). Thus, IgA targets C. albicans hyphae and intestinal B cell responses temper expression of this morphotype in the gut.
We have shown here that intestinal IgA preferentially binds to specific fungi, and previous studies have revealed a similar phenomenon for gut bacteria7. However, for both bacteria and fungi, little is known regarding the identity and function of epitopes that are targeted by IgA. To identify C. albicans genes that are required for IgA targeting, we screened two homozygous deletion mutant collections for strains with reduced IgA binding16,17. This analysis identified 13 strains with a reduction in IgA binding. The ahr1∆/∆ strain (adhesion and hyphae regulator 1) and other identified mutants correspond to transcription factors that promote C. albicans adhesion and biofilm formation18 (Fig. 2a, b, Supplementary Table 2). Notably, adhesion and biofilm formation are also central characteristics of hyphae, highlighting adhesion as a key process targeted by intestinal IgA responses.

a, Strains from the Noble16 (white bars) and Homann17 (grey bars) collections with an IgA binding z-score of ≤ −2. Intestinal wash from SW and B6 mice that were monocolonized with C. albicans. Strains in bold were identified in both collections (n = 2; one experiment). b, Gene Ontology (GO) biological process terms that were enriched in the strains from a. c, Small intestinal IgA. d, ALS1 quantitative PCR with reverse transcription (qRT–PCR) from the caecum contents of monocolonized mice. Normalized to PMA1 (n = 4 mice per group; one experiment (c, d)). e, Cell-wall fractions probed with intestinal-wash IgA from C. albicans-monocolonized mice. Als3 was identified only in the hyphal fraction using LC–MS/MS (representative of two experiments). f, IgA reactivity of monocolonized B6 (circles) or SW (squares) mice to the control S. cerevisiae, which expresses the CWP1 cell-surface scaffold but not the adhesin, or S. cerevisiae expressing the indicated C. albicans adhesins. Binding intensity was normalized to strains stained without faecal wash (FW) (n = 7 B6 and n = 8 SW mice; two experiments). g, Human faecal IgA reactivity to S. cerevisiae strains expressing the indicated C. albicans adhesins. Cg-Ad, CAGL0B00154g. Staining intensity for each human sample was normalized to the control S. cerevisiae strain expressing no adhesins (n = 70; one experiment). P values calculated using one-way ANOVA with Tukey’s test (c, d, g), or Holm–Sidak’s test (f). Data are mean ± s.d. (c, d, f, g).
The role of filamentation and adhesion in the induction of IgA was tested using a yeast-locked strain (TetOn-NRG1) that constitutively expresses the Nrg1 transcription factor; this strain blocks filamentation unless treated with anhydrotetractyline (aTC) to repress the expression of NRG119 (Extended Data Fig. 5a). The ahr1∆/∆ strain, which has defects in adhesion but is still capable of hyphal formation, was also used20. TetOn-NRG1 remained locked in the yeast state in monocolonized mice, and the ahr1∆/∆ strain produced hyphae to a similar level to wild-type C. albicans (Extended Data Fig. 5b). Analysis of IgA immune responses revealed that both TetOn-NRG1 and ahr1Δ/Δ induced significantly less intestinal IgA, with a trend towards fewer plasma cells in the colon, and fewer Peyer’s patch GC B cells and TFH cells, despite colonizing the intestine to a similar extent to wild-type C. albicans (Fig. 2c, Extended Data Fig. 5c–f). The function of Ahr1 in IgA stimulation was confirmed using a TetOff-AHR1 strain (Extended Data Fig. 5g–k). These data suggest that both Ahr1 and hyphae express molecules responsible for the induction of IgA immune responses.
Cell-surface adhesin proteins mediate C. albicans hyphae adherence to host tissue21 and Ahr1 directly promotes the expression of adhesin genes20,22. Notably, a key difference between C. glabrata and S. cerevisiae, which are otherwise genetically similar, is that C. glabrata encodes a large group of adhesin genes that facilitate host tissue association23. To test the hypothesis that fungal adhesins are targeted by IgA, cell-wall protein fractions from yeast and hyphal C. albicans were probed with intestinal IgA from mice that were monocolonized with C. albicans. An increase in IgA binding to a 245-kDa molecular weight region was detected in the hyphal cell-wall fraction. Liquid chromatography with tandem mass spectrometry (LC–MS/MS) analysis identified the Als3 adhesin as the most abundant protein specific to the hyphal fraction (Fig. 2e, Supplementary Table 3). In addition, quantitative PCR (qPCR) analysis revealed that the ALS1 adhesin was significantly reduced in expression in both the ahr1∆/∆ and the yeast-locked TetOn-NRG1 C. albicans strains (Fig. 2d, Extended Data Fig. 5l), together implicating Als1 and Als3 as adhesin targets of IgA. In line with this, constitutive expression of ALS1 (TetOn-ALS1 ahr1∆/∆) increased IgA binding to ahr1∆/∆ cells to a level even above that of the wild type, but repression of ALS1 (TetOff-ALS1 ahr1∆/∆) reduced IgA binding compared to the wild type (Extended Data Fig. 5m). Leveraging a collection of S. cerevisiae strains engineered to express C. albicans adhesins on the cell surface24 (Extended Data Fig. 6a), we observed that expression of C. albicans Als1, Als3 and Hwp1 adhesins was sufficient to promote IgA binding (Fig. 2f). Similarly, one predicted C. glabrata adhesin was recognized by C. glabrata-induced IgA (Extended Data Fig. 6b). The Als1 and Als3 adhesins are also directly targeted by human faecal IgA (Fig. 2g, Extended Data Fig. 5n), demonstrating that C. albicans adhesins are the dominant IgA epitopes. Of note, only Als1-expressing S. cerevisiae was able to induce IgA and IgG (Extended Data Fig. 7), indicating that although multiple adhesins are direct targets of the host IgA response, only specific adhesins are sufficient to induce antibody-promoting immune responses.
Adaptive immunity improves Candida fitness
GF Rag1−/− and wild-type mice were colonized with isogenic C. albicans strains expressing different fluorescent markers (called conditioned strains) to test whether adaptive immunity influences the colonization of C. albicans. Wild-type- and Rag1−/−-conditioned C. albicans were then competed in a group of C. albicans-naive wild-type or Rag1−/− mice (Fig. 3a). C. albicans strains conditioned for four weeks in wild-type mice had a significant competitive advantage over C. albicans conditioned in Rag1−/−mice (Fig. 3b). Supporting that IgA—which takes at least seven days to induce—is involved, this competitive advantage was lost when strains are conditioned for only two days (Fig. 3b). Notably, the wild-type-conditioned C. albicans fitness advantage did not require T and B cells in the recipient mice (Fig. 3b), and this fitness advantage persisted until 14 days (Extended Data Fig. 8). To test a role for hyphae during immune conditioning, the yeast-locked TetOn-NRG1 strain was used to repeat the four-week conditioning and competition experiment. In contrast to the wild-type C. albicans, immune conditioning of yeast-locked C. albicans did not increase its competitive fitness (Fig. 3c), indicating that the immune-mediated advantage depends on the ability to undergo the yeast to hyphal transition. In support of a role for adhesins in this process, constitutive expression of ALS3 (TetOn-ALS3) significantly reduced competitive fitness compared to the wild type, whereas repression of ALS3 expression (TetOff-ALS3) rescued competitive fitness (Fig. 3d). Together, these data suggest that expression of effectors enriched on hyphae are detrimental to the competitive fitness of C. albicans.

a, Schematic of intestinal conditioning of C. albicans in wild-type and Rag1−/− GF mice and subsequent competition in naive mice. b, Competitive index (CI) four days after inoculating antibiotic-treated mice with conditioned C. albicans (n = 2 B6 and n = 3 Rag1−/− mice (2 days); n = 3 mice per group (4 weeks); one experiment) c, Competitive index seven days after inoculating GF B6 mice with conditioned wild-type or TetOn-NRG1 (yeast-locked) C. albicans (n = 3 WT-colonized and n = 4 TetOn-NRG1-colonized mice; one experiment). The competitive index was normalized to that when strains were competed in wild-type and Rag1−/− mice directly from culture. d, Competitive index of wild-type versus TetOn-ALS3 or TetOff-ALS3 C. albicans during colonization of antibiotic-treated B6 mice that were untreated (UT) or treated with aTC (n = 4 mice per group; one experiment). P values calculated using two-way ANOVA with Tukey’s test (b), one-way ANOVA with Tukey’s test of area under the curve (d) or two-sided unpaired t-test (c). Data are mean ± s.d. (b–d). Silhouettes in a were created using BioRender.
Immune-targeted adhesins worsen colitis
Candida albicans and other Candida species are associated with IBD6, so we used the dextran sulfate sodium (DSS)-induced colitis model to test a role for filamentation during disease (Extended Data Fig. 8a) using vehicle (no C. albicans), wild-type C. albicans, yeast-locked C. albicans (TetOn-NRG1) and hyphal-locked C. albicans (TetOff-NRG1). Wild-type C. albicans and hyphal-locked C. albicans significantly exacerbated colon damage (Fig. 4a) whereas colitis was ameliorated in mice treated with the yeast-locked (TetOn-NRG1) strain (Fig. 4a, b), indicating that hyphae worsen colitis. The adhesin-deficient ahr1∆/∆ strain and the TetOff-AHR1 strain induced significantly less colon damage than wild-type C. albicans and the TetOn-AHR1 strain, respectively (Fig. 4c, Extended Data Fig. 9b). Constitutive expression of ALS1 in the ahr1∆/∆ background (TetOn-ALS1 ahr1∆/∆) significantly increased colon damage compared to the ahr1∆/∆ mutant, and this increase was reversed to ahr1∆/∆ levels upon repression of ALS1 expression (TetOff-ALS1 ahr1∆/∆) (Fig. 4c, Extended Data Fig. 9c.) These data suggest that IgA-targeted hyphal cells and the Als1 adhesin exacerbate colitis.

a, b, DSS histology images (a) and DSS histology scores (b) for mice treated with no C. albicans (No Ca), wild-type C. albicans (WT), TetOn-NRG1 C. albicans (yeast-locked) or TetOff-NRG1 (aTC-treated) C. albicans (hyphal-locked) (n = 5 mice per group; one experiment). Scale bar, 200 μm. c, DSS histology scores for mice treated with no C. albicans or with wild-type C. albicans, ahr1∆/∆ C. albicans, TetOn-ALS1 ahr1∆/∆ C. albicans or TetOff-ALS1 ahr1∆/∆ (aTC-treated) C. albicans (n = 10 (no Ca, WT and ahr1∆/∆), n = 5 (TetOn-ALS1) and n = 4 (TetOff-ALS1) mice per group; two experiments). d, IgA binding to faecal C. albicans in monocolonized alum or NDV-3A-vaccinated mice quantified by flow cytometry. e, Colon-tissue-associated colony-forming units (CFU). f, ALS3 and ALS1 expression quantified by qRT–PCR from the colon contents of C. albicans-monocolonized mice treated as in d (n = 5 mice per group; one experiment (d–f)). g, DSS histology scores for alum- or NDV-3A-vaccinated mice treated with no C. albicans or wild-type C. albicans (n = 10 mice per group; one experiment). P values calculated using two-way ANOVA with Sidak’s test (d), two-sided unpaired t-test (e, f) or one-way ANOVA with Tukey’s test (b, c, g). Data are mean ± s.d. (b–g).
To determine whether induction of an adhesin-specific immune response could prevent C. albicans-associated damage during colitis, the anti-Candida NDV-3A vaccine was used to immunize mice. NDV-3A induces an Als3-specific immune response that has been shown to be effective in preventing recurrent vaginal yeast infections in a human phase Ib/IIa trial25. Vaccination with NDV-3A induced faecal Als3-reactive IgG and IgA in GF mice (Extended Data Fig. 10), which bound C. albicans hyphae (Fig. 4d, Extended Data Fig. 10f). Vaccination did not affect C. albicans morphology or intestinal lumen fungal burden (Extended Data Fig. 10d, e, g, h). However, NDV-3A vaccination reduced tissue-associated C. albicans in the colon (Fig. 4e) and reduced the expression of C. albicans ALS1 (Fig. 4f). Notably, NDV-3A vaccination prevented C. albicans-associated damage in mice with colitis (Fig. 4g, Extended Data Fig. 10i–l). These data show that adaptive immune responses can be harnessed against a C. albicans adhesin to reduce C. albicans-associated damage during colitis.
Our studies reveal that host adaptive immune responses represent a force that promotes an expression program within commensal fungi that licenses their mutualism. For C. albicans, pathogenic hyphae and hyphae-associated virulence factors have been shown to be less fit for gut colonization1,2. Here we find that adaptive immune responses target and select against these cell types in the gut, improving their general commensal fitness. This example highlights a potential positive feedback loop between host and fungus that maintains homeostasis. This phenomenon may not be specific to interactions with C. albicans, as C. glabrata also provokes an adhesin-specific IgA response. Our study also provides a foundation to develop clinical interventions to restore homeostasis during disease. Human IgA deficiency is associated with IBD, although not associated with altered Candida levels26, may increase colitogenic effector expression. We have identified at least one antigen, Als1, as the first—to our knowledge—specific C. albicans effector that has been shown to contribute to intestinal colitis. Als1, and related adhesins (for example, Als3, which is around 84% identical to Als1 at the amino acid level27), are important virulence factors that promote mucocutaneous and disseminated infection28. Using a clinically tested Als-based vaccine25, we show that adhesin-specific immune responses can prevent intestinal disease and that vaccination strategies can be used to enhance commensal processes that already occur naturally. Altogether, these data reveal a mutualistic interaction between eukaryotes that comprises a bidirectional communication circuit involving fungal colonization factors and host immunity.
Methods
Mice
All mice used for experiments were between 6 and 12 weeks of age at the start of experiments, with the exception of the NDV-3A vaccination experiments in which 4–5-week-old mice were used. GF mouse experiments were either performed in sterile isolator bubbles or in sterile techniplast cages. When using techniplast cages, mice received antibiotic water (0.5 mg/ml ampicillin, chloramphenicol, gentamycin, erythromycin), replaced every 2 weeks, to prevent bacterial contamination. GF male C57BL/6 or male SW mice were used to quantify IgA targeting of GFP–C. albicans (YJB11522) and GFP–S. cerevisiae (JLR.KOf024). GF male C57BL/6 mice were used to assess IgA immune response to C. albicans (YJB11522), S. cerevisiae (JLR.KOf024), C. albicans iRFP-TetO-AHR1 (JLR.KOf204) versus Neon-SC5314, C. albicans Neon (JLR.KOf207) versus ahr1∆/∆ Neon (JLR.KOf258) versus TetOn-NRG1 Neon (JLR.KOf229), and C. glabrata (Cg1). GF SW females and males were used to assess IgA responses towards the Strope et al. 124 S. cerevisiae collection13 and the S. cerevisiae strains expressing C. albicans or C. glabrata adhesins. Male and female C57BL/6 wild-type (Jackson Laboratories) and Tcrb−/− (Jackson Laboratories) mice, cohoused by sex, were used to assess IgA targeting of C. albicans (YJB11522) in faeces. Age-matched male C57BL/6 wild-type, Rag1−/− and μMT−/− (Jackson Laboratories), and age-matched female GF C57BL/6 wild-type and Rag1−/− mice were used to analyse intestinal Neon–C. albicans (JLR.KOf207) morphology. Age-matched GF male wild-type and Rag1−/− (ULAM Germ Free Mouse Facility University of Michigan) mice were used for C. albicans (JLR.KOf207) RNA-seq and C. albicans morphology. Age-matched male and female C57BL/6 wild-type and Rag1−/−, antibiotic-treated or GF, were used to compare the competitive fitness of C. albicans conditioned in GF mice. All DSS experiments were performed on male C57BL/6 mice ordered directly from Jackson Laboratories, except for DSS experiments using TetO-AHR1, which were performed using age- and sex-matched male and female C57BL/6 mice bred in our mouse facility. Age-matched GF female C57BL/6 mice were used for the GF NDV-3A vaccination experiment. Mice colonized by fungi were gavaged once with 1 × 107–1 × 108 fungal cells. Conventional mice colonized with fungi received antibiotic water (0.5 mg/ml ampicillin, chloramphenicol, gentamycin, erythromycin) for 3–14 days before inoculation and for the duration of the experiment. Antibiotic water was changed weekly. For experiments involving Tet-repressible C. albicans strains, aTC (Cayman Chemical Company) was added to the drinking water at 100 μg/ml and replaced every 2 weeks for monocolonization experiments and every week for conventional mice. Mice were randomly assigned in within study groups. No statistical methods were used to predetermine sample size. In general, 4–5 mice were used per group per experiment. Sample sizes were largely based on availability of GF mice and age- and sex-matched controls. Analyses that relied on the investigators’ judgement were blinded: DSS colitis studies were not blinded to the investigator performing the experiment, but histology slides were randomized and blinded before scoring. Quantification of the percentage of C. albicans hyphae in the Rag1−/− and wild-type samples was performed by blinding microscopy files and quantifying the percentage of hyphae in each image. Mouse colonization and immunology experiments and human antibody quantification were performed unblinded as it was necessary for the investigators to know the identity of each experimental group. All mice were housed with a 12-h light–dark cycle per day, a temperature of 22 °C, and 22–30% relative humidity. All mouse experiments were performed in compliance with federal regulations and guidelines set forth by the University of Utah Institutional Animal Care and Use Committee.
Human sample collection
Age and sex information for human samples are in Supplementary Table 9. Human faecal study protocols were approved by the Institutional Review Board (HIC 1607018104) of the Yale School of Medicine. Informed consent was obtained from all participants and/or their legal guardians and all methods were performed in accordance with relevant guidelines and regulations. Patients with IBD (either Crohn’s disease or ulcerative colitis) were identified via the EPIC electronic medical record system and all individuals resided in the state of Connecticut. Demographics, medical history and other clinical variables were collected after enrolment. A heterogeneous population of healthy individuals was recruited through advertisements on the Yale medical campus and in the New Haven Public Library. To be eligible for participation, the individuals had to have never had a diagnosis of IBD or any other gastrointestinal disorder, an immunodeficiency or a family history of IBD, and also had to be at least 18 years of age. Healthy control individuals were clinically examined for disease. All faecal samples in this study were collected at home and stored on ice-packs at −20 °C before overnight shipment or direct drop-off the day after collection in the insulated container provided to each participant. Samples were then stored at −80 °C until use.
Human serum samples were obtained after obtaining informed consent in accordance with University of Utah protocol IRB-00072376. Blood samples from individuals with Crohn’s disease and healthy control individuals were drawn at the IBD clinic at the University of Utah and sent directly to the laboratory. Serum samples were obtained from consenting, randomly selected patients with Crohn’s disease who were seen in the IBD clinic regardless of age, gender, disease location, disease duration, medications, surgical history or disease activity. Thus, a selection bias is unlikely to influence the results. Healthy control individuals consisted of generally healthy volunteers that did not have an immunodeficiency, IBD or other gastrointestinal disorders. Healthy controls were clinically examined for disease.
Fungal strains, media and growth conditions
Fungal strains used in this study are listed in Supplementary Table 4. S. cerevisiae strains expressing C. albicans and C. glabrata adhesins are listed in Supplementary Table 5. Primers used for fungal strain creation and qRT–PCR are listed in Supplementary Table 6. Unless otherwise stated, fungi were propagated on yeast peptone dextrose medium (1% yeast extract (Thermo Fisher Scientific), 2% peptone (Thermo Fisher Scientific), 2% dextrose) at 30 °C. aTC (Cayman Chemical Company) was added to a concentration of 5 μg/ml for C. albicans TetOff strains. S. cerevisiae strains expressing C. albicans or C. glabrata adhesins were cultured in synthetic URA dropout medium at 30 °C. To induce C. albicans hyphal formation, strains were cultured at 37 °C in RPMI complete (RPMI 1640 with l-glutamine (Corning) supplemented with 10% fetal bovine serum (FBS), 1× MEM non-essential amino acids (Corning), 1 mM sodium pyruvate (Corning) and 1× 2-mercaptoethanol (Gibco)). For selection of NAT-resistant C. albicans transformants, nourseothricin (Jena Bioscience) was added to YPD medium at 200 μg/ml. For Hyg-resistant transformants, hygromycin B was added to YPD medium at 600 μg/ml.
Generation of the C. albicans and S. cerevisiae strains
C. albicans TetO strains were created using the transient CRISPR approach using the primers listed in Supplementary Table 6. The NAT promoter replacement cassette was amplified from pLC605 using the forward and reverse repair primers. The sgRNA fusion cassettes were made by PCR from pV1524 using KO160 and the reverse sgRNA primers (fragment A) and KO161 and forward sgRNA primers (fragment B), and then fusion PCR was performed on the fragments using the nested primers KO162 and KO163. CAS9 DNA was PCR-amplified from pV1525 with KO158 and KO159. The NAT-TetO cassette (40 μl), sgRNA (10 μl), and CAS9 DNA (10 μl) were transformed into C. albicans SC5314 wild type. Integration was tested using the reverse check primers and KO188. Lack of a wild-type allele was tested using forward and reverse check primers. For some strains, NAT sensitivity was restored by culture in YNB-BSA.
The ahr1∆/∆ strain was made using the transient CRISPR approach described above. The NAT replacement cassette was amplified from pLC605 using KO168 and KO169. The sgRNA fusion cassette was made as described for TetO-AHR1/TetO-AHR1. The NAT cassette, sgRNA and CAS9 DNA were transformed into C. albicans SC5314 wild type. Upstream integration was tested using KO167 and KO188. Lack of a wild-type allele was tested using KO166 and KO167 (upstream) and also KO134 and KO135 (downstream).
GFP–S. cerevisiae JLR.KOf024 strain was created by digesting pKO5 with sphI and sacI and transforming into RM11 (MATα lys2Δ0 ura3Δ0) and selected on synthetic URA dropout medium. Integration of ENO1-GFP-4peptide-ENO1term-URA3 was confirmed by PCR using KO030 and KO024. Positive transformants were then crossed with RM11 (MATα leu2Δ0) to create a prototrophic JLR.KOf024 GFP+ RM11 strain.
The S. cerevisiae + Als1 strain (JLR.KOf262) was created by transforming BY4741 with pKO20 and URA+ transformants were screened for surface HA expression using flow cytometry.
Vector creation
Vectors used in this study are listed in Supplementary Table 7. pKO5 was created using an In-Fusion HD Cloning Plus kit (Takara Bio). Five inserts were amplified using the primers listed in Supplementary Table 6 and cloned into pUC19. RM11 gDNA was used as a template for the following PCR reactions: KO033 and KO034; KO010 and KO048; KO013 and KO030. URA3 cassette was amplified using KO031 and KO032 from pML4329. GFP-4peptide was amplified from M436630.
pKO12 and pKO13 were created using an In-Fusion HD Cloning Plus kit (Takara Bio). HYG was amplified from pYM7031 using KO119 and KO120 and inserted into EcoRV digested NAT pENO1-Neon and pENO1-iRFP plasmids32.
pKO22 was created using an In-Fusion HD Cloning Plus kit (Takara Bio). Primers KO227 and KO226 were used to amplify the first 1,728 bp of ALS1 from SC5314 gDNA. This fragment was cloned into the pBC542 vector amplified with KO225 and KO228.
Processing of faecal or intestinal samples for total or fungal-binding antibodies, and assessing fungal burden
Faeces or intestinal contents were resuspended in sterile, cold PBS to a concentration of 100–500 mg/ml. Samples were homogenized by breaking up solid material with a pipette tip, followed by 1–2 min of vortexing. Homogenate was used for quantifying in vivo antibody binding to fungi using flow cytometry. Fungal burden was quantified by diluting 10−1–10−4 with sterile PBS and plating on YPD medium. To quantify total and fungal binding IgA in vitro, samples were spun at 13,000g for 10 min and the cleared supernatant was saved. Mouse IgA was quantified from 10−1–10−4 dilutions of intestinal supernatant using the Ready-SET-Go mouse IgA ELISA kit (Thermo Fisher Scientific).
Imaging C. albicans in faeces or in the gut
For imaging in the gut, intestinal sections were placed in tissue embedding cassette (Thermo Fisher Scientific) and samples were fixed for 3–4 h in Carnoy’s solution (Spectrum Chemical) at room temperature with shaking. Cassettes were then washed twice for 5 min with cold PBS, and then for 40 min in 40% ethanol. Samples were stored in 70% ethanol until sectioning by the ARUP Research histology laboratory. To stain intestinal sections, slides were de-paraffined in Coplin Staining Jars (VWR): two 6-min washes in xylenes followed by sequential 2-min washes in 100% ethanol, 100% ethanol, 95% ethanol, 70% ethanol and 40% ethanol. Slides were incubated in humidified staining chamber at room temperature for 20 min with 150 μl blocking buffer (PBS with 4% donkey serum). Slides were stained with 150 μl (1:500 dilution) of AF488 anti-Candida antibody (Meridian) in humified staining chamber overnight at 4 °C. Slides were washed twice for 5 min in PBS supplemented with 0.1% Tween-20 with shaking. Slides were rinsed twice in cold PBS before mounting and imaging.
To image C. albicans (JLR.KOf207) from faecal or intestinal contents, material was homogenized to 100 mg/ml in PBS and 10 μl was placed in a 96-well V-bottom plate. Samples were incubated in 100 μl blocking buffer at 4 °C for 20 min, and then stained at 4 °C with 100 μl 1:500 dilution AF488 anti-Candida antibody for 20 min. Cells were washed twice with 150 μl PBS and fixed for 10 min with 2% paraformaldehyde solution (Thermo Fisher Scientific). Staining of C. albicans using the anti-Candida antibody markedly amplified the brightness of C. albicans cells over simply using the Neon fluorescent marker expressed by the C. albicans strain used for these imaging studies.
All samples were mounted using Vectashield HardSet Antifade Mounting Medium (Vector) and images were taken at the University of Utah Imaging Core using a Nikon A1R Confocal microscope. Imaging analysis was done using Fiji33.
RNA isolation, qRT–PCR and RNA-seq
Faeces or intestinal contents (250–500 mg) were collected from mice, immediately frozen on dry ice and then stored at −80 °C until RNA isolation. RNA was isolated using the RNAeasy mini kit (Qiagen). For qRT–PCR experiments, cDNA was synthesized using qScript cDNA synthesis kit (Quanta Biosciences), and C. albicans transcripts were quantified using PowerUp SYBR Green Master Mix (Applied Systems). Primers used for qRT–PCR are listed in Supplementary Table 6. Transcripts were compared to C. albicans ACT1 or PMA1.
For RNA-seq of C. albicans (JLR.KOf207) colonizing GF wild-type and Rag1−/− mice, RNA was isolated from caecal contents after 4 weeks of colonization. NEBNext Ultra II Directional RNA library pep kit and the NEBNext Poly(A) mRNA Magnetic Isolation Module (NEB) were used to generate mRNA-seq libraries according to the manufacturer’s directions. Each sample’s library (n = 9) was barcoded with NEB-provided oligos and libraries were multiplexed before sequencing. Multiplexed libraries were sequenced on a single lane of a HiSeq 2500 with paired-end 125-cycle sequencing by the University of Utah Genomics Core facility, a part of the Health Sciences Cores at the University of Utah.
Raw Illumina fastq sequences were first quality-trimmed and adaptor-filtered using trim_galore’s implementation of cutadapt34. Sequences were trimmed when the quality score dropped below 20 and any remaining sequences with length less than 20 or for which the mate-pair did not pass quality checks was discarded. Quality filtered sequences were then aligned against the mouse transcriptome (GRCm38) with Bowtie2 to remove host reads. Sequence pairs that did not align concordantly to the mouse reference were then used as input to align against C. albicans reference. On average 66% ± 4% (mean ± s.e.m.) of reads mapped to the host transcriptome. Reads were mapped against the current C. albicans SC5314 transcriptome reference (assembly 22, http://candidagenome.org/) using the “_default_coding” version, which contains a haploid complement of all coding features without introns. Kallisto (v.0.45.0) was used to map and quantify transcript abundances and resulted in a final average of 6.7 million (±1.1 million) read pairs mapping per sample35. We used the “chromosomal_feature.tab” file provided by the Candida genome database (http://candidagenome.org/) for the assembly 22 version to create a map file to correlate each transcript to a gene, and also replace the systematic gene names implemented in assembly 22 with common or standard gene names where they were available. We then used the Sleuth R package to read in the Kallisto read quantification files and aggregate mapped transcripts by gene, providing the gene-name map file we created before36 Sleuth was further used to perform gene-level differential expression testing between the two mouse genotypes with Wald’s test. GO term enrichment analysis was performed on transcripts that were at least twofold (log2 ≥ 1) differentially regulated between groups with q < 0.05 using the ‘clusterProfiler’ package in R and an organism annotation package created with the ‘AnnotationForge’ R package from the NCBI-hosted genome37 The R package ‘fgsea’ was used to compare the published hyphal-upregulated gene set1 with the ranked list of differentially expressed genes identified with Sleuth38. Genes at least twofold induced (log2 ≥ 1) in hyphal-inducing conditions were classified as hyphal-upregulated. RNA-seq volcano plots using EnhancedVolcano39.
Quantification of IgA binding to cultured fungi in vivo
To quantify IgA binding to cultured fungi, 500 µl–1 ml of faecal or intestinal homogenate was filtered through a 40-μm or 70-μm filter into a 50-ml conical tube. Filters were rinsed with 10 ml cold PBS, filters were discarded and then samples were spun at 4,000 rpm for 5 min. Supernatants were discarded, and pellets were resuspended in 10 ml PBS and spun at 4,000 rpm for 5 min. Supernatants were discarded and pellets were vortexed in the residual PBS by vortexing. Subsequently, 10 µl of each sample was pipetted into a 96-well V-bottom plate. A well was prepared for an unstained control and IgA isotype control. Then, 200 µl of 10% FBS (v/v) in PBS was added to each sample and incubated on ice for 10 min. Samples were spun down at 3,000 rpm for 5 min, and the samples were stained with 100 μl 1:250 anti mouse IgA PE (eBioscience clone mA-6E1) and 5 μg/ml calcofluor white (CFW) (Sigma-Aldrich) in column buffer (PBS supplemented with 10mM HEPES (Corning) 2 mM EDTA (Corning), and 0.5% (v/v) FBS (Gibco BRL)). The isotype control sample was similarly stained, but with 1:500 rat IgG1 K isotype control PE. Samples were analysed on the BD LSR Fortessa and data analysed by FlowJo.
Imaging flow cytometry of C. albicans in faeces
Faecal samples from SW mice that had been monocolonized with GFP–C. albicans (YJB11522) for 3 weeks were used for imaging. Faecal samples were prepared and stained for CFW and IgA as described for flow cytometry analysis. Samples from three mice were analysed using the Amnis ImageStream Mk II using the 488-nm, 405-nm and 592-nm laser. Data were analysed using IDEAs 6.3 software. C. albicans were gated on GFP+ and CFW intermediate populations, and finally gated by IgA. Bright-field images of C. albicans populations were visually inspected to exclude non-C. albicans faecal particles. Data from the three mice were combined into a single file to analyse C. albicans circularity by IgA binding. Circularity of the IgA+ and IgA− populations were calculated using the CFW channel using the Shape Change wizard.
Assessing in vitro antibody binding to cultured fungi
Cultured fungi were normalized to an optical density at 600 nm (OD600) = 1–3 in PBS supplemented with 1% bovine serum albumin (Sigma-Aldrich) and 0.01% sodium azide (P/B/A). Then, 25 µl of cultured fungi was mixed with 25 μl cleared intestinal wash or diluted serum in 96-well V-bottom plates and incubated on ice, or at 4 °C for 45 min–1 h. Samples were spun at 3,000 rpm for 5 min, and washed twice with 150 μl P/B/A. Samples were stained in the dark at 4 °C with anti-mouse IgA (eBioscience clone mA-6E1) diluted 1:500 in column buffer (PBS supplemented with 10 mM HEPES (Corning) 2 mM EDTA (Corning) and 0.5% (v/v) fetal bovine serum (Gibco BRL)). An isotype control, stained with 1:500 rat IgG1 K isotype control PE, or a sample stained without serum or intestinal antibodies but with the fluorescent secondary antibody was included for each fungus tested as a negative control. For mouse serum samples, yeast were incubated with 25 μl serum diluted 1:25 and then stained with anti-IgA, anti-IgG1, anti-IgG2b, anti-IgG3 and anti-IgM antibodies (Supplementary Table 8) diluted 1:500. Samples were washed twice with 150 μl column buffer and IgA binding was quantified using either a BD LSR Fortessa or BD Celesta. Antibody-binding intensity was normalized to the isotype negative control or yeast stained with fluorescent secondaries but without faecal or serum antibodies.
Screening the Noble and Homann collections of mutant C. albicans strains for IgA binding
Intestinal wash from small intestinal contents pooled from four male C57BL/6 mice monocolonized with C. albicans (YJB11522) for 25 days, or whole-intestinal contents from female SW mice monocolonized with C. albicans (YJB11522) for 60 days, were used. For both intestinal wash samples, contents were homogenized in PBS to 100 mg/ml, cleared by spinning at 5,000 rpm for 15 min and then filtered through a 0.22-μm filter.
The Homann17 and Noble16 C. albicans homozygous deletion collections were purchased from the Fungal Genetics Stock Center (http://www.fgsc.net/). Mutant collections were cultured overnight in round-bottom 96-well plates in 200 μl YPD. Each strain was normalized to OD600 = 3 in sterile-filtered P/B/A. Then, 25 µl of each strain was mixed with 25 μl intestinal wash in 96-well V-bottom plates and incubated for 50 min on ice. Samples were spun at 3,000 rpm for 5 min and washed twice with 150 μl P/B/A. Samples were stained in the dark for 20 min on ice with 50 μl anti-mouse IgA PE (eBioscience clone mA-6E1), diluted 1:500 in column buffer (PBS supplemented with 10 mM HEPES (Corning), 2 mM EDTA (Corning) and 0.5% (v/v) fetal bovine serum (Gibco BRL)), and then washed twice with 150 μl column buffer. Each collection contains an isogenic background wild-type strain used as a positive control. As a negative control, the wild-type strains were incubated with intestinal wash, but then stained with the PE isotype control antibody (rat IgG1 K isotype control PE eBioscience) at a 1:500 dilution. For the Homann screen, IgA binding was quantified using the BD FACSCanto Analyzer using the 96-well high throughput sampler. The geometric mean IgA binding was quantified using FlowJo and binding intensity was divided by the geometric mean intensity of the isotype control. For the Noble collection, IgA binding was quantified using the BD Celesta using the 96-well high-throughput samples. When quantifying IgA binding intensity by FlowJo, we noticed that there was substantial plate-to-plate variability in the average IgA binding intensity. We therefore normalized binding intensity by dividing the geometric mean IgA binding intensity for each sample by the average geometric mean IgA value for corresponding plate. For both collections, normalized IgA binding intensity was averaged between duplicate samples, and then IgA binding z-scores were calculated by the following: z = (normalized IgA binding − average normalized IgA binding for whole collection)/(standard deviation of normalized IgA binding for the collection). For the Homann collection, both wells of the orf19.610∆/∆ (encoding EFG1) mutant were contaminated and this mutant was excluded from the figures and tables in this study. We isolated a pure culture of efg1∆/∆ and quantified IgA binding using the same protocol, finding no difference in IgA targeting compared to the wild-type strain. For the Noble collection, we did not acquire data from three mutant strains (orf19.191, orf19.1041, and orf19.6124) because the cells were lost during the processing of the samples.
Assessment of lamina propria and Peyer’s patch lymphocyte populations
Lamina propria cells were isolated as described previously40 with the following alterations. The epithelial dissociation step was performed with 10 ml HBSS (without Mg2+ and Ca2+) containing 30 mM 0.5 EDTA (Corning), 10 mM HEPES (Corning), and 1.5 mM DL-dithirothreitol (DTT) (Sigma). Cells were dissociated for 30 min at 37 °C with shaking at 37 °C for 30 min. Digestions were carried out as described previously40, but instead of percoll separation, digestion solution was spun at 800g for 10 min and washed one time in 10 ml cold PBS. Digests were resuspended in 5 ml RPMI complete (RPMI 1640 with l-glutamine (Corning) supplemented with 10% FBS, 1× MEM non-essential amino acids (Corning), 1 mM sodium pyruvate (Corning), 1× 2-mercaptoethanol (Gibco)), and counted. Peyer’s patches were isolated and prepared as described previously40.
A total of 5 × 105 to 1 × 106 live cells were stained for flow cytometry. All cells were first stained for with Ghost Dye Violet 510 (Tonbo) viability stain following the Ghost dye protocol. Extracellular and intracellular antibody staining was performed as described previously40 using the antibodies and concentrations listed in Supplementary Table 8. Data were collected using a BD LSR Fortessa and analysed using FlowJo software. All cell populations were first gated by FSC and SSC to exclude cellular debris, then doublets were gated out, and finally gating was performed for live (Ghost-negative) cell populations. Gates and analysis for all stains were set using fluorescent minus one (all antibodies except for one) controls for each antibody.
C. albicans cell-wall isolation and western blot
C. albicans (SC5314) was cultured overnight in 5 ml YPD at 30 °C. Then, 100 µl was added either to 5 ml YPD and rotated overnight at 30 °C (yeast) or to 5 ml RMPI complete and shaken at 150 rpm overnight at 37 °C (hyphae). Yeast and hyphal cultures were collected and washed once with 10 ml cold 10 mM Tris HCl pH 7.4, resuspended in 1 ml 10 mM Tris HCl and stored in bead beating tubes at −80 °C. Fungal cell walls were isolated as described previously41. Cell walls were normalized to 20 mg/ml in Tris HCl pH 7.4, and 50 μl of each sample was mixed with 10 μl 6× Laemmli sample buffer and boiled for 10 min. Then, 30 µl (500 μg) was loaded and run on two 4–20% 12-well Mini-PROTEAN TGX precast protein gels (BioRad). For one gel, proteins were transferred to a 0.45-nm nitrocellulose membrane, stained with intestinal wash from C57BL/6 mice monocolonized with C. albicans (cleared small intestinal wash diluted 100 μl in 5 ml TBST with 5% dried milk (v/v)), followed by staining with 1:1,000 dilution of goat anti-mouse IgA (μ chain) HRP secondary antibody (SouthernBiotech) diluted in TBST with 5% milk. The duplicate protein gel was stained with Coomassie. Two protein gel regions of greater than 254 kDa were cut from each lane and submitted to the Mass Spectrometry and Proteomics Core Facility at the University of Utah for protein identification using the procedure described below.
Digestion of in-gel proteins
Gel bands were first destained with 50 mM ammonium bicarbonate in 50:50 water:methanol. Proteins were reduced with DTT for 45 min at 60 °C and then alkylated with idodoacetamide for 30 min at room temperature in the dark. Gel spots were washed three times in 50 mM ammonium bicarbonate in water for 45 min per wash cycle. Gel spots were cut into small pieces and dehydrated using 100% acetonitrile. Proteins were digested overnight at 38 °C with a trypsin and LysC mixture. One μg pf trypsin was used per sample. The digestion was quenched by acidification with 1% formic acid to a pH of 2–3. Peptides were extracted from the gel using 50% acetonitrile/1% formic acid and then concentrated in a vacuum to a final volume of 5 μl.
LC–MS/MS analysis
Reversed-phase nano-LC–MS/MS was performed on an Eksigent Ekspert nanoLC 425 system (SciEx) coupled to a Bruker MAXIS ETD II QToF mass spectrometer equipped with a nanoelectrospray source. Concentrated samples were diluted with a 1:1 ratio of sample:0.1% formic acid in water. The samples (5 µl) were then injected onto the liquid chromatograph. A gradient of reversed-phase buffers (buffer A: 0.2% formic acid in water; buffer B: 0.2% formic acid in acetonitrile) at a flow rate of 150 μl/min at 60 °C was set up. The LC run lasted for 83 min with a starting concentration of 5% buffer B increasing to 55% over the initial 53 min and a further increase in concentration to 95% over 63 min. A nanocolumn of length 15 cm and inner-diameter 100 μm was used for chromatographic separation. The column was packed, in-house, with reverse-phase BEH C18 3.5 μm resin (Xbridge). MS/MS data were acquired using an auto-MS/MS method selecting the most abundant precursor ions for fragmentation. The mass-to-charge range was set to 350–1,800.
Analysis of MS/MS data
Mascot generic format (MGF) files were generated from the raw MS/MS data. Mascot (v.2.6) uses the MGF file for database searching and protein identification. For these samples a custom database was searched with Candida albicans taxonomy selected. The parameters used for the Mascot searches were: trypsin digest; two missed cleavages; carbamidomethylation of cysteine set as fixed modification; oxidation of methionine were set as variable modifications; and the maximum allowed mass deviation was set at 11 ppm.
Conditioning of C. albicans in wild-type and Rag1 −/− C57BL/6 GF mice and competitive colonization
Colonization of mice
To assess the competitive fitness of C. albicans conditioned in wild-type or Rag1−/− C57BL/6 GF mice, faecal samples from monocolonized mice were homogenized to 100 mg/ml and wild-type or Rag1−/− mice were pooled in equal volumes. Homogenates were kept on ice overnight while CFU were quantified from each pool by quantitative culture. Wild-type or Rag1−/− homogenates were normalized by CFU and mixed in equal volumes and gavaged into antibiotic-treated or GF wild-type and Rag1−/− mice. (see ‘Mice’ section for antibiotic treatment protocol). In addition, 100 µl of the combined homogenate was also plated on YPD and cultured at 30 °C. Faecal samples from each recipient mouse were homogenized in 1 ml sterile PBS and the entire sample was cultured on YPD plates at 30 °C. Cultured iRFP and Neon C. albicans strains were also competed in wild-type and Rag1−/− mice to control for inherent competitive colonization differences. Cultured Neon+ and iRFP+ strains were mixed 1:1 and 108 cells were gavaged into antibiotic-treated or GF wild-type and Rag1−/− recipient mice. Then, 100 µl of the inoculum and homogenized faecal samples was plated on YPD and cultured overnight at 30 °C. For competitions shown in Fig. 3b, Neon C. albicans (JLR.KOf207) was conditioned in wild-type mice and iRFP C. albicans (JLR.KOf206) was conditioned in Rag1−/− mice. For competitions in Fig. 3c, iRFP C. albicans (JLR.KOf263) or iRFP TetO-NRG1 (JLR.KOf228) were conditioned in wild-type mice, and Neon C. albicans (JLR.KOf207) or Neon TetO-NRG1 (JLR.KOf229) were conditioned in Rag1−/− mice.
Calculating normalized competitive index
C. albicans cultured from inoculum or from faecal samples were scraped and homogenized in 5 ml sterile H2O. Suspended C. albicans were diluted and the relative numbers of iRFP (Rag1−/−-conditioned) and Neon (wild-type-conditioned) were quantified using flow cytometry using a BD LSR Fortessa with FACS Diva and FlowJo analysis. Competitive index (CI) for iRFP- or Neon strains was calculated using the following formula described previously16: (Recovered count/total recovered count)/(inoculum count/total inoculum count). We normalized the CI values calculated from the competition of intestinal conditioned strains by dividing them by the CI values of the same strains competed in mice without first conditioning in GF mice (from culture).
CI was calculated for Neon C. albicans (JLR.KOf207) versus TetO-ALS3 (JLR.KOf250) using the same culture and flow quantification described above, except that strains were compared by Neon+ wild-type versus unstained (TetO-ALS3). Strains were competed in antibiotic treated C57BL/6 mice with and without aTC in the water, as described in the ‘Mice’ section.
Assessing fungal reactive antibodies and ASCAs from human faeces and serum
Human faecal samples were homogenized in sterile PBS to a concentration of around 100 mg/ml by disruption by pipette tip and vortexing for 2 min. Tubes were spun at 13,000g for 15 min and supernatants were stored in 100-μl aliquots at −80 °C. Total IgA was quantified by ELISA (coating SAB3701393, detection ab97215, and standards were human IgA1 and IgA2 (Athens Research & Technology). The top dilution of each faecal wash sample was normalized to between 1–9 μg/ml total IgA before probing cultured fungi. Human serum samples were diluted 1:75 in PBS. IgA, IgG and IgM binding to S. cerevisiae (RM11), C. albicans (SC5314), C. glabrata (Cg1), and C. tropicalis (MYA3404) was performed as described in ‘Assessing in vitro antibody binding to cultured fungi’. Samples were stained for IgM (anti-IgM Fc5μ goat polyclonal antibody AF488, Jackson ImmunoResearch), IgG (goat anti-human IgG antibody AF594, Jackson ImmunoResearch) and IgA (goat anti-human IgA α antibody AF647, Jackson ImmunoResearch). Each antibody was diluted according to the manufacturer’s recommendations and used at a 1:500 dilution. Staining intensity was quantified using a BD LSR Fortessa with FACS Diva and analysed by FlowJo. Geometric mean staining intensity was normalized between fungi so that the unstained controls (stained without faecal IgA but with the fluorescent secondaries) had the same baseline staining intensity. Geometric mean IgA binding to the S. cerevisiae strains expressing C. albicans adhesins was normalized as described above and then divided by the intensity of IgA binding to the control S. cerevisiae strain (UB2158).
IgA ASCA were quantified by ELISA (Eagle Bioscience) using 1:100 dilutions of human serum and undiluted cleared human faecal wash.
Investigating a collection of 124 S. cerevisiae strains for IgA induction in GF mice
The Strope et al. 124 S. cerevisiae collection13 was purchased from the Fungal Genetics Stocks centre (http://www.fgsc.net/). GF male and female SW mice were used to investigate the IgA response to the Strope et al. 124 S. cerevisiae strain collection. Individual cultured strains were normalized by OD600 and combined into 6 pools of 20–24 strains. Mice were gavaged with 1–3 × 108 cells every week for 3 weeks. All mice were kept in sterile techniplast cages and kept on antibiotic water (500 mg/l ampicillin, chloramphenicol, gentamycin, erythromycin) to prevent bacterial contamination. Caecal contents were used to assess IgA binding of S. cerevisiae in vivo and to quantify total IgA levels and IgA binding to the 20–24 strains present in the pool colonizing the mice (See ‘Assessing in vitro antibody binding to cultured fungi’).
DSS colitis experiments
Mice were given 2.5% dextran sodium sulfate salt (molecular weight (MW) = 36,000–50,000 kDa), colitis grade, MP Biomedicals) for 8 days. Mice were weighed daily and euthanized on day 8. Colons were removed, cleared of faecal material, measured, fixed in 10% buffered formalin (Fisher Chemical) for 1–2 days at room temperature and stored in 70% ethanol. The ARUP Research histology laboratory sectioned colons and performed haematoxylin and eosin (H&E) staining. Colon damage score was scored as described previously40.
NDV-3A vaccination
NDV-3A or alum vaccination of GF or conventionally colonized mice (SPF mice) was performed as described previously42, although with just one boost. Anti-Als3 IgA and IgG was assessed in the faeces using an ELISA as described previously42. See Extended Data Fig. 9a for a diagram of the vaccination experiments.
Statistical analysis
Figure creation and statistical analysis was performed with Prism 8 software. Specific statistical tests are indicated in the figure legends.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
Raw C. albicans RNA-seq reads have been deposited at the NCBI Sequence Read Archive under the BioProject accession number PRJNA728116. All other data needed to evaluate the conclusions in the paper are available within the Article or its Supplementary Information. Source data are provided with this paper.
Code availability
All code used for processing and mapping RNA-seq reads is available at https://github.com/RoundLab/Ost_CandidaRNASeq.
References
Witchley, J. N. et al. Candida albicans morphogenesis programs control the balance between gut commensalism and invasive infection. Cell Host Microbe 25, 432–443 (2019).
Tso, G. H. W. et al. Experimental evolution of a fungal pathogen into a gut symbiont. Science 362, 589–595 (2018).
Leonardi, I. et al. CX3CR1+ mononuclear phagocytes control immunity to intestinal fungi. Science 359, 232–236 (2018).
Zhai, B. et al. High-resolution mycobiota analysis reveals dynamic intestinal translocation preceding invasive candidiasis. Nat. Med. 26, 59–64 (2020).
Jain, U. et al. Debaryomyces is enriched in Crohn’s disease intestinal tissue and impairs healing in mice. Science 371, 1154–1159 (2021).
Li, X. V., Leonardi, I. & Iliev, I. D. Gut mycobiota in immunity and inflammatory disease. Immunity 50, 1365–1379 (2019).
Weis, A. M. & Round, J. L. Microbiota–antibody interactions that regulate gut homeostasis. Cell Host Microbe 29, 334–346 (2021).
Huertas, B. et al. Serum antibody profile during colonization of the mouse gut by Candida albicans: relevance for protection during systemic infection. J. Proteome Res. 16, 335–345 (2017).
Doron, I. et al. Human gut mycobiota tune immunity via CARD9-dependent induction of anti-fungal IgG antibodies. Cell 184, 1017–1031 (2021).
Bai, X.-D., Liu, X.-H. & Tong, Q.-Y. Intestinal colonization with Candida albicans and mucosal immunity. World J. Gastroenterol. 10, 2124–2126 (2004).
Millet, N., Solis, N. V. & Swidergall, M. Mucosal IgA prevents commensal Candida albicans dysbiosis in the oral cavity. Front. Immunol. 11, 555363 (2020).
Standaert-Vitse, A. et al. Candida albicans is an immunogen for anti-Saccharomyces cerevisiae antibody markers of Crohn’s disease. Gastroenterology 130, 1764–1775 (2006).
Strope, P. K. et al. The 100-genomes strains, an S. cerevisiae resource that illuminates its natural phenotypic and genotypic variation and emergence as an opportunistic pathogen. Genome Res. 25, 762–774 (2015).
Richardson, J. P., Ho, J. & Naglik, J. R. Candida–epithelial interactions. J. Fungi 4, 22 (2018).
Noble, S. M., Gianetti, B. A. & Witchley, J. N. Candida albicans cell-type switching and functional plasticity in the mammalian host. Nat. Rev. Microbiol. 15, 96–108 (2017).
Noble, S. M., French, S., Kohn, L. A., Chen, V. & Johnson, A. D. Systematic screens of a Candida albicans homozygous deletion library decouple morphogenetic switching and pathogenicity. Nat. Genet. 42, 590–598 (2010).
Homann, O. R., Dea, J., Noble, S. M. & Johnson, A. D. A phenotypic profile of the Candida albicans regulatory network. PLoS Genet. 5, e1000783 (2009).
Lohse, M. B., Gulati, M., Johnson, A. D. & Nobile, C. J. Development and regulation of single- and multi-species Candida albicans biofilms. Nat. Rev. Microbiol. 16, 19–31 (2018).
Braun, B. R., Kadosh, D. & Johnson, A. D. NRG1, a repressor of filamentous growth in C. albicans, is down-regulated during filament induction. EMBO J. 20, 4753–4761 (2001).
Ruben, S. et al. Ahr1 and Tup1 contribute to the transcriptional control of virulence-associated genes in Candida albicans. MBio 11, e00206-20 (2020).
De Groot, P. W. J., Bader, O., De Boer, A. D., Weig, M. & Chauhan, N. Adhesins in human fungal pathogens: glue with plenty of stick. Eukaryot. Cell 12, 470–481 (2013).
Askew, C. et al. The zinc cluster transcription factor Ahr1p directs Mcm1p regulation of Candida albicans adhesion. Mol. Microbiol. 79, 940–953 (2011).
Carreté, L. et al. Patterns of genomic variation in the opportunistic pathogen Candida glabrata suggest the existence of mating and a secondary association with humans. Curr. Biol. 28, 15–27 (2018).
Nobbs, A. H., Vickerman, M. M. & Jenkinson, H. F. Heterologous expression of Candida albicans cell wall-associated adhesins in Saccharomyces cerevisiae reveals differential specificities in adherence and biofilm formation and in binding oral Streptococcus gordonii. Eukaryot. Cell 9, 1622–1634 (2010).
Edwards, J. E. Jr et al. A fungal immunotherapeutic vaccine (NDV-3A) for treatment of recurrent vulvovaginal candidiasis—a phase 2 randomized, double-blind, placebo-controlled trial. Clin. Infect. Dis. 66, 1928–1936 (2018).
Fiedorová, K. et al. Bacterial but not fungal gut microbiota alterations are associated with common variable immunodeficiency (CVID) phenotype. Front. Immunol. 10, 1914 (2019).
Spellberg, B. J. et al. Efficacy of the anti-Candida rAls3p-N or rAls1p-N vaccines against disseminated and mucosal candidiasis. J. Infect. Dis. 194, 256–260 (2006).
Ibrahim, A. S. et al. Vaccination with recombinant N-terminal domain of Als1p improves survival during murine disseminated candidiasis by enhancing cell-mediated, not humoral, immunity. Infect. Immun. 73, 999–1005 (2005).
Voth, W. P., Richards, J. D., Shaw, J. M. & Stillman, D. J. Yeast vectors for integration at the HO locus. Nucleic Acids Res. 29, e59 (2001).
Igyártó, B. Z. et al. Skin-resident murine dendritic cell subsets promote distinct and opposing antigen-specific T helper cell responses. Immunity 35, 260–272 (2011).
Basso, L. R., Jr et al. Transformation of Candida albicans with a synthetic hygromycin B resistance gene. Yeast 27, 1039–1048 (2010).
Seman, B. G. et al. Yeast and filaments have specialized, independent activities in a zebrafish model of Candida albicans infection. Infect. Immun. 86, e00415-18 (2018).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12 (2011).
Bray, N. L., Pimentel, H., Melsted, P. & Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527 (2016).
Pimentel, H., Bray, N. L., Puente, S., Melsted, P. & Pachter, L. Differential analysis of RNA-seq incorporating quantification uncertainty. Nat. Methods 14, 687–690 (2017).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012).
Sergushichev, A. A. An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. Preprint at https://doi.org/10.1101/060012 (2016).
Blighe, K., Rana S. & Lewis, M. EnhancedVolcano: publication-ready volcano plots with enhanced colouring and labeling. https://github.com/kevinblighe/EnhancedVolcano (2018).
Kubinak, J. L. et al. MyD88 signaling in T cells directs IgA-mediated control of the microbiota to promote health. Cell Host Microbe 17, 153–163 (2015).
Plaine, A. et al. Functional analysis of Candida albicans GPI-anchored proteins: roles in cell wall integrity and caspofungin sensitivity. Fungal Genet. Biol. 45, 1404–1414 (2008).
Singh, S. et al. The NDV-3A vaccine protects mice from multidrug resistant Candida auris infection. PLoS Pathog. 15, e1007460 (2019).
Acknowledgements
We thank A. Weis and J. Hill for their edits of this manuscript; R. Wheeler for providing the pENO1-iRFP and pENO1-Neon C. albicans constructs; L. Cowen for the pLC605 TetO construct; J. Berman for the YJM11522 C. albicans strain; A. Nobbs for the S. cerevisiae strains expressing C. albicans adhesins; D. Stillman for S. cerevisiae strains and reagents; and the ULAM GF Mouse Facility at the University of Michigan for the GF Rag1−/− mice. Proteomics mass spectrometry analysis was performed at the Mass Spectrometry and Proteomics Core Facility at the University of Utah. Mass spectrometry equipment was obtained through a Shared Instrumentation Grant 1 S10 OD018210 01A1. This work was supported by the Helen Hay Whitney Foundation (K.S.O.), a University of Utah NRSA Microbial Pathogenesis T32 Training Grant (K.S.O.), a CCFA Senior Research Award (J.L.R.), NIDDK R01DK124336 (J.L.R.), the Edward Mallinckrodt Jr. Foundation (J.L.R.), a NSF CAREER award (IOS-1253278) (J.L.R.), a Packard Fellowship in Science and Engineering (J.L.R.), a Burroughs Welcome Investigator in Pathogenesis Award (J.L.R), the American Asthma Foundation (J.L.R.), the Margolis Foundation (J.L.R.), an MS Society Center grant (J.L.R.), NIAID R01046223 (B.C.), an NIH New Innovator Award DP2GM111099-01 (R.M.O.), NHLBI R00HL102228-05 (R.M.O.), an American Cancer Society Research Grant (R.M.O.), a Kimmel Scholar Award (R.M.O.), R01AG047956 (R.M.O.) and NIAID R01AI141202 (A.S.I.). This work was supported by the University of Utah Flow Cytometry Facility in addition to the National Cancer Institute through award number 5P30CA042014-24. The support and resources from the Center for High Performance Computing at the University of Utah are gratefully acknowledged.
Author information
Authors and Affiliations
Contributions
K.S.O. conceived the study, performed most experiments and helped to write the manuscript. T.R.O. helped with experimental design and fungal strain creation, and edited the manuscript. W.Z.S. analysed the RNA-seq data, helped with experimental design and edited the manuscript. T.C. helped with the immune profiling experiments and edited the manuscript. H.Z. helped to perform the C. albicans IgA screens and edited the manuscript. J.P. helped with fungal IgA-binding assays and edited the manuscript. R.B. managed the GF mouse experiments, helped with the immune profiling experiments and edited the manuscript. J.R.C., D.S. and N.W.P. provided the collection of human faecal samples, provided guidance on human antibody experiments and edited the manuscript. D.H.C. and K.A.C. guided the imaging flow cytometry experiments and edited the manuscript. E.H.-W. and B.C. created the S. cerevisiae strains expressing the C. glabrata adhesin-like proteins and edited the manuscript. K.E.H. edited the manuscript and provided the clinical C. glabrata strains. R.M.O. provided guidance on immunological experiments. S.M.N. provided C. albicans strains, provided guidance on fungal genetics experiments and edited the manuscript. A.S.I. and S.S. provided the NDV-3A vaccine and edited the manuscript. J.F.V. provided the human serum samples. J.L.R conceived the study, guided the experiments, analysed data and helped to write the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature thanks Gordon Brown and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Human faecal and serum anti-fungal antibodies.
a, Human faecal antibody binding to cultured fungi and quantified by flow cytometry after staining with fluorescent secondary antibodies (n = 70). Staining intensity normalized to fungi stained with secondary antibodies but without human faecal wash. Box plots show minimum 25% quartile and maximum 75% quartile around the median and whiskers show range. b, IgA binding to cultured fungi from serial dilutions of human faecal wash (n = 30 healthy, n = 23 Crohn’s disease and n = 17 UC). Geometric mean with 95% CI c, d, Human serum antibody binding to cultured fungi. Serum diluted 1:75. (n = 12, n = 4 healthy and n = 8 Crohn’s disease). Mean ± s.d. e, Faecal ASCA IgA levels from undiluted faecal wash (n = 30 healthy, n = 18 Crohn’s disease and n = 14 UC). Median with 95% CI f, Serum ASCA IgA levels from human serum samples diluted 1:100 (n = 4 healthy and n = 8 Crohn’s disease). Median with 95% CI. P values calculated using two-way ANOVA with Tukey’s test (a), one-way ANOVA with Dunn’s test (c) or two-sided Mann–Whitney U-test (f, d).
Extended Data Fig. 2 IgA targets Candida species but not S. cerevisiae.
a, IgA-bound faecal fungi gating strategy. b, Peyer’s patch GC B cell and TFH cell gating strategy. c, Colon LP IgA plasma cell gating strategy (n = 4 mice per group 30 days after inoculation; representative of two experiments for b, c). d, IgA binding to faecal GFP+ S. cerevisiae and GFP+ C. albicans in monocolonized SW mice. e, Total IgA levels from monocolonized SW mice. f, Flow cytometry quantification of SW IgA binding to cultured S. cerevisiae and C. albicans (n = 4 C. albicans-colonized and n = 5 S. cerevisiae-colonized, one experiment for d–f). g, h, Serum antibody binding to cultured C. albicans or S. cerevisiae from SW (g) or B6 (h) GF or monocolonized mice. Antibody quantified by flow cytometry from serum diluted 1:25 (SW: GF n = 4, Sc-colonized n = 5, Ca-colonized n = 5; B6: GF n = 3, Sc n = 5, Ca-colonized n = 3). i, Lumen and tissue-associated fungal burden in monocolonized B6 mice 30 days after inoculation (n = 4 mice per group; one experiment; representative of two experiments). j, Whole-intestinal IgA four weeks after inoculation. k, Caecal wash IgA binding to cultured C. glabrata measured by flow cytometry. l, Peyer’s patch TFH cells four weeks after inoculation. m, Peyer’s patch GC B cells (n = 4 mice per group; one experiment for j–m). n, IgA binding to cultured C. glabrata, S. cerevisiae and C. albicans from faecal wash from GF, C. albicans-monocolonized or C. glabrata-monocolonized intestinal wash (n = 2 C. albicans, n = 3 C. glabrata and n = 3 S. cerevisiae faecal washes) o, Percentage of IgA binding and binding intensity of faecal C. albicans during colonization of antibiotic-treated wild-type and Tcrb−/− mice (n = 6 Tcrb−/− and n =8 wild-type mice from two experiments). P values calculated using two-way ANOVA (d, o), with Sidak’s test (b, e, f, g, h, n), or two-sided unpaired t-test (j, k, l, m). Mean values ± s.d. for b, d–o.
Extended Data Fig. 3 An IgA response is not induced by 124 distinct S. cerevisiae strains.
a, IgA binding to the 20–24 strains from each pool was assessed by flow cytometry. Mice were gavaged weekly with the indicated pool for three weeks and caecal wash from mice was used as a source of IgA. C. albicans bound by IgA from C. albicans-monocolonized mice is shown in red. b, Total IgA in caecum contents quantified by ELISA. Mean values ± s.d. c, IgA binding to S. cerevisiae (pre-gated on CFW intermediate) populations from caecal material. (n = 3 mice per group representative of two experiments).
Extended Data Fig. 4 Fungal burden and GO term enrichment analysis of RNA-seq comparison of C. albicans in monocolonized wild-type and Rag1−/− mice.
a, Fungal burden in wild-type and Rag1−/− mice monocolonized with C. albicans four weeks after inoculation. Mean values ± s.d. b, c, Biological process (b) or molecular function (c) GO term enrichment in genes with q ≤ 0.05 and log2-transformed fold change ≥ 1 or ≤ −1. d, Volcano plot of the ratio of C. albicans transcripts in wild-type and Rag1−/− mice with active transmembrane transporter activity genes labelled in red (n = 5 wild type and 4 Rag1−/− mice for a–d; one experiment). e, C. albicans morphology in colon contents from monocolonized wild-type or Rag1−/− mice four weeks after colonization. Mean values ± s.d. (n = 3 mice per group; one experiment). f, IgA binding to C. albicans in the faeces of antibiotic-treated wild-type and μMT−/− mice four weeks after inoculation. Mean values ± s.d. (n = 5 mice per group; one experiment). P values calculated using two-way ANOVA with Sidak’s multiple comparisons test (a, f) or two-sided unpaired t-test (e).
Extended Data Fig. 5 Filamentation and Ahr1 promote intestinal IgA responses.
a, Morphology of indicated C. albicans strains incubated for 4 h in RPMI with 10% FBS, YPD or YPD + 5 μg ml−1 aTC). TetO-NRG1 constitutively expresses NRG1 when untreated (TetOn), but aTC repressed NRG1 expression (TetOff). b, C. albicans in caecum contents stained with AF488 anti-Candida antibody. c, Intestinal fungal burden (mean values ± s.d.). d, Peyer’s patch TFH cells (ICOS+PD-1+CD4+CD3+ live cells) (mean values ± s.d.). e, Peyer’s patch GC B cells (GL-7+Fas+IgD−CD19+ live cells) (mean values ± s.d.). f, Colon LP IgA+ plasma cells (IgA+CD138+CD45+CD3−CD19− live cells) (mean values ± s.d.) quantified from mice monocolonized for four weeks (for c–f, n = 4 mice per group; one experiment). g, Faecal AHR1 qPCR in aTC-treated mice monocolonized with wild-type or TetO-AHR1 (TetOff-AHR1) (wild type n = 3 and TetOff-ALS1 n = 5; one experiment). Mean values ± s.d. h, Fungal burden of wild-type- and TetOff-AHR1-monocolonized mice. i, IgA from wild-type- or TetOff-AHR1-monocolonized mice, j, k, Peyer’s patch TFH cells (j) and Peyer’s patch GC B cells (k) from mice monocolonized with wild type or TetOff-AHR1. l, qRT–PCR from the small intestinal contents of monocolonized mice (for h–l, wild type n = 8 and TetOff-ALS1 n = 10 mice per group from two experiments). m, Intestinal IgA (from C. albicans-monocolonized mice) binding to strains that were cultured untreated or were treated with aTC. n, Human IgA binding to indicated strains cultured without aTC (wild type, ahr1∆/∆, ahr1∆/∆ TetOn-ALS1) or with 5 μg ml−1 aTC (ahr1∆/∆ TetOff-ALS1). IgA binding quantified by flow cytometry (healthy n = 13 and IBD n = 22; one experiment. Samples chosen had enough C. albicans-reactive IgA to bind at least 10% of cultured wild-type C. albicans). P values calculated using one-way ANOVA with Tukey’s test (c–f), two-way ANOVA with Sidak’s test (i), two-sided unpaired t-test (j, k, l), two-sided Mann–Whitney U-test (g) or Friedman test with Dunn’s test (n).
Extended Data Fig. 6 C. albicans- and C. glabrata-induced IgA targets adhesins or adhesin-like proteins.
a, Anti-HA staining of the control S. cerevisiae expressing the Cwp1 scaffold control and the S. cerevisiae strains expressing HA-tagged C. albicans adhesins. b, Anti-HA and IgA binding to S. cerevisiae strains expressing HA-tagged C. glabrata adhesins after incubation in caecal wash from mice monocolonized with C. glabrata. SC104, SC106, SC97 and SC27 express adhesins not tagged by HA. HA and IgA binding quantified by flow cytometry.
Extended Data Fig. 7 Antibody induction by S. cerevisiae strains expressing Candida adhesins.
GF SW mice were monocolonized with the indicated strains or left GF. Colonized mice were gavaged three times per week with cultured strains. The control S. cerevisiae expresses the CWP1 cell surface scaffold but not an adhesin. a, Weekly faecal IgA levels normalized by faecal weight. b, c, Intestinal IgA (b) and IgG (c) levels at day 28 normalized by material weight. d, Colon lamina propria IgG1 plasma cells (live IgG1+IgA−CD138+CD19−CD3−CD45+ live cells). e, Colon lamina propria IgA plasma cells (live IgA+IgG1−CD138+CD19−CD3−CD45+ live cells) (for a–e, GF n = 6, control Sc n = 4, Sc + Als1 n = 5, Sc + Als3 n = 5, Sc + Hwp1 n = 4, Sc + CAGL0B00154g n = 5 mice per group; one experiment). P values calculated using one-way ANOVA with Tukey’s test (d, e) or two-way ANOVA with Tukey’s test (a–c). All data are mean ± s.d.
Extended Data Fig. 8 Immune-enhanced fitness diminishes after 14 days.
Competitive index (CI) of C. albicans conditioned for four weeks in indicated GF recipient mice. B6-conditioned C. albicans was iRFP+ and Rag1−/−-conditioned C. albicans was Neon+. CI normalized to the CI when strains were competed in wild-type and Rag1−/− mice directly from culture (competition mice, n = 3 B6 and n = 4 Rag1−/− mice from one experiment). P values calculated using two-way ANOVA with Sidak’s test. Data are mean ± s.d.
Extended Data Fig. 9 AHR1 exacerbates DSS colitis.
a, Schematic of DSS colitis experiments. b, Histology images and scores for mice treated with no C. albicans or with TetO-AHR1 with and without aTC (no-Ca UT, no-Ca aTC and TetOn-AHR1 UT n = 7 mice per group, TetOff-AHR1 aTC n = 8 mice per group from two independent experiments). Data are mean ± s.d. c, DSS histology images for mice treated with no C. albicans or with wild-type C. albicans, ahr1∆/∆ C. albicans, TetOn-ALS1 ahr1∆/∆ C. albicans and TetOff-ALS1 ahr1∆/∆ C. albicans (aTC-treated). P values calculated using two-way ANOVA with Tukey’s test (b).
Extended Data Fig. 10 NDV-3A induces an intestinal anti-Als3 antibody response.
a, model of monocolonization and DSS experiment in vaccinated mice. b, c, ELISA quantification of Als3-specific IgA (b) and IgG (c) from the faeces of GF mice one week after boost with alum of NDV-3A vaccine. d, e, Faecal (d) and intestinal (e) lumen CFU of C. albicans in monocolonized alum or NDV-3A vaccinated mice. Intestinal CFU quantified 12 days after colonization. f, Imaging flow cytometry images of IgA+ C. albicans from caecum of NDV-3A vaccinated mice. g, Percentage of hyphae quantified using an AF488 anti-Candida antibody to visualize morphology from indicated intestinal region 12 days after monocolonization. h, HWP1 and HYR1 transcripts quantified by qRT–PCR from colon C. albicans 12 days after monocolonization. (for b–h, n = 5 mice per group; one experiment). i, j, ELISA quantification of Als3-specific IgA (i) and IgG (j) in the faeces of conventionally colonized mice used for the DSS experiment. k, C. albicans CFU in colon contents after DSS treatment (for i–k, n = 10 mice per group, one experiment). l, Example H&E-stained histology images from the NDV-3A DSS experiment. P values calculated using two-way ANOVA with Sidak’s test (b, c, i, j). All data are mean ± s.d. Silhouettes in a were created using BioRender.
Supplementary information
Supplementary Information
This file contains Supplementary Figure 1.
Supplementary Table 1
RNAseq differential gene expression analysis.
Supplementary Table 2
IgA binding screen of Noble and Homann knock-out collections.
Supplementary Table 3
IgA targeted hyphae vs. yeast cell wall proteomics analysis.
Supplementary Table 4
Fungal strains used in this study.
Supplementary Table 5
S. cerevisiae strains expressing C. albicans or C. glabrata adhesins or adhesin-like domains.
Supplementary Table 6
Primers used for this study.
Supplementary Table 7
Plasmids used in this study.
Supplementary Table 8
Flow cytometry antibodies used in this study.
Supplementary Table 9
Human fecal and serum metadata.
Source data
Rights and permissions
About this article

Cite this article
Ost, K.S., O’Meara, T.R., Stephens, W.Z. et al. Adaptive immunity induces mutualism between commensal eukaryotes. Nature 596, 114–118 (2021). https://doi.org/10.1038/s41586-021-03722-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-021-03722-w
- Springer Nature Limited
This article is cited by
-
Antibody Fc-receptor FcεR1γ stabilizes cell surface receptors in group 3 innate lymphoid cells and promotes anti-infection immunity
Nature Communications (2024)
-
A mouse protozoan boosts antigen-specific mucosal IgA responses in a specific lipid metabolism- and signaling-dependent manner
Nature Communications (2024)
-
The hyphal-specific toxin candidalysin promotes fungal gut commensalism
Nature (2024)
-
A comprehensive guide to assess gut mycobiome and its role in pathogenesis and treatment of inflammatory bowel disease
Indian Journal of Gastroenterology (2024)
-
Veränderte T-Zell-Reaktion gegen Hefepilze bei chronischer Darmentzündung
BIOspektrum (2024)