

Abstract
Gut microorganisms modulate host phenotypes and are associated with numerous health effects in humans, ranging from host responses to cancer immunotherapy to metabolic disease and obesity. However, difficulty in accurate and high-throughput functional analysis of human gut microorganisms has hindered efforts to define mechanistic connections between individual microbial strains and host phenotypes. One key way in which the gut microbiome influences host physiology is through the production of small molecules1,2,3, yet progress in elucidating this chemical interplay has been hindered by limited tools calibrated to detect the products of anaerobic biochemistry in the gut. Here we construct a microbiome-focused, integrated mass-spectrometry pipeline to accelerate the identification of microbiota-dependent metabolites in diverse sample types. We report the metabolic profiles of 178 gut microorganism strains using our library of 833 metabolites. Using this metabolomics resource, we establish deviations in the relationships between phylogeny and metabolism, use machine learning to discover a previously undescribed type of metabolism in Bacteroides, and reveal candidate biochemical pathways using comparative genomics. Microbiota-dependent metabolites can be detected in diverse biological fluids from gnotobiotic and conventionally colonized mice and traced back to the corresponding metabolomic profiles of cultured bacteria. Collectively, our microbiome-focused metabolomics pipeline and interactive metabolomics profile explorer are a powerful tool for characterizing microorganisms and interactions between microorganisms and their host.
Similar content being viewed by others

Main
The human gut microbiota encodes diverse metabolic pathways. Gut microorganisms, which express numerous anaerobic pathways that process diverse diet- and host-derived molecules, produce numerous previously undescribed compounds with relevance for human health and that have untapped therapeutic potential. Many of these microbial products in the gut subsequently enter the tissue and circulation of the host, where additional metabolic steps can add to the chemical diversity1,2,3. Several recent studies have shown that microbiota-dependent metabolites (MDMs) influence immune function4, metabolism5,6, cardiovascular health7, and cognition and behaviour8. In many cases, MDMs exert these effects on host biology by binding to specific host receptors9 and activating downstream signalling pathways10. Discovery of how individual prevalent human gut microorganisms mechanistically contribute to host phenotypes has been hampered by the difficulty in accurately monitoring the diversity of molecules produced by gut microorganisms. To address this gap, recent studies have leveraged improvements in high-resolution mass spectrometry11 as well as growing mass-spectrometry and compound databases12 (for example, Mass Bank of North America (MoNA), Metabolite Link (METLIN13), Human Metabolome Database (HMDB14), and Kyoto Encyclopedia of Genes and Genomes (KEGG15). Nevertheless, because of fundamental differences between anaerobic metabolism in the gut versus aerobic biochemistry, as well as the underrepresentation of anaerobic microbial products in existing databases, the full metabolic capability of the microbiota remains understudied. Here we present a microbiome-focused, integrated mass-spectrometry pipeline to facilitate the identification of MDMs in diverse sample types, and to associate these metabolites with microbial strains and genetic pathways.
Microbiome-focused metabolomics
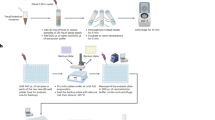
To enable the interrogation of microbiome metabolism, we (1) constructed a mass-spectrometry-based reference library to detect anaerobic biochemistry and an analytical pipeline to integrate large metabolomics datasets; (2) validated our methods to ensure applicability to the broader scientific community; and (3) enabled interactive, public access to our datasets (https://sonnenburglab.github.io/Metabolomics_Data_Explorer) (Fig. 1, Methods, Extended Data Figs. 1–3 and Supplementary Tables 1–4).

Schematic of our metabolomics workflow, consisting of mass-spectrometry reference library construction and validation, producing in vitro and in vivo metabolomic profiles across diverse sample types. Our entire dataset is publicly accessible through a web-based, interactive Metabolomics Data Explorer (https://sonnenburglab.github.io/Metabolomics_Data_Explorer).
Next, we leveraged this tool to create a reference dataset of metabolomic profiles for individual bacterial strains to enable multiple modes of analysis and discovery. We acquired 178 individual prevalent human gut microorganisms representing 130 species and spanning 6 phyla from ATCC, DSMZ and BEI (Supplementary Tables 5, 6). To create the most comparable dataset of metabolism, we cultured all supported strains (158 out of 178) in mega medium—a rich, undefined medium known to support the growth of diverse bacteria—and collected the culture supernatant between the mid-log and stationary phase (Extended Data Fig. 4a, b and Supplementary Methods). The remaining 20 strains were grown in 9 additional media as described in Supplementary Table 6, and 29 strains were grown and analysed across multiple types of media (Extended Data Fig. 4c and Supplementary Table 7).
To assess large-scale metabolite production and consumption patterns, we hierarchically clustered individual bacterial strains (Extended Data Fig. 4d–f and Supplementary Table 7). In some cases, two closely related species exhibited distinct metabolomic profiles punctuated with metabolite-level similarities (for example, Clostridium sporogenes and Clostridium cadaveris) (Extended Data Fig. 5a, b). In other cases, phylogenetic proximity is accompanied by similarity in metabolic patterns (for example, four strains of Bacteroides fragilis, Pearson r > 0.80 for all pairwise comparisons) (Extended Data Fig. 5a, b). Conversely, hierarchical clustering of species by metabolomic profile distance reveals unexpectedly shared metabolic patterns among phylogenetically distant species (for example by Atopobium parvulum, phylum Actinobacteria, and Catenibacterium mitsuokai, phylum Firmicutes) (Extended Data Fig. 6a–c).
In addition to the large-scale metabolic patterns, we discovered unique high producers or consumers of specific metabolites within our strain collection. For example, Enterococcus faecalis and Enterococcus faecium produce high levels of tyramine (Extended Data Fig. 4e)—a biogenic amine known to modulate host neurological functions16. By contrast, C. cadaveris consumes high levels of pantothenic acid (vitamin B5) (Extended Data Fig. 4f), a molecule that is associated with inflammatory bowel diseases17. This large-scale in vitro screen enables us to identify numerous high-abundance, variably conserved, microbially derived metabolites that can be tracked in vitro and in vivo (Extended Data Fig. 6d).
Metabolonomy distinct from phylogeny
We next addressed large-scale relationships between strain metabolism (metabolonomy) and phylogeny—a complex topic that has been addressed with different approaches in previous studies18,19,20,21. Bacterial metabolism is a product of the genetic metabolic toolkit and the chemical environment of a microorganism. Comparing metabolomic and phylogenetic trees for the same set of 158 strains grown in mega medium revealed a broadly conserved topology with the strains most often clustering by phyla (Fig. 2a, Extended Data Figs. 6a, 7a and Supplementary Methods). However, this similarity is punctuated by considerable divergences in which the relative location of specific strains in the two trees differs substantially (magenta and gold coloured branches in Fig. 2a). Notably, these patterns of clustering are preserved when metabolites are weighted by chemical similarity (Mantel test, r2 = 0.863, P = 0.001) (Extended Data Fig. 7b, c).

a, Comparison of tree topology constructed based on phylogenetic (left) and metabolomic profile (fold-change data, right) distance matrices of 158 strains grown in mega medium spanning five phyla. Data are from 1–3 independent experiments, each with n ≥ 3 biological replicates. b, Metabolite accumulation patterns across all 158 strains grown in mega medium, clustered based on phylogenetic distance. Dot size, mean production levels of 1–3 independent experiments, each with n ≥ 3 biological replicates. For each metabolite, the largest dot represents the highest production level for that metabolite. Full names of the bacterial species listed in a: Clostridium saccharolyticum, Clostridium symbiosum, Bifidobacterium adolescentis, Holdemania filiformis, and in b: Escherichia coli, Citrobacter youngae, Enterobacter cancerogenus, Citrobacter portucalensis, Providencia stuartii, Proteus penneri, Collinsella aerofaciens, Slackia exigua, Eggerthella lenta, Bacillus sp., Facklamia sp., Enterococcus faecalis, Enterococcus faecium, Lactobacillus reuteri, Streptococcus parasanguinis, Lactococcus lactis, Fusobacterium nucleatum, Fusobacterium nucleatum subsp. animalis, Fusobacterium ulcerans, Mitsuokella multacida, Veillonella sp., Clostridium sporogenes, Clostridium cadaveris, Clostridium perfringens.
To quantify these differences, we compared the metabolomic distance between strains to their evolutionary distance (Extended Data Fig. 7d and Supplementary Table 7). Using a phylogeny derived from the V4 16S region, the relationship between phylogenetic distance and metabolomic distance is linear (r2 = 0.30, P < 1 × 10−92) below around 0.11 branch-length units, approximating a difference of taxonomic ‘class’ in our data. Above a branch length of 0.11, the 16S distance explains almost none of the variance in the metabolomic distance (r2 = 0.02, P < 1 × 10−9). These patterns are robust to data transformation and evolutionary distance derived from full-length 16S genes (Extended Data Fig. 7e–j). Comparing the metabolic distance of bacteria grouped by taxonomic rank alone (for example, the distance between different strains of the same species) reveals a similar pattern of saturation (Extended Data Fig. 7d and Supplementary Table 7). These data indicate that when two strains are grown in the same complex medium, differences in the detected microbial metabolism are smaller on average than what would be extrapolated linearly from evolutionary or taxonomic relationships, particularly for distantly related bacteria. Notably, the high variance in metabolic distance between microorganisms of any relatedness (taxonomic or phylogenetic) reaffirms the use of metabolite profiles when comparing specific strains.
We next leveraged our strain-resolved metabolomic and genomic data to examine the correlation between bacterial genetic and metabolic variations in the context of a single pathway: polyamine biosynthesis (Fig. 2b and Extended Data Fig. 7k). Gut microbially derived putrescine and its precursor ornithine have both been implicated in influencing aspects of host physiology22,23. Their biosynthetic enzymes have been functionally characterized in select bacterial species (for example, ornithine-producing arc genes24 and putrescine-producing spe genes25).
We discovered two groups of phylogenetically distant strains in two phyla, Firmicutes and Actinobacteria (Fig. 2b, phyla with orange and purple borders, respectively), that accumulate high levels of ornithine and citrulline in the absence of substantial downstream polyamine production. We performed comparative genomics starting with the ornithine-producing arc genes described in Lactococcus lactis and found their conserved presence (Extended Data Fig. 7k) among the ornithine-accumulating strains, such as the Lactobacillales (Fig. 2b, strain names highlighted in orange). Notably, these genes are not detectable in the non-ornithine-accumulating phylogenetic neighbours in both Lactobacillales and Actinobacteria. These examples illustrate that, when metabolic phenotypes depart from phylogeny, orthologous gene–metabolite relationships may be preserved. We next identified strains that accumulate high levels of downstream putrescine and/or agmatine within three phyla: Proteobacteria, Fusobacteria and Firmicutes (Fig. 2b, phyla with green, red and orange borders, respectively). Although several putrescine-accumulating Proteobacteria strains (Fig. 2b, strain names highlighted in green) share the putrescine-producing spe gene cluster described in Escherichia coli (Extended Data Fig. 7k), these genes are not detectable in the Fusobacteria. These data indicate the limited ability of phylogeny- or genome-based prediction of metabolic functions in bacterial strains and highlight the utility of measuring metabolic phenotypes to identify strains and genes that produce specific metabolites that have the potential to affect host biology.
Metabolic phenotype-to-gene discovery
Metabolite production and consumption have long been used as mechanisms to group and identify organisms (for example, indole production). Here, we used our comprehensive metabolomic dataset constructed from strains grown in mega medium along with simple machine learning (random forest) models to identify sets of metabolites that could distinguish different taxonomic groups. Simple random forest models could accurately classify the taxonomic origin of microbial supernatants (Fig. 3a and Supplementary Methods). Although the total metabolome is not clearly predictive of taxonomy (Fig. 2a and Extended Data Fig. 7d), these random forest models revealed subsets of the chemical features that were highly conserved and predictive of taxonomic identity (Extended Data Fig. 8a).

a, Classification accuracy of random forest models at each taxonomic level, based on metabolomic profiles of 158 bacterial strains grown in mega medium from 1–3 independent experiments, each with n ≥ 3 biological replicates. Phylum (n = 5), class (n = 11), order (n = 15), family (n = 27), genus (n = 45), species (n = 115) and strain (n = 158). b, Amino acid production or consumption levels by Bacteroidetes strains from 1–3 independent experiments, each with n ≥ 3 biological replicates. Data shown are log2-transformed. Only uniquely detected (non-co-eluting) amino acids are shown. a, b, Boxes, median, 25th and 75th percentiles; whiskers, Tukey’s method. c, Phylogenetic tree of Bacteroidetes strains, growth curve maximum optical density (OD (600 nm)), and percentage of protein sequence identity for E. coli asparagine-consuming, ammonium-liberating enzymes. d, Representative growth curves of wild-type and mutant B. thetaiotaomicron (2757−3983−) in modified Salyer’s minimal medium from one experiment with n = 3 biological replicates. c, d, Nitrogen sources included ammonia (NH4), glutamine (Gln) and asparagine (Asn).
The most discriminating features selected by the random forest models for differentiating phyla included an overrepresentation in amino acid metabolism (Extended Data Fig. 8a). Notably, Bacteroidetes were differentiated by their consumption of most of the glutamine (median consumption, 83%) and asparagine (median consumption, 96%) in the mega medium (Fig. 3b). Previous studies showing that Bacteroides could not use free amino acids as the sole nitrogen source did not test asparagine and glutamine26. On the basis of the data from the 60 Bacteroidetes taxa in the collection, we hypothesized that glutamine and asparagine could serve as the sole nitrogen source. To test this, we grew all 60 Bacteroides and Parabacteroides species in a minimal medium that lacked free ammonium, but contained 10 mM glutamate, glutamine or asparagine. Notably, asparagine or glutamine sufficed as the nitrogen source for 50 out of 60 Bacteroidetes taxa tested (Fig. 3c and Extended Data Fig. 8b, c). To determine the genetic basis of asparagine utilization, we searched the Bacteroidetes genomes for homologues of E. coli enzymes that consume asparagine and release ammonia (Fig. 3c, red rows). For taxa with available genomes, an l-asparaginase II homologue (ansB; >59% identity) strongly correlated (Pearson r = 0.91) with the maximum optical density when grown on asparagine. Using a transposon mutant in the Bacteroides thetaiotaomicron type strain (B. thetaiotaomicron VPI 5482 2757−3983−), we confirmed that this l-asparaginase II homologue was necessary for growth with asparagine as the sole nitrogen source (Fig. 3d). The effect that we observed was not dependent on the presence of cysteine; B. thetaiotaomicron VPI 5482 and B. thetaiotaomicron VPI 5482 2757−3983− both grew with sodium sulfide substituted as a reduced sulfur source and the pattern of growth was maintained (Extended Data Fig. 8d). We next examined the amino acid consumption patterns of Bacteroides in vivo. In the caecum of mice monocolonized with B. thetaiotaomicron VPI 5482, asparagine was the most depleted amino acid (median decrease of 86.9%) compared with germ-free control mice (Extended Data Fig. 8e). This observation is consistent with in vivo asparagine utilization by B. thetaiotaomicron, but does not exclude colonization-dependent changes in host asparagine utilization. These findings demonstrate the power of combining strain-resolved metabolomics with simple statistical models—in this case, to discover a major metabolic capacity for nitrogen assimilation for the most abundant genus in the industrialized microbiota.
Metabolomic effect of community and host
Mechanistic studies in microbiome science can be aided by reverse translation of findings from complex communities (humans or conventionally colonized animals) into highly controlled (for example, gnotobiotic) models. We have recently demonstrated the use of our in vitro strain metabolite profiles in reverse translation by recreating metabolic phenotypes of interest to study mechanisms involved in the development of inflammatory bowel disease27. On the basis of two metabolites detected in human biological fluids (biofluids)28 and conventionally colonized mice, we asked whether we could reconstitute the production of microbially derived metabolites in the host gut and/or circulation by colonizing mice with the highest in vitro producing strain in our collection. One candidate, agmatine, is a polyamine with neuroprotective roles in mammals29 and a substrate for transporters in kidney and liver cells30. The other candidate, α-ketoglutaric acid, is a tricarboxylic acid cycle intermediate that extends the lifespan of the nematode Caenorhabditis elegans and increases autophagy in mammalian cells31.
Consistent with our in vitro observations, agmatine and α-ketoglutaric acid levels were both significantly increased in the faeces of mice mono-colonized with a high in vitro producer: Citrobacter portucalensis and Anaerostipes sp., respectively (Fig. 4a and Extended Data Fig. 9a). Furthermore, mono-colonization increased the levels of agmatine in the host circulation (for example, urine) relative to the germ-free control mice (Fig. 4a). These examples provide a proof-of-concept application of our in vitro dataset to reconstitute specific microbially derived metabolism in a mouse model, enabling potential mechanistic studies that are relevant to host physiology.

a, Quantification of agmatine levels. Data are mean ± s.e.m. of two independent experiments, each with n = 4 (germ-free) or n = 5 (Citrobacter mono-colonized) individual mice. b, Significantly produced metabolites associated with C. sporogenes or B. thetaiotaomicron in serum (left) or urine (right). Top, violin plots show median and quartiles; bottom, data are mean ± s.e.m. of one experiment with n = 4 (C. sporogenes, serum), n = 3 (C. sporogenes, urine), n = 5 (B. thetaiotaomicron, serum), n = 4 (B. thetaiotaomicron, urine), n = 7 (six-member, serum), n = 6 (six-member, urine) and n = 9 germ-free mice pooled from both mono-colonization (n = 4) and community (n = 5) experiments. c, Serum metabolite levels in mice colonized with the six-member community (with C. sporogenes (+Cs)) or the five-member community (without C. sporogenes (−Cs)). Metabolites shown represent a panel of significantly elevated or reduced metabolites (≥4-fold, corrected P < 0.05) in the six-member community. Data are mean ± s.e.m. of one experiment with n = 7 (six-member community) and n = 8 (five-member community) mice. Venn diagram of significantly elevated or reduced metabolites in different host biofluids based on the same threshold defined above. a–c, Data shown are log2-transformed. P values were calculated using two-tailed Student’s t-tests with Benjamini–Hochberg correction for multiple comparisons. *P < 0.05, **P < 0.01, ***P < 0.001.
We leveraged our strain-resolved metabolomic dataset combined with gnotobiotic colonization (Supplementary Table 8) and asked whether specific in vivo gut-bacteria-derived metabolites serve as biomarkers for a given taxonomic group. Among the 34 significantly produced metabolites in both colonized mice and individual strain cultures, we found several phylum-specific metabolites (for example, 5-aminopentanoic acid and indolepropionic acid by Firmicutes; malic acid and melatonin by Bacteroidetes) (Extended Data Fig. 9b and Supplementary Table 9). These data highlight that taxa-specific metabolites may serve as biomarkers for aspects of microbiome composition.
We next assessed the extent to which metabolites produced in vitro are reconstituted in gnotobiotic mice colonized with the same microorganisms. At the metabolomic profile level, faeces and caecal contents from mice mono-colonized with C. sporogenes or B. thetaiotaomicron correlated with C. sporogenes or B. thetaiotaomicron in vitro culture when compared against 158 taxa grown in mega medium (C. sporogenes, top 1%; B. thetaiotaomicron, top 10%) (Extended Data Fig. 9c). The lack of correlation in serum and urine (average Spearman ρ = 0.058, Extended Data Fig. 9c) is probably due to the inability of the bacterial culture to recapitulate host-encoded metabolism (for example, phase I/II enzymes). At the individual metabolite level, 8 out of 20 (40%, C. sporogenes) and 3 out of 29 (10%, B. thetaiotaomicron) significantly produced caecal metabolites in vivo were also produced by the same strain in vitro (Extended Data Fig. 9d). Furthermore, when assessing a six-species defined microbiota, 15 out of 46 (33%) significantly produced caecal metabolites were also produced by one or more of the six species in vitro (Extended Data Fig. 9d). Collectively, these data illustrate that metabolites produced in a standard rich medium can inform a portion of the microbially derived metabolites produced in the gut environment.
To better understand whether and how microorganism-dependent metabolites in the gut can inform circulating metabolites in the host, we examined enteric and systemic metabolic contributions of C. sporogenes and B. thetaiotaomicron in the host. We measured metabolite profiles of four sample types (faeces, caecal contents, serum and urine) in different colonization states (Fig. 4b). Principal component analyses reveal that metabolomic profiles cluster by sample type (for example, caecal contents versus serum) from mice colonized with the same microorganism, as well as by colonization state (for example, C. sporogenes mono-colonization versus a C. sporogenes-containing six-member community) (Extended Data Fig. 9e, f). We identified a distinct set of known and candidate host–microbial co-metabolites that are significantly elevated in the serum and/or urine, and are strongly associated with the presence of either C. sporogenes or B. thetaiotaomicron in the gut (Fig. 4b and Extended Data Fig. 9g, h). Notably, in both serum and urine, accumulation of N-(cinnamoyl)glycine is dependent on C. sporogenes, whereas accumulation of indoxyl sulfate is dependent on B. thetaiotaomicron (Fig. 4b and Extended Data Fig. 9g, h). Our systematic and high-throughput detection of microorganism-derived and host–microorganism metabolites across different sample types (for example, from caecum to serum) enables the identification of intermediates within known or candidate host–microbial co-metabolism pathways (Extended Data Fig. 10a).
To determine whether enteric presence of C. sporogenes is necessary for the increase or decrease in specific metabolites in the host circulation, we omitted C. sporogenes from the original six-member community. Metabolites shown are significantly increased or decreased by at least fourfold in the serum, urine or caecal contents of mice with the six-member community, relative to germ-free control mice (Fig. 4c and Extended Data Fig. 10b, c). By contrast, the five-member community that lacks C. sporogenes either abrogated the production or restored the depletion of a subset of these metabolites in the serum or urine, indicating that the enteric presence of C. sporogenes is necessary for modulating levels of these metabolites in the host circulation (Fig. 4c and Extended Data Fig. 10b) and illustrating the potential of microbiome editing to alter MDMs that circulate in the host blood.
Discussion
Untargeted metabolomics has led to many discoveries of microbiota-dependent metabolic pathways9,10 and metabolites linked to host diseases17,32,33,34, yet there is considerable untapped potential. Here we present a customizable and expandable method of constructing a chemical standard library-informed metabolomics pipeline tailored to detecting products of gut anaerobic biochemistry. Using this method, we construct an atlas of gut-microbiota-dependent metabolic activities in vitro and in vivo, enabling functional studies of gut microbial communities. Complementary to recent studies using phylogenetic (16S)35 or metagenomic comparisons36 to predict gene functions, we used strain-resolved metabolomics to provide expansive biochemical profiles of individual strains. These profiles demonstrate that substantial metabolic variation is common even between closely related strains. Our findings, along with emerging studies on microbiome-focused metabolomics37,38,39 and gut microbial metabolism40,41, reinforce the limits of phylogeny or genome-scale analysis to provide direct measurement or prediction of metabolic phenotypes and the molecules that link the microbiota to host physiology. Our existing strain-specific genome-by-metabolic profile data provides a rich resource for the comparative discovery of genes and pathways that underlie bacterial phenotypic variation. Furthermore, these data and this approach can be used as a direct reference or as a readily implemented platform for improving MDM identification in biological samples. Adding previously undescribed microbially derived metabolites, along with new strains such as those isolated from diverse human populations, will uncover new mediators of the interactions between the host and microbiota as well as molecular targets for therapeutic interventions.
Methods
Data reporting
No statistical methods were used to predetermine sample size. The experiments were not randomized and the investigators were not blinded to allocation during experiments and outcome assessment.
Metabolomics pipeline construction logic
The accurate identification and analysis of diverse small molecules in complex biological samples (for example, those present in the mammalian gut) are challenging due to a variety of technical factors, including chemical structural diversity, matrix effects and linearity of ion detection. To ensure that our liquid chromatography–mass spectrometry (LC–MS) pipeline is relevant for biological samples and that it is useful to the broader scientific community, we highlight six key points of our approach: (1) detectability of diverse chemical classes of compounds that characterize bacterial and host metabolism using three complementary analytical methods42,43 (Extended Data Figs. 1d, 3a–c); (2) retention time (RT) shifts that occur in divergent matrices (for example, culture supernatant versus host serum) to determine whether metabolites in a biological sample could be faithfully identified using RT data from our m/z-RT reference library (Extended Data Fig. 3d, e and Supplementary Table 2); (3) linearity of signal over a large range of concentrations, a prerequisite for performing sample comparisons and determining differences in the fold change (Extended Data Fig. 3f and Supplementary Table 2); (4) use of MS/MS fragmentation to validate the high-abundance metabolites identified in biological samples (Extended Data Fig. 1e and Supplementary Table 2); (5) construction of an MS/MS reference library of 750+ authentic standards on two distinct types of MS instrument (qTOF and Q Exactive) at multiple standard collision energies (Supplementary Table 3), enabling level-1 confidence annotation when used in conjunction with our m/z-RT reference library; and (6) implementation of our m/z-RT reference library on different types of MS instruments following minimal nonlinear RT correction44 (Extended Data Fig. 3g and Supplementary Table 4). For data analysis, we constructed an integrated pipeline combining (1) MS analysis tools45 that leverage our reference library for compound identification (Extended Data Fig. 1f) and (2) a custom bioinformatics pipeline that enables the computation and statistical analysis of large datasets (Extended Data Fig. 2).
Authentic chemical standard collection
The authentic metabolite standard collection is composed of individually curated and commercially available standards (Mass Spectrometry Metabolite Library of Standards, IROA Technologies). Individually curated metabolites (303 metabolites) were weighed (2 mg minimum) and transferred from the original manufacturer’s stock bottles (for example, Sigma, Fisher, Acros and so on) to 2-ml Eppendorf tubes and reconstituted with 50% LC–MS grade methanol to reach a stock concentration of 10 mM. Additional compounds (284 metabolites) were purchased as 10-mg stocks from MetaSci (MetaSci Custom Library). Dried power from company stock tubes were transferred (2 mg minimum) into 2-ml tubes and reconstituted with 50% methanol to a concentration of 10 mM. Metabolites from the IROA metabolite standard library (634 metabolites), which were supplied in much smaller amounts (around 5 μg per well), were reconstituted with various amount of methanol in water (v/v) as per the manufacturer’s instructions, but owing to the limited mass, their concentrations were less precise. Individual pools (12–30) of metabolite standards, which do not share the same molecular mass, were generated by combining stocks and diluted with 50% methanol to reach a final concentration of 200 μM. A subset of these pools (377 metabolites) was also serially diluted in 50% methanol. Individual metabolite pools and dilutions were analysed using three LC–MS analytical methods.
LC–MS methods
Instrumental and chromatographic settings
Compounds were separated using an Agilent 1290 Infinity II UPLC (binary pumps) and detected using an Agilent 6545 LC–MS Quadrupole Time-of-Flight (qTOF) instrument equipped with a dual jet stream electrospray ionization source (ESI) operating under extended dynamic range (1,700 m/z) in the positive (ESI+) or negative (ESI−) ionization modes. Published C18 methods42 and HILIC method43 were used with minor modifications. See Supplementary Methods for details.
Sample preparation for metabolomics
Five different sample types were processed with a similar sample preparation protocol as described in the Supplementary Methods. In brief, samples were homogenized and proteins were precipitated in a methanol-based recovery buffer that contains the extraction standards. Samples were then centrifuged, their supernatant was collected and evaporated, and a reconstitution buffer containing internal standards was added. Reconstituted samples were filtered and subsequently analysed by three analytical methods on the LC–MS-qTOF.
m/z-RT reference library
The exact m/z of each metabolite standard was calculated by combining the monoisotopic mass of the metabolite (PubChem) and adding or subtracting the mass of a proton (1.007276 Da) depending on the default adduct ion ([M + H]+ for ESI+ and [M − H]− for ESI−). The Agilent MassHunter Qualitative Data Analysis software (Qual, v.B.07.00) was used to match individual extracted-ion chromatogram peaks within a ±10-ppm window from the predicted m/z of each metabolite standard. Alternative adducts ions were identified using ‘Search by Molecular Feature’ in Qual; when multiple adducts were identified, the adduct ion with the greatest area under the curve was used in the reference library. An RT was assigned to a metabolite when a single extracted-ion chromatogram peak was identified. When multiple chromatographic peaks were identified, which probably resulted from degradation products, different isotopes or adducts of other molecules in the mixture, a subsequent injection of that metabolite standard alone was conducted to identify the RT for that metabolite. For metabolites run in dilution series, RTs at all concentrations at which the same metabolite was detected were used to produce an averaged RT for this metabolite in the reference library. The averaged RT was used to (1) increase the accuracy by averaging small injection-to-injection variations; and (2) distinguish the true signal from background noise by validating the peaks for which the ion counts proportionally increase with the concentration.
To address how the same reference library performed on different instruments, we compared two different LC–MS systems: an Agilent 6545 qTOF, the instrument with which the original library was constructed, and a second instrument, an Agilent 6530 qTOF or a Thermo Orbitrap Q Exactive (QE). Although these different instruments shared the same chromatographic conditions (for example, analytical methods, solvents and columns), they differed in resolution and ESI ion source parameters optimized to support each instrument. To compare inter-instrumental RT shifts, a subset of the full reference library (219 metabolite standards spanning diverse RTs) was reconstructed on the second qTOF instrument, and 773 metabolite standards were reconstructed on the QE instrument. For each analytical method, RT correction was done by cubic polynomial transformation of the original library44 based on inter-instrumental RT shifts of 10–20 robustly detected metabolites (for example, internal standards) that span the detected RT range. For each analytical method, using the corrected library with a RT tolerance window of 0.2 min, around 99% for the 219 metabolites tested on the second qTOF instrument, and approximately 94% of the 773 metabolites tested on the QE instrument, were correctly identified.
MS/MS library construction
MS/MS raw data were collected from individual pools (12–24 compounds per pool) for 833 authentic library standards, using three liquid chromatography methods applied to two distinct types of MS instruments (Agilent qTOF 6545 and Thermo Orbitrap QE). For qTOF, auto-MS/MS-preferred ion settings with an individual input list of m/z and RT information specific to the compounds in each pool were used to collect spectra at three collision energies (10 eV, 20 eV and 40 eV). For QE, full MS/dd-MS2 settings with a single shared inclusion list containing the m/z and RT information for all of the compound pools were used for data collection at the stepped normalized collision energy of 20–30–40%. A scan range of 60–900 m/z was used to collect centroid type data. On both instruments, ±0.5 min was used as an RT search window for MS1 peak selection, based on the RTs provided by the qTOF reference library. Accurate mass windows were ±10 ppm on both instruments. RTs identified during the MS1 peak selection for the 773 compounds detected on the QE instrument are reported in the m/z-RT library in the ‘QE_rt’ column (Supplementary Table 1).
MS/MS spectra were extracted from MS/MS raw data files (mzml format) with an automated Python script (extract_ms2_spectra.ipynb) using the pymzML parsing library46. For each compound, the intensity of each spectral fragment was normalized to the fragment with the highest intensity (set to 1,000). Spectral fragments with intensities below 0.5% relative to the highest intensity fragment were filtered out. Compound metadata (for example, InChIKey and collision energy) and fragmentation information (for example, m/z and intensity) are reported for each compound. Spectra from the same compound collected using different analytical methods (for example, C18-positive and C18-negative) are all reported. In limited instances, spectra from the same compound were collected multiple times due to representation in multiple compound pools. All of the information above was compiled in Supplementary Table 3, and is publicly available in the MoNA spectrum database under query phrase ‘Sonnenburg Lab MS2 library’. In summary, spectra from 750 and 773 unique compounds were collected on the qTOF and QE instrument, respectively.
MS experimental validations
Linear dynamic range
For large-scale metabolomics experiments, it is typically assumed that instrument response varies linearly with analyte concentration. To test the concentration linearity objectively, we constructed dilution series of 377 metabolites (from pools generated as above), in threefold serial dilutions spanning five orders of magnitude (from 1 nM to 200 μM). These diluted compound pools were then analysed using the three analytical methods. Linear regression of log-transformed concentrations versus log-transformed ion counts was performed and the coefficient of determination (r2) was calculated. Across all metabolites, the average r2 and slope (on log–log plots) were both very close to 1 (0.99 and 0.92, respectively), providing a strong indication of linearity.
Matrix effects
The biochemical complexity of biological samples such as faeces and serum may alter the RT and/or detected signal of individual metabolites. To determine whether accurate identification was significantly affected by RT shifts in multiple matrices, we spiked in 132 metabolite standards into five distinct biological matrices (germ-free mouse faeces, serum and urine, human charcoal-stripped serum and mega medium) and a library control condition (50% methanol, v/v) at a final concentration of 10 μM, and analysed each matrix using all three analytical methods. Three biological replicates for each matrix were used, and the RT and ion count for each spiked-in metabolite standard in each of these matrices were determined. The difference in RT between a biological matrix and the library control condition was calculated (50% methanol in water, v/v) for individual spike-in metabolites. For all 132 metabolites in all five matrices, differences in RTs were minimal, falling within a conservative ±0.1-min window. Changes in total ion count (area under the curve) between a biological matrix and the library control condition were determined by first removing matrix-specific background ion counts for a small number of metabolites present in specific matrix before spike-in. Next, the ratio between spike-in metabolite ion counts in biological matrices and those in library blank controls was calculated (relative fold change, log2-transformed). The majority of spiked-in metabolites exhibit less than fourfold change in ion counts relative to those detected at the library control condition (97% in mouse faeces, 83% in mouse serum, 95% in mouse urine, 88% in human serum and 71% in mega medium). See code details in ‘calculate_biological_matrix_effect.ipynb’. The relatively minor influence of different biological matrices on RTs of the reference library metabolites helped to establish the identification parameter (±0.1-min RT window) for our subsequent biological experiments.
MS/MS validation
To verify the accuracy of compound identification obtained by our MS1 m/z-RT library built from authentic standards, we unbiasedly searched MS/MS spectra of m/z-RT-matched individual metabolites against the MoNA spectrum database. MoNA-reported similarity scores based on spectrum comparisons were recorded (Supplementary Table 2). For each analytical method, using the auto-MS/MS-preferred ions settings of the qTOF, MS/MS spectra were generated at three collision energies (10 eV, 20 eV and 40 eV) from MS1 peaks identified by m/z and RT from our reference library. For biological samples, MS/MS spectra were collected for 162 high-abundance metabolites identified in quality-control samples from in vitro (bacterial supernatants) and in vivo experiments (B. thetaiotaomicron- and C. sporogenes-mono-colonized mouse samples: serum, urine and faecal/caecal contents). Quality-control samples were generated on a per-experiment basis by pooling equal volumes from each biological replicate from the same experiment (3–8 biological replicates per condition across the entire 96-well plate) to provide a representation of the highest number of metabolites in that experiment. To establish a baseline of MoNA similarity scores, MS/MS spectra were also collected from a corresponding set of library authentic standards.
MS/MS spectra were extracted using an automated Python script by first extracting MS/MS spectra for individual m/z-RT-matched metabolites using pymzML46, and then searching individual extracted spectra against the MoNA spectrum database. The search results were restricted to spectra generated using (1) LC–MS instruments and (2) ESI+ ionization mode (for C18-positive and HILIC-positive spectra) or ESI− ionization mode (for C18-negative spectra). Each spectral search used the MoNA-default similarity score threshold of 500, and returned the top-five matches with the highest similarity scores computed by the built-in MoNA algorithm. Among these top matches, the highest similarity score with the correct metabolite name was recorded (Supplementary Table 2). Because MoNA search results contained data from various LC–MS instrument platforms such as qTOF, Orbitrap and Triple-Quadrupole, in some cases there are data collected from multiple MS platforms or multiple collision energies, we would opt for the qTOF and a similar collision energy to our search spectra. Each MS/MS spectral comparison corresponding to the recorded score was also manually inspected. For individual metabolites repeatedly detected in the same sample type (for example, bacterial supernatant or faeces) in more than one experiment, an averaged similarity score among MS/MS spectra for the same metabolite was calculated and recorded in the summary table (Supplementary Table 2). Collectively, all similarity scores between our MS/MS spectra and MoNA spectra for the same set of metabolites have a median score of 992 (library standards, s.d. = 36.78) and 923 (biological samples, s.d. = 114) relative to a perfect score of 1,000, indicating good agreement between our data and what has previously been reported.
Data analysis
MS-DIAL analysis
The MS-DIAL software45 (v.3.83) was used for analysing all in vitro and in vivo data on a per-experimental run and per-analytical method basis. Quality-control samples from each experimental run were used for peak alignment. Chemical assignment of molecular features in samples was performed by comparing the recorded RT and m/z information to our reference library constructed from authentic standards. Tolerance windows were set to 0.1 min RT and 0.01 Da m/z for the C18 methods and 0.2 min RT and 0.01 Da m/z for the HILIC method. When a large RT shift was observed in the internal standards (for example, after instrument repair), a library RT correction was done before MS-DIAL analysis, through a polynomial transformation of the library based on inter-instrumental RT shifts of 10–20 robustly detected metabolites (for example, internal standards). The minimal peak count (height) filter was set to 3,000 for all experiments except for select experiments in which the MS exhibited reduced sensitivity. The MS-DIAL analysis generated a list of m/z, RT and ion counts (area under the curve) for high-confidence annotations (matched to the reference library) as well as unknown molecular features. On the basis of the list of annotations for each experiment, each set of aligned peaks was manually checked using the MS-DIAL graphical user interface. Select metabolite features were removed from this list when: (1) two adjacent but distinct peaks were concurrently assigned to a single molecular feature; (2) odd curvature/shape of the peak led to the integration of several ‘peaks’ from separate sections of the same peak; or (3) features were detected only in the blank controls. Annotated peaks that passed this inspection were included in the final output file.
Custom bioinformatics
After MS-DIAL analysis, data were analysed with a set of custom bioinformatics pipelines. In brief, these pipelines implemented a set of filtration and normalization procedures with the goal of reducing technical variability and controlling for batch effects. The pipelines, including all code for the in vitro and in vivo sample data cleaning and standardization, are described in the Supplementary Methods.
Distance calculations and classifiers
Comparisons between metabolomic and phylogenetic distances (Fig. 2a and Extended Data Fig. 7) and metabolite-based classification (Fig. 3a and Extended Data Fig. 8a) were done with custom Python code described in the Supplementary Methods. For all these analyses, the metabolomic distance matrix used Euclidean distance generated from log2-transformed, medium-blank, delta and variance-filtered fold change data. Only the 158 strains that grew in mega medium were used for these analyses to prevent conflation of metabolic and starting medium differences.
Bacterial culture
The bacterial strains and associated metadata (such as taxonomy, original repository and 16S sequence) used in this work are reported in Supplementary Table 6. All bacterial inoculation and growth occurred in a Coy Laboratories anaerobic chamber kept at an atmosphere of approximately 80%:15%:5% (N2:CO2:H2). All incubations occurred at 37 °C, all bacterial stocks were stored at −80 °C, and all ODs were recorded at 600 nm using a BioTek Epoch 2 plate reader.
Stock preparation
Bacterial strains were acquired from various culture collections including ATCC, DSMZ, NCTC and BEI. Source cultures were plated on a rich medium, single colonies were picked, cultured in rich medium and stored as 1-ml frozen cultures (25:25:50 v/v glycerol:H2O:culture) in ThermoFisher Matrix Tubes. The solid and liquid media used for stock generation are described in Supplementary Table 6 (worksheet ‘media’). Source cultures that exhibited multiple morphologies on agar plates were purified and morphologies separated and retained if the 16S sequence matched the expected 16S sequence. For all cultures, the purity of the final cultures was checked by 16S rRNA sequencing (Supplementary Methods).
Bacterial media
All media used in this study are included in Supplementary Table 6 (worksheet ‘media’). Note that in some cases we grew and recorded metabolites from taxa in multiple media. For the media used for particular supernatant samples and metabolomics, see Supplementary Table 7 (worksheet ‘aggregated_md’).
Mega medium was prepared according to the protocol described in the Supplementary Methods. The recipe is slightly adapted from a previous publication47. In our usage of mega medium, each batch was autoclaved, moved into the anaerobic chamber and allowed to become anaerobic for at least 24 h before use. For taxa that would not grow in mega medium, a different medium was selected based on the literature. In each case, we referenced an ATCC, DSMZ or media manufacturer (for example, Hardy Diagnostics) recipe as outlined in Supplementary Table 6 (worksheet ‘media’). In all cases, these media were prepared for use similarly to mega medium. Specifically, the adjustment of the pH was done before autoclaving, filter-sterilized vitamins and sterile blood were added after autoclaving, and media were moved immediately from the autoclave to the anaerobic chamber and allowed to become fully anaerobic for at least 24 h before use.
For identification of nitrogen utilization in Bacteroidetes, Salyer’s minimal medium (SMM) was prepared (Supplementary Methods), the preparation of which was slightly modified from published protocols26,48. In brief, SMM base was prepared (SMM without haematin, nitrogen source or reduced sulfur source) and allowed to become anaerobic in foil-covered bottles. SMM was prepared without nitrogen source to avoid spontaneous glutamine degradation49. Immediately before use, the SMM base was amended with filter-sterilized solutions of haematin (final concentration 0.5 mg per 100 ml), nitrogen source (glutamine, asparagine, glutamic acid or ammonium sulfate, final concentration of 10 mM) and reduced sulfur source (cysteine or sodium sulfide, final concentration of 4.12 mM). Taxa were plated (mega medium or brain heart infusion with blood) and a single colony picked into freshly prepared SMM. Preculture for 24 h was followed by subculture in freshly prepared SMM for 12–36 h. OD readings were taken as described above.
In vitro growth for metabolomics
Bacterial supernatants included in the in vitro data were generated according to the following protocol. Cultures were inoculated in anaerobic medium (around 4 μl:1,600 μl) in triplicate in 2-ml 96-well blocks and incubated for 24–72 h depending on the taxa selected. Therefore, a single biological replicate from the bacterial culture experiments represents an individual well or tube of bacterial culture growth from an independent 4-μl aliquot from a frozen glycerol culture stock. These pre-cultures were subcultured into mega medium (around 4 μl:1,600 μl) and similarly incubated for 12–60 h. Then, 200 μl of subculture was incubated in a plate reader so that OD readings could be taken to monitor growth phase. The remaining cell cultures were collectd when the OD readings showed the late log or early stationary phase. The collected culture was immediately removed from the anaerobic chamber, centrifuged to pellet the cells (5,000g, 10 min) and the cell-free supernatant was either frozen at −80 °C or immediately extracted as described in the Supplementary Methods.
For details of the purity analysis, sequencing protocol and phylogenetic tree reconstruction, see Supplementary Methods.
Mouse experiments
Mouse experiments were performed with gnotobiotic Swiss–Webster germ-free mice (male, 10–14 weeks of age, n = 3–8 per group for all experiments) or Swiss-Webster excluded flora mice (‘conventional mice’; male, 10–14 weeks of age, n = 3 per group) that were maintained in aseptic isolators and originally obtained from Taconic Bioscience. Mice were maintained on a 12-h light/dark cycle at 69 °F (20.6 °C) in ambient humidity, fed ad libitu, and maintained in flexible film gnotobiotic isolators for the duration of all experiments (Class Biologically Clean). For mono-colonization experiments, mice were colonized with B. thetaiotaomicron VPI 5482, Clostridium sporogenes ATCC 15579, C. portucalensis BEI HM-34 or Anaerostipes sp. BEI HM-220 by oral gavage (200 μl, around 1 × 107 colony-forming units (CFU)) and were maintained on a standard chow (LabDiet 5K67). For the defined-community experiment, mice with a six-member community were colonized with a 200-μl mixture consisting of equal volumes from saturated cultures of B. thetaiotaomicron VPI 5482 (8.7 × 109 CFU), C. sporogenes ATCC 15579 (1.4 × 108 CFU), Edwardsiella tarda ATCC 23685 (3.6 × 1010 CFU), Collinsella aerofaciens ATCC 25986 (1.4 × 109), Eubacterium rectale ATCC 33656 (6.9 × 106 CFU) and Parabacteroides distasonis ATCC 8503 (1.5 × 109 CFU). Mice with a five-member community were colonized with all cultures mixed at the same volumes as described above except for C. sporogenes ATCC 15579, which was not included. Successful colonization and stable community members were determined by 16S amplicon sequencing of the V4 (515F, 806R) region of microbial populations that were present in the faeces and caecal contents of individual mice.
For all experiments, mice were euthanized by CO2 asphyxiation 9 days (mono-colonization with C. portucalensis BEI HM-34 or Anaerostipes sp. BEI HM-220) or 4 weeks (all other experiments) after colonization, and four sample types (serum, urine, faeces and caecal contents) were collected from each mouse. A single biological replicate in the mouse experiments represents a specific sample type (for example, serum) collected from an individual mouse (that is, each biological replicate is from a different mouse). Before euthanization, urine and faeces were collected. Whole blood was collected by cardiac puncture and serum was obtained using microcontainer serum separator tubes from Becton Dickinson following the manufacturer’s instructions. The intact caecum was collected and snap-frozen in liquid nitrogen. A single caecal sample was obtained for mono-colonization and conventional experiments, and three samples at three different sections of the caecum were obtained for the defined-community experiment. All mouse experiments were conducted under a protocol approved by the Stanford University Institutional Animal Care and Use Committee.
Comparative genomics
Genome annotation and database
Bacterial isolates from the culture collection were manually linked up to their respective NCBI BioProject ID numbers. The Rentrez package (https://cran.r-project.org/package=rentrez) was used to link BioProject ID numbers with existing GenBank or RefSeq assemblies or with reads from the Sequence Read Archive (SRA) for isolates that were previously sequenced but not assembled. Isolates lacking assembly accession numbers (Supplementary Table 6 (worksheet ‘full_taxonomy’)) were assembled using previously described methods50. In brief, reads were trimmed using Trimmomatic51 and assembled using SPAdes v.3.9.152 using the following parameters: k = 21,33,55 --careful --cov-cutoff auto. Contigs smaller than 1,500 bp were removed, and assemblies were gene-called and annotated using prokka v.1.14.553. MultiGeneBlast54 (v.1.1.13) was used to build a database containing all of the assembled and downloaded genomes listed in Supplementary Table 6.
Gene and gene cluster searches
The arc gene cluster from Lactococcus lactis and the spe gene cluster from E. coli were used as the query to search publicly available, assembled genomes of strains within our collection. Comparative genomics analyses were conducted using the ‘Architecture Search’ feature of the MultiGeneBlast software (v.1.1.13) with default parameters with one modification, which set the ‘maximum distance between genes in locus (kb)’ to 40 kb. For identification of Asparaginase-containing genomes, the custom BLAST database described above was queried for homologues of E. coli genes (ansA, ansB and aspA) that encode asparagine-consuming enzymes.
Metabolomics Data Explorer
The Metabolomics Data Explorer (https://sonnenburglab.github.io/Metabolomics_Data_Explorer) was constructed in JavaScript and was used to generate scatter plots of our in vitro and in vivo fold-change data based on user input. In vitro and in vivo metadata and fold-change data files were used as data input and were parsed using the Papa Parse library to extract the data and populate the dropdown menus on each page. The dropdown menus enable users to pick the desired taxonomy, metabolite and medium (in vitro), and colonization, metabolite and sample type (in vivo). The Nivo library was used to render interactive scatter plots of the fold change data relative to medium blank controls (in vitro) or to germ-free controls (in vivo). Each dot represents an independent biological replicate, and all metabolites (uniquely identified or co-eluting) are shown. In rare cases, the same metabolite may appear twice in the scatter plot if it is uniquely identified in one analytical method while co-eluting with other metabolites in another analytical method. The scatter plot presents all biological replicates from all independent experiments available in the dataset and provides label details when hovering over the data points to enable easy identification.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
All raw data from metabolomics are publicly available from the Metabolomics Workbench under study number ST001683 for in vivo data and study number ST001688 for in vitro data. MS/MS libraries generated using the qTOF and QE instruments are publicly accessible in the MoNA spectrum database (https://mona.fiehnlab.ucdavis.edu) and can be queried using the keywords ‘Sonnenburg Lab MS2 Library’.
Code availability
Custom Python code was written to enable the construction of the MS/MS libraries, the processing and visualization of the in vitro and in vivo LC–MS data, the optical density and growth curve data, the bioinformatics analysis of 16S and whole genomes, and the analysis of the metabolomic data. Full code for each of these steps is available at https://doi.org/10.5281/zenodo.4890994. The JavaScript code supporting the interactive, web-based Metabolomics Data Explorer is available at https://doi.org/10.5281/zenodo.4890999.
References
Koppel, N., Maini Rekdal, V. & Balskus, E. P. Chemical transformation of xenobiotics by the human gut microbiota. Science 356, eaag2770 (2017).
Koh, A., De Vadder, F., Kovatcheva-Datchary, P. & Bäckhed, F. From dietary fiber to host physiology: short-chain fatty acids as key bacterial metabolites. Cell 165, 1332–1345 (2016).
Donia, M. S. & Fischbach, M. A. Small molecules from the human microbiota. Science 349, 1254766 (2015).
Rooks, M. G. & Garrett, W. S. Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 16, 341–352 (2016).
Cani, P. D. Microbiota and metabolites in metabolic diseases. Nat. Rev. Endocrinol. 15, 69–70 (2019).
Sonnenburg, J. L. & Bäckhed, F. Diet-microbiota interactions as moderators of human metabolism. Nature 535, 56–64 (2016).
Kasahara, K. & Rey, F. E. The emerging role of gut microbial metabolism on cardiovascular disease. Curr. Opin. Microbiol. 50, 64–70 (2019).
Lynch, J. B. & Hsiao, E. Y. Microbiomes as sources of emergent host phenotypes. Science 365, 1405–1409 (2019).
Nemet, I. et al. A cardiovascular disease-linked gut microbial metabolite acts via adrenergic receptors. Cell 180, 862–877.e22 (2020).
Koh, A. et al. Microbially produced imidazole propionate impairs insulin signaling through mTORC1. Cell 175, 947–961.e17 (2018).
Rinschen, M. M., Ivanisevic, J., Giera, M. & Siuzdak, G. Identification of bioactive metabolites using activity metabolomics. Nat. Rev. Mol. Cell Biol. 20, 353–367 (2019)
Blaženović, I., Kind, T., Ji, J. & Fiehn, O. Software tools and approaches for compound identification of LC-MS/MS data in metabolomics. Metabolites 8, 31 (2018).
Guijas, C. et al. METLIN: a technology platform for identifying knowns and unknowns. Anal. Chem. 90, 3156–3164 (2018).
Wishart, D. S. et al. HMDB 4.0: the human metabolome database for 2018. Nucleic Acids Res. 46 (D1), D608–D617 (2018).
Kanehisa, M., Sato, Y., Furumichi, M., Morishima, K. & Tanabe, M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 47 (D1), D590–D595 (2019).
Sampson, T. R. & Mazmanian, S. K. Control of brain development, function, and behavior by the microbiome. Cell Host Microbe 17, 565–576 (2015).
Lloyd-Price, J. et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 569, 655–662 (2019).
Goldford, J. E. et al. Emergent simplicity in microbial community assembly. Science 361, 469–474 (2018).
Kamneva, O. K. Genome composition and phylogeny of microbes predict their co-occurrence in the environment. PLOS Comput. Biol. 13, e1005366 (2017).
Borenstein, E., Kupiec, M., Feldman, M. W. & Ruppin, E. Large-scale reconstruction and phylogenetic analysis of metabolic environments. Proc. Natl Acad. Sci. USA 105, 14482–14487 (2008).
Plata, G., Henry, C. S. & Vitkup, D. Long-term phenotypic evolution of bacteria. Nature 517, 369–372 (2015).
Tofalo, R., Cocchi, S. & Suzzi, G. Polyamines and gut microbiota. Front. Nutr. 6, 16 (2019).
Qi, H. et al. Lactobacillus maintains healthy gut mucosa by producing l-ornithine. Commun. Biol. 2, 171 (2019).
Zúñiga, M., Pérez, G. & González-Candelas, F. Evolution of arginine deiminase (ADI) pathway genes. Mol. Phylogenet. Evol. 25, 429–444 (2002).
Tabor, C. W. & Tabor, H. Polyamines in microorganisms. Microbiol. Rev. 49, 81–99 (1985).
Varel, V. H. & Bryant, M. P. Nutritional features of Bacteroides fragilis subsp. fragilis. Appl. Microbiol. 28, 251–257 (1974).
Mars, R. A. T. et al. Longitudinal multi-omics reveals subset-specific mechanisms underlying irritable bowel syndrome. Cell 182, 1460–1473.e17 (2020).
Wastyk, H. C. et al. Gut microbiota-targeted diets modulate human immune status. Preprint at https://doi.org/10.1101/2020.09.30.321448 (2020).
Kotagale, N. R., Taksande, B. G. & Inamdar, N. N. Neuroprotective offerings by agmatine. Neurotoxicology 73, 228–245 (2019).
Winter, T. N., Elmquist, W. F. & Fairbanks, C. A. OCT2 and MATE1 provide bidirectional agmatine transport. Mol. Pharm. 8, 133–142 (2011).
Chin, R. M. et al. The metabolite α-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature 510, 397–401 (2014).
Zhou, W. et al. Longitudinal multi-omics of host–microbe dynamics in prediabetes. Nature 569, 663–671 (2019).
Watrous, J. D. et al. Directed non-targeted mass spectrometry and chemical networking for discovery of eicosanoids and related oxylipins. Cell Chem. Biol. 26, 433–442.e4 (2019).
Dumas, M. E. et al. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc. Natl Acad. Sci. USA 103, 12511–12516 (2006).
Langille, M. G. I. et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821 (2013).
Sberro, H. et al. Large-scale analyses of human microbiomes reveal thousands of small, novel genes. Cell 178, 1245–1259.e14 (2019).
Quinn, R. A. et al. Global chemical effects of the microbiome include new bile-acid conjugations. Nature 579, 123–129 (2020).
Zimmermann, M., Zimmermann-Kogadeeva, M., Wegmann, R. & Goodman, A. L. Mapping human microbiome drug metabolism by gut bacteria and their genes. Nature 570, 462–467 (2019).
Wu, G. D. et al. Comparative metabolomics in vegans and omnivores reveal constraints on diet-dependent gut microbiota metabolite production. Gut 65, 63–72 (2016).
Kim, S. G. et al. Microbiota-derived lantibiotic restores resistance against vancomycin-resistant Enterococcus. Nature 572, 665–669 (2019).
Maini Rekdal, V., Bess, E. N., Bisanz, J. E., Turnbaugh, P. J. & Balskus, E. P. Discovery and inhibition of an interspecies gut bacterial pathway for Levodopa metabolism. Science 364, eaau6323 (2019).
Wikoff, W. R. et al. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl Acad. Sci. USA 106, 3698–3703 (2009).
Showalter, M. R. et al. Obesogenic diets alter metabolism in mice. PLoS ONE 13, e0190632 (2018).
Cajka, T., Smilowitz, J. T. & Fiehn, O. Validating quantitative untargeted lipidomics across nine liquid chromatography-high-resolution mass spectrometry platforms. Anal. Chem. 89, 12360–12368 (2017).
Tsugawa, H. et al. MS-DIAL: data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 12, 523–526 (2015).
Kösters, M. et al. pymzML v2.0: introducing a highly compressed and seekable gzip format. Bioinformatics 34, 2513–2514 (2018).
Wu, M. et al. Genetic determinants of in vivo fitness and diet responsiveness in multiple human gut Bacteroides. Science 350, aac5992 (2015).
Shepherd, E. S., DeLoache, W. C., Pruss, K. M., Whitaker, W. R. & Sonnenburg, J. L. An exclusive metabolic niche enables strain engraftment in the gut microbiota. Nature 557, 434–438 (2018).
Tritsch, G. L. & Moore, G. E. Spontaneous decomposition of glutamine in cell culture media. Exp. Cell Res. 28, 360–364 (1962).
Dodd, D. et al. A gut bacterial pathway metabolizes aromatic amino acids into nine circulating metabolites. Nature 551, 648–652 (2017).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477 (2012).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014).
Medema, M. H., Takano, E. & Breitling, R. Detecting sequence homology at the gene cluster level with MultiGeneBlast. Mol. Biol. Evol. 30, 1218–1223 (2013).
Acknowledgements
We thank C. Khosla, Y. Dai and the Stanford ChEM-H Metabolomics Knowledge Center for use of the LC–MS-qTOF instrument; A. Shiver, K. C. Huang and A. Cheng for sharing bacterial strains; T. Meyers and T. Cowan for sharing chemical standards; H. C. Wastyk and G. K. Fragiadakis for sharing their metabolite data before publication; J. K. Yang for consultation on web design tools; and T. Le for discussion on MS methods. This work is supported by R01-DK085025, DP1-AT009892, R01-DK101674, gifts from M. and J. Pasquesi, H. Buhr and J. Feiber, a Stanford Discovery Innovation Fund Award (J.L.S.), Chan Zuckerberg Biohub (M.A.F. and J.L.S.), DP1-DK113598 and P01-HL147823 (M.A.F.), K08DK110335 (D.D.), Stanford Dean’s Postdoctoral Fellowship and NRSA F32AG062119 (S.H.), NSF-GRFP DGE-114747 (W.V.T.) and 5T32AI007328-32 (L.G.).
Author information
Authors and Affiliations
Contributions
S.H., W.V.T., D.D., M.A.F. and J.L.S. designed this study. S.H., W.V.T. and S.K.H. performed all bacterial culture and gnotobiotic mouse experiments. S.H. constructed MS qTOF and QE m/z-RT and MS2 spectrum libraries, performed MS experimental validation and MS-DIAL data analysis, conducted comparative genomics and metabolomic distance analyses, constructed the in vivo metabolomics pipeline and database, and designed the Metabolomics Data Explorer. W.V.T. constructed the bacterial strain library, performed phylogenetic analysis, built the in vitro bioinformatics pipeline and database, performed phylogenetic versus metabolomic distance comparisons, and built random forest models. C.R.F. and L.G. conducted chemoinformatics analyses of reference library compounds. B.D.M. built the strain-resolved comparative genomics database. S.H., B.C.D. and J.M.S. developed qTOF and QE MS2 methods and collected data. D.D., L.A.F. and C.R.F. set up the MS1 MS methods. S.H., D.D. and L.A.F. built the authentic compound collection and developed metabolomics sample preparation methods. All authors provided intellectual contributions. S.H., W.V.T. and J.L.S. wrote the paper, and all authors provided feedback. S.H. and W.V.T. contributed equally with author order determined by coin-flip.
Corresponding authors
Ethics declarations
Competing interests
M.A.F. is a co-founder and director of Federation Bio and Viralogic, a co-founder of Revolution Medicines, and a member of the scientific advisory boards of NGM Bio and Zymergen.
Additional information
Peer review information Nature thanks Gary Siuzdak and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Summary statistics for the MS reference library metabolites, their detection and validation.
a, Chemical similarity network of the compound library. Network nodes, library compounds coloured by their superclasses. Node size, monoisotopic mass. Edges between nodes, substructure similarity values above a z-score threshold of 1 s.d. from the mean. b, Scatter plots and histograms of chemical properties of 833 library metabolites. c, Venn diagram of library compounds that are detected by each of the three methods. d, Venn diagram of compounds (by PubChem CID) identified in the reference compound library (Supplementary Table 1), in vitro conditions (Supplementary Table 7, ‘count.ps’) and in vivo conditions (Supplementary Table 8, ‘istd_corr_ion_count_matrix’). In vitro conditions include all medium types, and in vivo conditions include all sample types: urine, serum, faeces and caecal contents, and all colonization states. e, Scatterplot of all pairwise similarity scores (biological sample versus library) of the same compound searched against the MoNA spectrum database. All library standards (median similarity score = 992) and 97.3% of the corresponding compounds from biological samples (median similarity score = 923) had similarity scores of ≥600, and 2.7% of those compounds from biological samples scored below 600. Confidence levels were determined based on both similarity scores and visual validation of the MS/MS spectra. f, Schematic of the data collection and analysis workflow of the metabolomics pipeline. Panel created with Biorender.com.
Extended Data Fig. 2 Schematic of a custom bioinformatics analysis pipeline that generates a metabolite fold-change matrix.
The pipeline integrates data across multiple experimental runs and minimizes intra-replicate, intra-experiment and inter-experiment variability. The four steps detailed here are explained in depth in the Supplementary Methods (see ‘Custom bioinformatics: in vitro pipeline’ section). Step 1, a database recording sample metadata (organism, media, growth data, and so on) and MS-DIAL output files are integrated into data matrices that are specific to each analytical method. Step 2, all data are grouped by replicate (biological sample groups (BSGs)) and analysed to remove replicates with low intra-replicate correlation. Replicates are then grouped by experiment (EXPs) to assess inter-experiment variability. Transformations reducing inter-experiment variability are identified and compared. For metabolites that are detected by multiple methods, their ion counts are compared on a per-replicate and per-experiment basis to identify one or more methods that consistently detect these metabolites. Step 3, using an internal standard-based correction, ion counts for individual samples are adjusted and transformed into different fold-change data matrices. Step 4, data matrices corresponding to each method are combined into a single data matrix representing all detected metabolites.
Extended Data Fig. 3 High-throughput identification and analysis of diverse metabolites in complex biological matrices.
a, Number of unique compounds (by PubChem CID) within distinct chemical superclasses detected in the m/z-RT reference library (n = 815, 11 superclasses), in vitro dataset (n = 458, 9 superclasses) or in vivo dataset (n = 551, 9 superclasses), excluding internal standards. Nine of the eleven chemical superclasses in the reference library are represented in the metabolites detected in vitro and in vivo. The two remaining library superclasses (organosulfur and organometallic compounds) not represented in the experimental data contain one compound each. b, Diverse classes of metabolites identified in the conventional mouse caecum. Representative metabolites shown are significantly elevated (≥4-fold, corrected P < 0.05) in conventional mice versus germ-free controls in one experiment with n = 3 (conventional) and n = 4 (germ-free) mice. P values were calculated using two-tailed Student’s t-tests with Benjamini–Hochberg correction for multiple comparisons. c, Examples of precursors, intermediates and products from the tryptophan fermentation pathway that were identified by our methods both in vitro (C. sporogenes culture supernatant) and in vivo (C. sporogenes mono-colonization caecal contents). Extracted ion chromatogram peaks representing relative ion counts for each metabolite are shown. d, e, Histograms of changes in RT (d) and total ion count (e) for 132 spike-in metabolites in five complex biological matrices using three analytical methods. All spiked-in metabolites show minimal change in RT, falling within a conservative ±0.1-min search window from their RTs as determined in the library control condition (d). The majority of spiked-in metabolites (for example, 97% in faeces) exhibit less than fourfold change in ion counts relative to those detected in the library control condition (e). Representative examples of RT shifts (d) and changes in total ion counts (e) in individual metabolites in the mouse faecal matrix are shown. Data are mean ± s.e.m. of one experiment with n = 3 biological replicates. f, Histograms of linear ranges of 377 reference library metabolites measured in serial dilutions. A representative linear range of 5-hydroxyindole is shown. g, Violin plots (median, quartiles) of differences in RTs measured by three analytical methods between distinct MS instruments: the qTOF 6454, with which the library was built, was compared with a second instrument: a qTOF 6530 for a shared panel of 219 reference library metabolites (top) or a Orbitrap QE for a shared panel of 773 reference library metabolites (bottom). Mean RT differences (in min) between two instruments by each method (C18-positive, C18-negative, HILIC-positive, respectively) were as follows: qTOF versus qTOF, pre-correction: 0.238, 0.044, −0.110; post-correction: −0.023, −0.020, 0.015; qTOF versus QE, pre-correction: 0.151, 0.027, 0.196; post-correction: −0.040, −0.021, 0.026). Per method, RT correction was performed by polynomial transformation of the library based on inter-instrumental RT shifts of 10–20 robustly detected metabolites. Per method, using the corrected library with a RT tolerance window of 0.2 min, around 99% of the 219 metabolites tested on the second qTOF and about 94% of the 773 metabolites tested on the QE were correctly identified.
Extended Data Fig. 4 Conserved and unique metabolomic signatures across bacterial taxa.
a, Schematic of our high-throughput bacterial culture and sample collection workflow. Panel created with Biorender.com. b, Intra-replicate Pearson correlation coefficients (triplicates and greater) stratified by fourteen independent bacterial culture experiments and three analytical methods. For each experiment, Pearson correlation r values were calculated for all supernatant and medium sample replicate groups: n = 346 (C18-positive), n = 344 (C18-negative) and n = 344 (HILIC-positive). Total ion count data were corrected by internal standards and log-transformed, standardized and scaled, before computing Pearson correlation values. Box, median, 25th and 75th percentiles; whiskers, Tukey’s method. c, Left, number of medium-specific or common metabolites detected in the same bacterial strain grown in two different media (29 strains cultured in two or more of the 12 different media). Each dot represents the total number of metabolites from a single comparison between two media in which a strain has been grown: n = 58 (co-detected in two media), n = 116 (detected in one of the two media), n = 33 (detected in the mega medium) and n = 16 (detected in polyamine-free medium). Box, median, 25th and 75th percentiles; whiskers, minimum and maximum. Right, agmatine production levels by B. eggerthii. Data are mean ± s.e.m. from 2–3 independent experiments, each with n = 3 biological replicates. P values, two-tailed t-test with Benjamini–Hochberg correction for multiple comparisons. d, Heat map of metabolomic profiles of 158 bacterial strains grown in mega medium, clustered by 16S phylogenetic distance. Individual metabolites are hierarchically clustered (Ward’s method) using Euclidean distance between the fold-change (log2-transformed) values across all taxonomies. Metabolites shown are detected in at least 50% of the 158 taxonomies to enable Ward clustering. e, f, Production or consumption patterns of tyramine and pantothenic acid across 158 strains grown in mega medium. Data are mean ± s.e.m. from 1–3 independent experiments (identified by dot colour), each with n ≥ 3 biological replicates.
Extended Data Fig. 5 Metabolic profile variation among related bacteria.
a, Pairwise metabolomic profile comparisons between two closely related strains grown in mega medium: C. sporogenes ATCC 15579 and C. cadaveris HM-1039 (subpanel 1), and among four strains of Bacteroides fragilis (subpanels 2–7): HM-710, HM-711, HM-714 and HM-20. Each dot represents an averaged fold-change value (log2-transformed) from 1–3 independent experiments, each with n = 3 biological replicates. Pearson correlation r values of pairwise metabolomic profile comparisons, performed on standardized and scaled data: ATCC 15579 versus HM-1039 (r = 0.063), HM-711 versus HM-710 (r = 0.859), HM-714 versus HM-710 (r = 0.866), HM-714 versus HM-711 (r = 0.880), HM-20 versus HM-710 (r = 0.829), HM-20 versus HM-711 (r = 0.845) and HM-20 versus HM-714 (r = 0.807). b, Metabolic similarities and variations among closely related species of C. sporogenes and C. cadaveris, and among different strains of the same species of B. fragilis grown in mega medium. Taxonomies shown are clustered by 16S phylogenetic distance, and are coloured according to the distinct phyla. Data are mean ± s.e.m. from 1–3 independent experiments, each with n = 3 biological replicates.
Extended Data Fig. 6 Relationships between phylogeny, taxonomy and metabolome.
a, Metabolomic profiles of 158 bacterial strains grown in mega medium. Individual taxonomies are clustered by metabolomic profile distances (fold change, log2-transformed) across all metabolites. Individual metabolites are hierarchically clustered (Ward’s method) using Euclidean distance between the fold-change (log2-transformed) values across all taxonomies. Metabolites shown are detected in at least 50% of the 158 taxonomies to enable Ward clustering. b, Metabolic similarities between two phylogenetically distant species grown in mega medium. Taxonomies are clustered by metabolomic profile distances (fold change, log2-transformed) across all metabolites. Data are mean ± s.e.m. of one experiment with n = 3 biological replicates. c, Scatter plot of pairwise metabolomic profile comparison between two phylogenetically distant species. Each dot represents an averaged fold-change value (log2-transformed) of one experiment with n = 3 biological replicates. Pearson correlation of pairwise metabolomic profile comparison between these two species, performed on standardized and scaled fold-change data, r = 0.7090. d, Venn diagram of unique and overlapping compounds (by PubChem CID) identified in the culture supernatant of 158 mega-medium grown strains and caecal contents of conventional mice.
Extended Data Fig. 7 Multiple data transformations identify nonlinear relationship between phylogenetic and metabolomic distance.
a, Heat map showing the comparison of phylogenetic and metabolomic tree topologies. Cells record the number of tips for which the neighbourhoods share more overlap than expected (P < 0.05; one-sided permutation test). Data are stratified by fractional overlap of neighbourhoods and permutation probability (see Supplementary Methods, ‘Distance comparisons’). b, Histogram of chemical similarity scores (based on Tanimoto 2D structures) between each unique pair of compounds (by PubChem CID) detected in the in vitro dataset. For this pairwise comparison, 359 non-co-eluting compounds were used. c, Metabolomic distance tree with each metabolite weighted based on their chemical similarity (left) or unweighted control metabolomic distance tree (right). The weighted and unweighted matrices were calculated using uniquely detected, non-co-eluting compounds in the in vitro dataset, for which a unique PubChem CID identifier can be assigned to each compound. Two-sided Mantel test for comparison between the weighted and unweighted distance matrices: r2 = 0.863, P = 0.001. d, Left, correlation of phylogenetic and metabolomic distance across pairs of strains coloured by lowest shared taxonomic rank with a LOESS fit shown. Dashed vertical line occurs at x = 0.11 as referenced in the text. Right, Metabolomic distance between pairs of strains binned by the lowest shared taxonomic rank. Species (n = 111), genus (n = 1,386), family (n = 159), order (n = 1,222), class (n = 34), phylum (n = 1,442) and kingdom (n = 8,442). Box, median, 25th and 75th percentiles; whiskers, Tukey’s method. e–i, Internal-standard-corrected fold-change data (e–g) and internal-standard-corrected total ion count data (h, i) were log-transformed and used to calculate pairwise metabolomic distances between microbial taxa. These distances were compared to the corresponding pairwise phylogenetic distances generated from a tree built with the V4 region of 16S (left) or the full-length 16S gene (right). Data are plotted with a LOESS fit. Set 1, microorganisms grown in at least one experiment simultaneously. Set 2, microorganisms grown in the same experiment only. j, Phylogenetic tree constructed using the full 16S sequences of a subset of the strains grown in mega medium. Only strains with available full 16S sequences are shown (Supplementary Table 6). k, Left, schematic of the pathway that synthesizes citrulline and ornithine, or synthesizes agmatine and/or putrescine. Right, the top six matches identified by the comparative genomics tool MultiGeneBlast within a 40-kb search window, when searched against a genomic database of our strain collection with sequenced genomes. Horizontal dashed lines between genes represent multiple other genes present within the search window.
Extended Data Fig. 8 Asparagine and glutamine can be used as sole nitrogen sources by most tested Bacteroidetes.
a, Top, an example decision tree from a forest that can differentiate Bacteroidetes versus bacteria from the other four represented phyla with >97% accuracy. For each decision node, phylum-level increases and decreases based on metabolite levels are shown (relative fold change compared to the bacterial medium controls, log2-transformed). Actinobacteria (n = 20), Bacteroidetes (n = 57), Firmicutes (n = 83), Fusobacteria (n = 3) and Proteobacteria (n = 10). Dashed line, metabolite threshold. Box, median, 25th and 75th percentiles; whiskers: Tukey’s method. Bottom, the 10 most important features differentiating the five tested phyla. Data are shown as median metabolite log2-fold-change values for each phylum; metabolites and phyla are ordered by Ward linkage distance. b, Representative growth curves from two independent experiments, each with n = 3 biological replicates for a subset of Bacteroides spp. using modified SMM with the indicated nitrogen source. Legend colours for the sole nitrogen source are the same in b–d. c, Representative growth curves of one experiment with n = 5 biological replicates for 60 Bacteroidetes using modified SMM with the indicated nitrogen sources. d, Growth curves of wild-type and mutant B. thetaiotaomicron (Bt) grown in defined minimal media with either cysteine (top) (one experiment, n = 3 biological replicates) or sodium sulfide (Na2S, bottom) as sole reduced sulfur sources (one experiment, n = 3 biological replicates). e, Amino acid production and consumption levels in gnotobiotic mice mono-colonized with B. thetaiotaomicron (one experiment, n = 5 mice). Box, median, 25th and 75th percentiles; whiskers, Tukey’s method. Numeric labels in b and c correspond to the following: 1, B. acidifaciens DSMZ 15896; 2, B. caccae ATCC 43185; 3, B. caccae BEI HM-728; 4, B. cellulosilyticus BEI HM-726; 5, B. cellulosilyticus DSMZ 14838; 6, B. coprophilus DSMZ 18228; 7, B. dorei BEI HM-29; 8, B. dorei BEI HM-717; 9, B. dorei BEI HM-718; 10, B. dorei BEI HM-719; 11, B. dorei DSMZ 17855; 12, B. eggerthii ATCC 27754; 13, B. eggerthii DSMZ 20697; 14, B. finegoldii BEI HM-727; 15, B. finegoldii DSMZ 17565; 16, B. fragilis BEI HM-20; 17, B. fragilis BEI HM-710; 18, B. fragilis BEI HM-711; 19, B. fragilis BEI HM-714; 20, B. fragilis NCTC 9343; 21, B. intestinalis DSMZ 17393; 22, B. ovatus ATCC 8483; 23, B. ovatus BEI HM-222; 24, B. pectinophilus ATCC 43243; 25, B. plebeius DSMZ 17135; 26, B. salyersiae BEI HM-725; 27, Bacteroides sp. BEI HM-18; 28, Bacteroides sp. BEI HM-189; 29, Bacteroides sp. BEI HM-19; 30, Bacteroides sp. BEI HM-22; 31, Bacteroides sp. BEI HM-23; 32, Bacteroides sp. BEI HM-258; 33, Bacteroides sp. BEI HM-27; 34, Bacteroides sp. BEI HM-28; 35, Bacteroides sp. BEI HM-58; 36, B. stercoris ATCC 43183; 37, B. stercoris BEI HM-1036; 38, B. thetaiotaomicron 3730; 39, B. thetaiotaomicron 3731; 40, B. thetaiotaomicron 633; 41, B. thetaiotaomicron 7330; 42, B. thetaiotaomicron 7853; 43, B. thetaiotaomicron 8702; 44, B. thetaiotaomicron 8713; 45, B. thetaiotaomicron 8736; 46, B. thetaiotaomicron 940; 47, B. thetaiotaomicron VPI 5482; 48, B. thetaiotaomicron WH302; 49, B. thetaiotaomicron WH305; 50, B. uniformis ATCC 8492; 51, B. vulgatus ATCC 8482; 52, B. vulgatus BEI HM-720; 53, B. xylanisolvens DSMZ 18836; 54, P. distasonis ATCC 8503; 55, P. distasonis BEI HM-169; 56, P. johnsonii BEI HM-731; 57, P. johnsonii DSMZ 18315; 58, P. merdae ATCC 43184; 59, P. merdae BEI HM-729; 60, P. merdae BEI HM-730.
Extended Data Fig. 9 Metabolic contribution by individual gut microorganisms in a multi-species community.
a, α-Ketoglutaric acid levels in faeces of mice mono-colonized with Anaerostipes sp. BEI HM-220. Data are mean ± s.e.m. of two independent experiments, each with n = 4 mice (germ-free) or n = 5 or 7 mice (Anaerostipes mono-colonized). b, Left, MDMs were associated with specific bacterial phyla leveraging both in vivo and in vitro metabolomic data. Right, number of bacterial strains grown in mega medium by phylum that produce MDMs identified in the caecal contents of mice colonized with B. thetaiotaomicron (Bt, n = 5) or C. sporogenes (Cs, n = 3), or with a six-member community (n = 3). Numbers of strains that produce at least one of these metabolites in vitro by phylum: Bacteroidetes, n = 52; Firmicutes, n = 60; Proteobacteria, n = 8; Actinobacteria, n = 16; and Fusobacteria, n = 3. Each metabolite shown was significantly produced both in vitro and in vivo (≥4-fold, corrected P < 0.05). Uniquely detected (non-co-eluting) metabolites are shown (Supplementary Table 9). c, Spearman correlation between metabolomic profiles (standardized and scaled, log2-transformed, fold-change data) of individual B. thetaiotaomicron- or C. sporogenes-mono-colonizesd host biofluids (caecal contents, faeces, serum or urine) and individual bacterial culture (158 strains grown in mega medium). Coloured dots, Spearman’s ρ values calculated by comparing metabolomic profiles of individual bacterial culture versus individual biofluid of either B. thetaiotaomicron- or C. sporogenes-mono-colonized mice. Black dots, Spearman’s ρ calculated using metabolomic profiles of B. thetaiotaomicron or C. sporogenes, the same strains used for mono-colonization in mice. d, Venn diagram of overlapping metabolites that are significantly produced (≥4-fold, corrected P < 0.05) in culture and in the caecum of colonized mice. e, Principal component analysis separates metabolomic profiles of identified metabolites by sample type in each colonization state. P values on metabolomic profile comparisons between different sample types of the same colonization state were determined using PERMANOVA: six-member community (P = 0.073) and all other colonization states (P = 0.001). f, Principal component analysis separates metabolomic profiles of identified metabolites by colonization states. P values on metabolomic profile comparisons between different colonization states of the same sample type were determined using PERMANOVA: P = 0.001 for all four sample types. g, h, Example chemical structures of significantly produced metabolites (≥4-fold, corrected P < 0.05) in serum (g) or urine (h) by each colonization state corresponding to Fig. 4b. a, b, d, g, h, P values were determined using two-tailed Student’s t-tests with Benjamini–Hochberg correction for multiple comparisons.
Extended Data Fig. 10 Metabolic contribution of multi-species communities in gnotobiotic mice.
a, Proposed host–microbial co-metabolism pathways that could lead to the synthesis of specific host–microbial co-metabolites in the urine and serum of mice colonized with the six-member community. b, c, Metabolite levels in urine (b) and caecal contents (c) of mice colonized with the six-member community (+Cs) or the five-member community (−Cs). Metabolites shown represent a panel of significantly elevated or reduced metabolites (≥4-fold, corrected P < 0.05) in the six-member community. Superscript ‘1’ in metabolite names, co-eluting metabolites as annotated in the MS reference library (Supplementary Table 1). Superscript ‘2’ in metabolite names, co-eluting isomeric metabolites with truncated names in the figure (2-hydroxy-3-methylpentanoic acid, 2-hydroxy-4-methylpentanoic acid; and α-galactose 1-phosphate, α-glucose 1-phosphate, glucose-6-phosphate, mannose 6-phosphate). Data are mean ± s.e.m. of one experiment with n = 6 (urine, six-member community), n = 7 (urine, five-member community) and n = 3 (caecal, both six-member and five-member communities). b, c, P values were calculated using two-tailed Student’s t-tests with Benjamini–Hochberg correction for multiple comparisons. *P < 0.05, **P < 0.01, ***P < 0.001. b, Venn diagram (right) of significantly elevated and reduced metabolites in individual host biofluids (caecal contents, serum and urine) using the same threshold in b (left).
Supplementary information
Supplementary Information
This file contains Supplementary Methods and a full guide to Supplementary Tables 1-9. The Supplementary Methods include mass spectrometry LC/MS methods, data analysis, bacterial sequencing and phylogenetics, distance comparisons and classifiers, the Mega Medium (MM) preparation protocol and Salyer’s Minimal Medium (SMM) preparation protocol.
Supplementary Table 1
Mass spectrometry compound m/z-RT reference library.
Supplementary Table 2
MS/MS validation of biological metabolites and library standards, matrix effects on ion count and RT, and linear range of individual metabolites.
Supplementary Table 3
MS/MS spectra library constructed on qTOF and QE instruments.
Supplementary Table 4
Inter-instrumental retention time shift and correction.
Supplementary Table 5
In vitro sample metadata.
Supplementary Table 6
Culture and strain information.
Supplementary Table 7
In vitro data matrices.
Supplementary Table 8
The in vivo database and in vivo pipeline output data matrices.
Supplementary Table 9
List of “phylum-associated” metabolites.
Rights and permissions
About this article

Cite this article
Han, S., Van Treuren, W., Fischer, C.R. et al. A metabolomics pipeline for the mechanistic interrogation of the gut microbiome. Nature 595, 415–420 (2021). https://doi.org/10.1038/s41586-021-03707-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-021-03707-9
- Springer Nature Limited
This article is cited by
-
Resource competition predicts assembly of gut bacterial communities in vitro
Nature Microbiology (2024)
-
The microbial metabolite agmatine acts as an FXR agonist to promote polycystic ovary syndrome in female mice
Nature Metabolism (2024)
-
Western diets and chronic diseases
Nature Medicine (2024)
-
High-throughput identification of gut microbiome-dependent metabolites
Nature Protocols (2024)
-
A multi-glycomic platform for the analysis of food carbohydrates
Nature Protocols (2024)